
Abstract
Background
The mitochondrial genome of Metazoa is usually a compact molecule without introns. Exceptions to this rule have been reported only in corals and sea anemones (Cnidaria), in which group I introns have been discovered in the cox1 and nad5 genes. Here we show several lines of evidence demonstrating that introns can also be found in the mitochondria of sponges (Porifera).
Results
A 2,349 bp fragment of the mitochondrial cox1 gene was sequenced from the sponge Tetilla sp. (Spirophorida). This fragment suggests the presence of a 1143 bp intron. Similar to all the cnidarian mitochondrial introns, the putative intron has group I intron characteristics. The intron is present in the cox1 gene and encodes a putative homing endonuclease. In order to establish the distribution of this intron in sponges, the cox1 gene was sequenced from several representatives of the demosponge diversity. The intron was found only in the sponge order Spirophorida. A phylogenetic analysis of the COI protein sequence and of the intron open reading frame suggests that the intron may have been transmitted horizontally from a fungus donor.
Conclusion
Little is known about sponge-associated fungi, although in the last few years the latter have been frequently isolated from sponges. We suggest that the horizontal gene transfer of a mitochondrial intron was facilitated by a symbiotic relationship between fungus and sponge. Ecological relationships are known to have implications at the genomic level. Here, an ecological relationship between sponge and fungus is suggested based on the genomic analysis.
Similar content being viewed by others


Background
Sponges (Porifera) are the first diverging metazoans. They are thus a key phylum in the understanding of the genomic characteristics of the metazoan ancestor [1]. For example, recent findings indicate that the sponge mitochondria possess ancestral characters that have been lost in other metazoans, such as additional genes, minimally modified genetic code, or bacteria-like rRNA structure [2, 3]. One intriguing finding is that most metazoan mitochondrial genomes lack introns. Mitochondrial introns may be present in large numbers and in many genes in the sister clades of Metazoa: Choanoflagellida [4], Ichthyosporea [4] and Fungi [5]. For example, the mitochondrial genome of the fungi Podospora anserina (accession number: NC_001329) contains 33 introns located in nine different genes, including 15 introns in the cox1 gene, which encode the subunit 1 of cytochrome c oxidase (COI). However, none of these introns are obligatory and some fungi do not include introns in their mitochondrial genome (e.g., Schizophyllum commune, NC_003049; Harpochytrium sp. NC_004760 and NC_004623). In Metazoa, mitochondrial introns have only been described in Cnidaria of the subclass Zoantharia [6, 7]. In sea anemones (order Actinaria), two mitochondrial introns have been found, one in the cox1 gene and one in the NADH dehydrogenase subunit 5 gene (nd5). In stony corals (order Scleractinia), however, only the nd5 gene contains an intron. The relative position of this intron is conserved between sea anemone and stony corals. Other cnidarians (e.g., jellyfish, hydras) do not seem to possess mitochondrial introns [8, 9].
Mitochondrial introns are self-splicing ribozymes. Self-splicing introns are divided into either Group I or Group II depending on their secondary structure. While Group II introns are prevalent in plants, the mitochondrial introns of Cnidaria, Choanoflagellida and Ichtyosporea are all of group I. In the mitochondria of fungi both types of introns can be found, though group I is more prevalent [5]. Self-splicing introns are mobile genetic elements [5, 10, 11]. They often encode homing endonucleases and/or maturases. Homing endonucleases cleave chromosomes and exploit the recombinational repair system of the cell for their multiplication. Maturases act as cofactors that bind the precursor RNA containing their intron to facilitate its folding and splicing [12]. It should be noted that enzymes of the LAGLIDADG family (which are frequently encoded within group I introns) can function as endonuclease, as maturase, or perform both functions. However, not all introns encode homing endonuclease or maturase. For example, the peculiar intron located in the nd5 gene of cnidaria does not encode a homing endonuclease, although it encodes other mitochondrial genes [6, 7].
Although three mitochondrial genomes of sponges have been recently sequenced [2, 3], no intron was found in these genomes. We report here that a sponge mitochondrial gene contains a group I intron, which encodes a putative LAGLIDADG member. We also provide phylogenetic evidence suggesting that the sponge intron was acquired by horizontal gene transfer.
Results
We amplified cox1 genes from nine demosponge species. All sponge species yielded similar cox1 PCR products (1206 bp) except Tetilla sp. (Spirophorida), whose product was much longer (2349 bp). Sequencing this gene revealed a putative intron of 1143 bp. This suggests that introns can also be found in the mitochondrial genome of sponges. Interestingly, the intron was located in the middle of the reverse primer used by Nichols et al. [13] to amplify cox1 gene of sponges. It is highly unlikely that the Tetilla cox1 sequence is a nuclear copy or a contamination, for three reasons: i. we extracted enriched mitochondrial DNA to avoid amplification of nuclear copies of mitochondrial sequences (Numts) [14]; ii. no frameshift mutations were noticeable in the cox1 sequence; and iii. an identical sequence was obtained from two individuals collected from separate locations.
Characteristics of the predicted Tetilla sp. intron
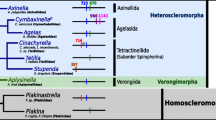
A study of the relative position of introns in the cox1 gene (Figure 1) revealed that the Tetilla sp. intron is inserted at position 850 of the alignment provided in Additional file 1. This insertion point corresponds to positions 672–673 of the cox1 coding sequence (CDS) of Tetilla. Unlike the Tetilla intron, the sea anemone intron is located at position 1017 of the alignment (positions 835–836 of the cox1 CDS of Tetilla). In fact, no intron (among fungal, choanoflagellate and ichtyosporean cox1 sequences) was found to be inserted at exactly the same nucleotide position as the predicted sponge intron. However, introns are located at positions 834, 846 and 859 of the alignment (i.e., 8 bp after the Tetilla intron insertion point), thus suggesting a hot spot for intron insertion (Figure 1).

Relative location of introns in the cox1 gene. DNA sequences were aligned base on protein sequence alignment. cox1 sequences are indicated by boxes. Intron locations are indicated by vertical bars. Numbers indicate the relative position of the introns in the alignment and not their position in their corresponding sequence. All introns are group I introns in these taxa. Introns aligned in Figure 3 are indicated in red. Introns containing a putative LAGLIDADG homing endonuclease are in black or red. Introns containing a putative GIY-YIG homing endonuclease are in green. Introns containing an unknown ORF are in blue. The alignment is provided in Additional file 1.
Secondary structure predictions show that the Tetilla intron can be folded into a canonical group I intron, except for the absence of the paired region P2 (Figure 2). A BLAST analysis reveals that the predicted intron sequence shows low primary sequence similarity with sequences present in the data banks. However, the conserved P, Q, R, and S regions, which form the main core of group I introns, can be aligned between the Tetilla intron and fungal mitochondrial introns. In Figure 3 we compare the core region of the Tetilla intron with the core region of eight other introns. Five of these introns were chosen because their encoded LAGLIDADG was closely related to the Tetilla's LAGLIDADG (see below). We also included in the comparison the three available animal introns with published secondary structure [6, 7]. Interestingly, the introns that include closely related LAGLIDADGs (i.e., Agrocybe aegerita cox1 intron 2, Emericella nidulans cox1 intron 3, Schizosaccharomyces pombe cox1 intron 2, Penicillium marneffrei cox1 intron 3) do not include the paired region P2. Unfortunately, the presence or absence of P2 could not be reliably determined for the cox1 intron 4 of Smittium culisetae. The absence of P2 paired-region in closely related introns suggests that the absence of P2 is genuine in the Tetilla intron and not an artifact produced by the method used. The cnidarian introns, in contrast, contain a P2 region, suggesting that they are less related to the Tetilla intron than the fungal introns mentioned above.

Predicted secondary structure of the cox1 intron of Tetilla sp. The exon and intron sequences are in lower-case and in upper-case, respectively. The conserved sequences (P, Q, R, S) of the intron core and the base-paired regions P1–P9 are shown according to the standard scheme for group I introns [53].

Alignment of the Tetilla intron with introns that include closely-related LAGLIDADG and with other metazoan introns. The conserved regions P, Q, S, R are indicated by yellow boxes; the base-paired regions P1–P9 are underlined; brackets indicate the number of nucleotides; blue boxes indicate the location of the ORF. The core regions and ORF positions of Agrocybe aegerita cox1 intron 2, Emericella nidulans cox1 intron 3, Schizosaccharomyces pombe cox1 intron 2, Penicillium marneffrei cox1 intron 3, Metridium senile cox1 and nd5 introns, and Acropor a tenuis nd5 intron were derived from publications [6, 7, 54–57]. The core region of the cox1 intron 4 of Smittium culisetae was determined using the program CITRON [45]. As suggested in the program manual the parameters that constrain the minimal definition of (P3, P3') were lowered, since no putative intron was found using the default parameters. Specifically, T10 was set to 3 and T11 to 15. The location of the P1 paired region and the presence or absence of the P2 paired region were not determined for Smittium.
Mitochondrial introns often encode various proteins [7, 15]. Hence, the predicted Tetilla intron was translated in all six reading frames using the coelenterate/mold mitochondrial genetic code (i.e., the genetic code of sponge mitochondria). The translation revealed an open reading frame (ORF) of 1029 bp starting from the first nucleotide of the intron (the initiation codon is TTA). The main difference between the standard and the sponge mitochondrial genetic codes is that TGA codes for a stop codon in the standard genetic code while in the sponge mitochondria it codes for tryptophan. The intron ORF includes three TGA codons in positions 100, 169, and 631 of the intron sequence, indicating that the coding sequence of the intron is not of a nuclear origin.
The BLAST analysis of the ORF suggests that it encodes an enzyme from the LAGLIDADG endonuclease-maturase family. LAGLIDADG is the largest family of homing endonucleases, whose members are characterized by the presence of the conserved motif LAGLIDADG in one or two copies. Crystal structures of LAGLIDADG endonucleases-maturases have revealed that single-motif proteins function as homodimers while double-motif enzymes are monomers [10]. Two LAGLIDADG motifs were identified in the Tetilla ORF (LAGLIEGDG and LAGFLDADG) located in positions 101–109 and 212–220 of the ORF protein sequence.
Phylogenetic tree of the COI protein sequence
To confirm that the Tetilla sequence was not a fungal contamination, a phylogenetic tree of Metazoa and its closest sister clades (i.e., Fungi and Choanoflagellida) was reconstructed based on COI protein sequences (Figure 4) and was rooted with fungal sequences. The resulting phylogeny is in agreement with previous phylogenetic trees based on mitochondrial sequences [2]. As expected, animals are monophyletic (Bootstrap percentage, BP = 100, Posterior probability PP = 1.0) and divided into two clades: Diploblastica (Porifera+Cnidaria; BP = 99, PP = 1.0) and Bilateria (BP = 100, PP = 1.0). The monophyly of diploblasts contradicts the RNA-based phylogenies that place sponges at the base of Metazoa [16–19]. The monophyly of diploblasts is likely to be the consequence of a long branch artefact resulting from the high rate of evolution of Bilateria [2]. The relationships within these two clades are not highly supported. Bilateria are divided into Protostomia (BP = 52 PP = 0.74) and Deuterostomia (BP = 67 PP = 0.98). Diploblasts are divided into Cnidaria (BP = 89 PP = 1.0) and Demospongiae (BP = 71). The Bayesian reconstruction does not support the monophyly of Porifera but instead places Cnidaria as the sister clade of Xestospongia (PP = 0.56). Thus, although the Bayesian and Maximum Likelihood (ML) trees support slightly different topologies, those differences only involve weakly supported nodes (i.e., nodes with BP<50% or PP<0.95; data not shown). Among Porifera, Tetilla sp. (order Spirophorida) clusters with Geodia (order Astrophorida) with high support values (BP = 100 PP = 1.0). This relationship is in agreement with traditional morphology-based classification and rRNA sequence analyses, which group the orders Astrophorida and Spirophorida together [20–22]. Sponge relationships show very low support value. Among the three orders represented by two species each (Poescilosclerida, Hadromerida, Verongida) only Verongida appears as monophyletic (BP = 100 PP = 1.0). Because relationships within sponges are not highly supported in both ML and Bayesian analyses, it is unknown whether the paraphyly of these genera reflects sponge history or lack of phylogenetic signal in the COI sequences.

Maximum likelihood phylogenetic trees of COI protein sequence. Support values are indicated near corresponding nodes in the following order: ML bootstrap support/Bayesian posterior probabilities. Sponge taxa are indicated by grey rectangles and species with one or more introns in their mitochondrial genome are indicated in bold. The accession numbers of the sequences are given in Table 1.
Phylogenetic tree of the LAGLIDADG
LAGLIDADG genes are present in all branches of the tree of life. Among eukaryotes they are widespread in the chloroplast and mitochondrial genomes and usually associated with group I introns although they have also been found to be encoded by group II introns [23]. They are less common in the nuclear genome and in this case they are associated with self-splicing inteins rather than introns [10, 15, 24]. The most similar sequences to the intron ORF of Tetilla, according to the BLAST search, are the LAGLIDADG sequences located in the cox1 gene of fungi. Among the 85 non-identical sequences analyzed, only 17 sequences (20%) were not cox1 sequences (Figure 5). These 17 sequences originated either from chloroplastic introns of algae (6 sequences) or from mitochondrial introns located in the NADH dehydrogenase subunit 5 (nd5; 8 sequences), small-subunit rRNA (srrna; 1 sequence), ATP synthase subunit 1 (atp1; 1 sequence), and cytochrome b (cytb; 1 sequence) genes. Similarly, only 16 sequences did not originate from fungi but from green algae (9 sequences), Embryophyta (3 sequences), Choanoflagellida (2 sequences), Ichthyosporea (1 sequence), and Metazoa (1 sequence).

Maximum likelihood phylogenetic trees of LAGLIDADG protein sequences. The circular tree presentation has been adopted to highlight the fact that the tree is unrooted. Most of the sequences were located in introns present in mitochondrial cox1 genes of Fungi. Sequences originating from introns located in other mitochondrial genes are indicated by white circles and chloroplast sequences are indicated by black circles. The gene at the origin of these sequences is indicated after the species name. Black triangles characterize animals and their closest outgroups (i.e., Monosiga brevicollis, Choanoflagellida; and Amoebidium parasiticum, Ichthyosporea). White triangles characterize plant sequences and grey squares indicate sequences from green algae. All the remaining sequences are from fungi. Stars indicate nodes supported by a ML bootstrap support (BP) ≥ 50% and a Bayesian posterior probability (PP) ≥ 0.90. Support values are indicated for the key node Tetilla + Smittium.
Phylogenetic reconstructions indicate a complicated evolutionary history for these homing endonuclease sequences (Figure 5). Because the LAGLIDADG protein tree has little to do with the species tree it was impossible to define an outgroup. In the tree, the predicted Tetilla sequence is associated with the LAGLIDADG sequence located in the 4th cox1 intron of Smittium culisetae. However, these two sequences show only 57% of identity in the conserved part of the LAGLIDADG alignment and the support values are weak for this grouping (BP = 54 and PP = 0.94). Similarly, the LAGLIDADG sequence present in the sea anemone Metridium senile does not show any close relationship with any of the sequences present in the data bank. More generally, intron sequences present in the same gene or in closely-related organisms do not form monophyletic groups. For example, neither chloroplast sequences nor animal sequences clustered together. Only the plant sequences (BP = 100, PP = 1.0) and five nd5 sequences (BP = 97, PP = 1.0) form two coherent groups.
Discussion
Insertion and deletion of mobile introns are common evolutionary events resulting in a sporadic distribution of these elements [5]. Consequently, the presence of an intron in a lineage but not in its sister clades can always be explained by independent losses. However, three lines of evidence suggest that the predicted Tetilla sp. intron arose by horizontal gene transfer rather than by independent losses. First, parsimony analysis of intron presence and absence favors a scenario of a recent intron introduction in Spirophorida since all other ten sponges for which the cox1 gene was determined lack an intron (Figure 4). Second, the cnidarian intron is not located at the same position as the sponge intron, suggesting an independent evolutionary origin (Figure 1). Third, phylogenetic analyses clearly show that the intron-encoded LAGLIDADG sequence and the cox1 exonic sequence share different phylogenetic histories (Figures 4, 5). The Tetilla COI protein is of sponge origin and its phylogeny agrees with previous sponge molecular phylogeny [21, 25], while the LAGLIDADG-based phylogenetic tree indicates that the Tetilla intron ORF is closer to fungal than to cnidarian or choanoflagelate sequences (Figure 5). Thus, the intron might be of fungal origin.
It is unlikely that the intron was transferred from the nuclear genome. The ORF can be translated with the sponge/mold/cnidarian mitochondrial genetic code but not with the nuclear genetic code. The mitochondrial origin of this sequence is also supported by the fact that the ORF sequence is more similar to cox1 LAGLIDADG than to ribosomal LAGLIDADG (Figure 5).
We predicted the first sponge mitochondrial intron and our phylogenetic analyses indicate that it might have a fungal origin. A fungal origin implies that the sponge and the donor interacted in such a way as to allow the transfer of the intron. This suggests the existence of a symbiosis between Tetilla sp. and a fungus donor of the intron.
There is an increasing interest recently in marine fungi as a source of novel bioactive-compounds [26]. More than 500 species of marine fungi, mainly Ascomycota, have been described [27, 28] and the number is constantly rising [29–31]. Unfortunately, no cox1 gene has yet been sequenced from marine derived fungi. In the marine environment, fungi have been isolated from sediments, algae, plants, fish, crabs, tunicates, corals, and sponges [26, 32]. In spite of the fact that many fungi had been isolated from sponges [32–35], the existence of a sponge-fungus symbiosis is under debate. No fungi had been observed within a sponge and it was therefore supposed that only dormant fungi propagules are present within sponges. The first clear case of an endosymbiotic yeast was recently discovered in sponges of the genus Chondrilla [36]. Additionally, another recent molecular study gave the first proof that sponges have the ability to recognize fungi in their surrounding environment [37]. Our results thus introduce additional evidence in favor of a sponge-fungus symbiosis.
Because horizontal gene transfers of group I introns encoding LAGLIDADG are frequent among fungi, we could not determine which lineage of fungi was at the origin of the sponge intron. The sponge LAGLIDADG sequence clusters with the LAGLIDADG present in the fourth cox1 intron of Smittium culisetae. However, the location of the LAGLIDADG ORF is different in these two introns (Figure 3). Most of the ORF is located in the paired region P8 in Tetilla while in Smittium it is located before the paired region P3. This suggests that the ORF and the rest of the intron (i.e., the ribozyme component) have independent origins, thus complicating our understanding of the sponge intron origin. Because the diversity of marine organisms is still poorly known and because sponges are remarkable for their widespread symbiosis with various organisms [38, 39], we cannot exclude the hypothesis that an unknown unicellular eukaryote was the donor of the intron. The accumulation of new data on marine fungi mitochondrial genomes is likely to shed additional light on the sponge intron origin and perhaps also on the origin of the LAGLIDADG sequence in Cnidaria.
Conclusion
Our analysis suggests that a cross-kingdom horizontal gene transfer event occurred in the sponge mitochondrial genome. Such events are remarkable from the evolutionary point of view, because they demonstrate an unexpected plasticity of the mitochondrial genomes of basal Metazoa compared to the more conserved genomes of Bilateria. Porifera and Cnidaria mtDNAs have been characterized by the presence of additional horizontally-transferred genes [6, 40], introns [6, 7], tRNA duplications [3], and tRNA losses [7, 41]. Our results suggest that a better sampling of these animals might improve our understanding of the evolution of this genome.
There are many exciting evolutionary events in marine organisms that are only now starting to be discovered, and these events will provide new insights concerning the evolution of the animal kingdom. Ecological relationships are known to have implications at the genomic level. Here, an ecological relationship between a sponge and a fungus (or an unknown eukaryote) is suggested, based on the genomic analysis.
Methods
DNA extraction and amplification
Eight sponge species were collected and identified by traditional morphological taxonomy based on their general morphology and skeletal organization. Total DNA extractions from the sponge species Xestospongia proxima, Biemna fistulosa, Cliona sp., Verongula giganthea, and Aplysina lacunosa were performed following Steindler et al. [42]. For Tetilla sp., Negombata magnifica, and Chondrosia reniformis, an enriched fraction of mitochondrial genomes was extracted following Arnason et al. [43]. This protocol reduces the chance of Numt contamination.
The primers LCO1490 [44] and COX1-R1 (5'-TGTTGRGGGAAAAARGTTAAATT-3') were used to amplify the cox1 gene. The conditions of PCR amplifications were: 1 cycle at 94°C for 2 min, 50°C for 1 min, 72°C for 2 min; 30 cycles at 94°C for 50 sec, 50°C for 50 sec, 72°C for 2 min; and a final elongation at 72°C for 10 min. Amplified fragments were directly sequenced on an ABI PRISM 3100 (Applied Biosystems). Internal primers are provided in Additional file 2.
Intron structure
Intron location and structure were inferred in silico. The core structure of the intron was inferred with the program CITRON [45] and manually aligned with fungal and metazoan introns on the basis of secondary structure prediction. The fungal species chosen were the closest based on the phylogenetic analyses of LAGLIDADG sequences. The structure of other regions (i.e., P1, P5, P6, P9) was predicted using the program Mfold [46].
Phylogenetic analysis
Two protein data sets were analyzed. The first data set comprised COI protein sequences of representative fungi and metazoans together with the sponge sequences obtained in this work (Table 1). Only sequences longer than 400 amino-acids were considered. The second data set contained homing endonuclease sequences of the LAGLIDADG family. These sequences were retrieved following a BLAST search using for query the ORF located in the cox1 intron of Tetilla sp. Following Hall [[47] p.16] all sequences with an E-value ≤ 10-5 were taken into account.
Sequences were aligned using ProbCons [48] with three consistency steps and 500 iterative refinement repetitions. The alignments were then corrected by hand and gaps present in more than 25% of the taxa were removed from the analyses. The COI corrected alignment comprised 24 species and 400 characters while the LAGLIDADG data set comprised 89 sequences and 263 characters. Both alignments are provided as Additional files 3 and 4. For each data set two analyses were conducted: a maximum likelihood analysis with the program PHYML v2.4.4 [49] and a Bayesian analysis with the program MrBayes3.1 [50]. Both analyses were done using the mtREV amino-acid replacement model [51]. Among-site rate variation was represented by a gamma distribution [52] with eight categories and a proportion of invariant sites for the COI data set. The proportion of invariant sites was set to zero for the LAGLIDADG analysis because preliminary analysis with PHYML had estimated the proportion of invariant sites to be very small. For maximum likelihood analyses, bootstrap percentages were computed using 1000 replicates for the COI data set and 500 replicates for the LAGLIDADG data set. The Bayesian analyses were performed with two independent runs. For each run, four chains were sampled every 100 generations. Each chain was run for 5,000,000 or 6,000,000 generations for the COI and the LAGLIDADG data set respectively. Clade posterior probabilities (PP) were calculated after removal of the first 12,500 trees for the COI analysis (burnin). In this case, the average standard deviation of split frequencies was below 0.01 before the burnin threshold, and the potential scale reduction factors of the parameters were equal to 1. This indicates that the run had probably converged. For the LAGLIDADG analysis, the average standard deviation of split frequencies was below 0.01 after 5,100,000 generations. Consequently, the first 51,000 trees were removed before computation of the clade posterior probabilities.
References
Müller WEG: Review: How was metazoan threshold crossed? The hypothetical Urmetazoa. Comp Biochem Physiol A-Mol Integr Physiol. 2001, 129 (2-3): 433-460. 10.1016/S1095-6433(00)00360-3.
Lavrov DV, Forget L, Kelly M, Lang BF: Mitochondrial genomes of two demosponges provide insights into an early stage of animal evolution. Mol Biol Evol. 2005, 22 (5): 1231-1239. 10.1093/molbev/msi108.
Lavrov DV, Lang BF: Transfer RNA gene recruitment in mitochondrial DNA. Trends Genet. 2005, 21 (3): 129-133. 10.1016/j.tig.2005.01.004.
Burger G, Forget L, Zhu Y, Gray MW, Lang BF: Unique mitochondrial genome architecture in unicellular relatives of animals. Proc Natl Acad Sci U S A. 2003, 100 (3): 892-897. 10.1073/pnas.0336115100.
Haugen P, Simon DM, Bhattacharya D: The natural history of group I introns. Trends Genet. 2005, 21 (2): 111-119. 10.1016/j.tig.2004.12.007.
Beagley CT, Okada NA, Wolstenholme DR: Two mitochondrial group I introns in a metazoan, the sea anemone Metridium senile: one intron contains genes for subunits 1 and 3 of NADH dehydrogenase. Proc Natl Acad Sci U S A. 1996, 93 (11): 5619-5623. 10.1073/pnas.93.11.5619.
van Oppen MJH, Catmull J, McDonald BJ, Hislop NR, Hagerman PJ, Miller DJ: The mitochondrial genome of Acropora tenuis (Cnidaria : Scleractinia) contains a large group I intron and a candidate control region. J Mol Evol. 2002, 55 (1): 1-13. 10.1007/s00239-001-0075-0.
Pont-Kingdon G, Vassort CG, Warrior R, Okimoto R, Beagley CT, Wolstenholme DR: Mitochondrial DNA of Hydra attenuata (Cnidaria): a sequence that includes an end of one linear molecule and the genes for l-rRNA, tRNA(f-Met), tRNA(Trp), COII, and ATPase8. J Mol Evol. 2000, 51 (4): 404-415.
Beaten MJ, Roger AJ, Cavalier-Smith T: Sequence analysis of the mitochondrial genome of Sarcophyton glaucum: conserved gene order among octocorals. J Mol Evol. 1998, 47 (6): 697-708. 10.1007/PL00006429.
Chevalier BS, Stoddard BL: Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 2001, 29 (18): 3757-3774. 10.1093/nar/29.18.3757.
Burt A, Koufopanou V: Homing endonuclease genes: the rise and fall and rise again of a selfish element. Curr Opin Genet Dev. 2004, 14 (6): 609-615. 10.1016/j.gde.2004.09.010.
Belfort M: Two for the price of one: a bifunctional intron-encoded DNA endonuclease-RNA maturase. Genes Dev. 2003, 17 (23): 2860-2863. 10.1101/gad.1162503.
Nichols SA: An evaluation of support for order-level monophyly and interrelationships within the class Demospongiae using partial data from the large subunit rDNA and cytochrome oxidase subunit I. Mol Phylogenet Evol. 2005, 34 (1): 81-96. 10.1016/j.ympev.2004.08.019.
Bensasson D, Zhang DX, Hartl DL, Hewitt GM: Mitochondrial pseudogenes: evolution's misplaced witnesses. Trends Ecol Evol. 2001, 16 (6): 314-10.1016/S0169-5347(01)02151-6.
Haugen P, Bhattacharya D: The spread of LAGLIDADG homing endonuclease genes in rDNA. Nucleic Acids Res. 2004, 32 (6): 2049-2057. 10.1093/nar/gkh520.
Borchiellini C, Manuel M, Alivon E, Boury-Esnault N, Vacelet J, Le Parco Y: Sponge paraphyly and the origin of Metazoa. J Evol Biol. 2001, 14 (1): 171-179. 10.1046/j.1420-9101.2001.00244.x.
Medina M, Collins AG, Silberman JD, Sogin ML: Evaluating hypotheses of basal animal phylogeny using complete sequences of large and small subunit rRNA. Proc Natl Acad Sci USA. 2001, 98 (17): 9707-9712. 10.1073/pnas.171316998.
Peterson KJ, Eernisse DJ: Animal phylogeny and the ancestry of bilaterians: inferences from morphology and 18S rDNA gene sequences. Evol Dev. 2001, 3 (3): 170-205. 10.1046/j.1525-142x.2001.003003170.x.
Zrzavý J, Mihulka S, Kepka P, Bezdek A, Tietz D: Phylogeny of the Metazoa based on morphological and 18S ribosomal DNA evidence. Cladistics. 1998, 14 (3): 249-285. 10.1006/clad.1998.0070.
Marshall W: Ideen über die Verwandtschaftsverhältnisse der Hexactinelliden. Z wiss Zool. 1876, 27: 113-136.
Borchiellini C, Chombard C, Manuel M, Alivon E, Vacelet J, Boury-Esnault N: Molecular phylogeny of Demospongiae: implications for classification and scenarios of character evolution. Mol Phylogenet Evol. 2004, 32 (3): 823-837. 10.1016/j.ympev.2004.02.021.
van Soest RWM: Demosponge higher taxa classification re-examined. Fossil and Recent Sponges. Edited by: Reitner J, Kreupp H. 1991, Berlin , Springer-Verlag, 54-71.
Toor N, Zimmerly S: Identification of a family of group II introns encoding LAGLIDADG ORFs typical of group I introns. Rna. 2002, 8 (11): 1373-1377. 10.1017/S1355838202023087.
Dalgaard JZ, Klar AJ, Moser MJ, Holley WR, Chatterjee A, Mian IS: Statistical modeling and analysis of the LAGLIDADG family of site-specific endonucleases and identification of an intein that encodes a site-specific endonuclease of the HNH family. Nucleic Acids Res. 1997, 25 (22): 4626-4638. 10.1093/nar/25.22.4626.
Erpenbeck D, McCormack GP, Breeuwer JAJ, van Soest RWM: Order level differences in the structure of partial LSU across demosponges (Porifera): new insights into an old taxon. Mol Phylogenet Evol. 2004, 32 (1): 388-395. 10.1016/j.ympev.2004.02.014.
Biabani MAF, Laatsch H: Advances in chemical studies on low-molecular weight metabolites of marine fungi. J Prakt Chem-Chem Ztg. 1998, 340 (7): 589-607. 10.1002/prac.19983400702.
Hyde KD, Sarma VV, Jones EBG: Morphology and taxonomy of higher marine fungi. Marine mycology — A practical approach. Edited by: Hyde KD, Pointing SB. 2000, Hong Kong , Fungal Diversity Press, 172–204 -
Höller U, Wright AD, Matthée GF, Konig GM, Draeger S, Aust HJ, Schulz B: Fungi from marine sponges: diversity biological activity and secondary metabolites. Mycol Res. 2000, 104 (11): 1354-1365. 10.1017/S0953756200003117.
Abdel-Wahab MA: Diversity of marine fungi from Egyptian Red Sea mangroves. Bot Marina. 2005, 48 (5-6): 348-355. 10.1515/BOT.2005.047.
Jones EBG, Abdel-Wahab MA: Marine fungi from the Bahamas Islands. Bot Marina. 2005, 48 (5-6): 356-364. 10.1515/BOT.2005.048.
Pilantanapak A, Jones EBG, Eaton RA: Marine fungi on Nypa fruticans in Thailand. Bot Marina. 2005, 48 (5-6): 365-373. 10.1515/BOT.2005.049.
Morrison-Gardiner S: Dominant fungi from Australian coral reefs. Fungal Divers. 2002, 9: 105-121.
Hiort J, Maksimenka K, Reichert M, Perovic-Ottstadt S, Lin WH, Wray V, Steube K, Schaumann K, Weber H, Proksch P, Ebel R, Muiller WEG, Bringmann G: New natural products from the sponge-derived fungus Aspergillus niger. J Nat Prod. 2004, 67 (9): 1532-1543. 10.1021/np030551d.
Proksch P, Ebel R, Edrada RA, Schupp P, Lin WH, Sudarsono, Wray V, Steube K: Detection of pharmacologically active natural products using ecology. Selected examples from indopacific marine invertebrates and sponge-derived fungi. Pure Appl Chem. 2003, 75 (2-3): 343-352.
Jadulco R, Brauers G, Edrada RA, Ebel R, Wray V, Sudarsono, Proksch P: New metabolites from sponge-derived fungi Curvularia lunata and Cladosporium herbarum. J Nat Prod. 2002, 65 (5): 730-733. 10.1021/np010390i.
Maldonado M, Cortadellas N, Trillas MI, Rützler K: Endosymbiotic yeast maternally transmitted in a marine sponge. Biol Bull. 2005, 209 (2): 94-106.
Perović-Ottstadt S, Adell T, Proksch P, Wiens M, Korzhev M, Gamulin V, Müller IM, Müller WEG: A (1 -> 3)-beta-D-glucan recognition protein from the sponge Suberites domuncula - Mediated activation of fibrinogen-like protein and epidermal growth factor gene expression. Eur J Biochem. 2004, 271 (10): 1924-1937. 10.1111/j.1432-1033.2004.04102.x.
Thakur NL, Müller WEG: Sponge-bacteria association: A useful model to explore symbiosis in marine invertebrates. Symbiosis. 2005, 39 (3): 109-116.
Hentschel U, Usher KM, Taylor MW: Marine sponges as microbial fermenters. FEMS Microbiol Ecol. 2006, 55 (2): 167-177. 10.1111/j.1574-6941.2005.00046.x.
Pont-Kingdon G, Okada NA, Macfarlane JL, Beagley CT, Watkins-Sims CD, Cavalier-Smith T, Clark-Walker GD, Wolstenholme DR: Mitochondrial DNA of the coral Sarcophyton glaucum contains a gene for a homologue of bacterial MutS: A possible case of gene transfer from the nucleus to the mitochondrion. J Mol Evol. 1998, 46 (4): 419-431. 10.1007/PL00006321.
Beagley CT, Okimoto R, Wolstenholme DR: The mitochondrial genome of the sea anemone Metridium senile (Cnidaria): introns, a paucity of tRNA genes, and a near-standard genetic code. Genetics. 1998, 148 (3): 1091-1108.
Steindler L, Huchon D, Avni A, Ilan M: 16S rRNA phylogeny of sponge-associated cyanobacteria. Appl Environ Microbiol. 2005, 71 (7): 4127-4131. 10.1128/AEM.71.7.4127-4131.2005.
Arnason U, Gullberg A, Widegren B: The complete nucleotide sequence of the mitochondrial DNA of the fin whale, Balaenoptera physalus. J Mol Evol. 1991, 33 (6): 556-568. 10.1007/BF02102808.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R: DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994, 3 (5): 294-297.
Lisacek F, Diaz Y, Michel F: Automatic identification of group-I intron cores in genomic DNA-sequences. J Mol Biol. 1994, 235 (4): 1206-1217. 10.1006/jmbi.1994.1074.
Zuker M: Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31 (13): 3406-3415. 10.1093/nar/gkg595.
Hall BG: Phylogenetic trees made easy: a how-to manual, second edition. 2004, Sunderland , Sinauer Associates, Inc., 221-Second edition
Do CB, Mahabhashyam MSP, Brudno M, Batzoglou S: ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res. 2005, 15 (2): 330-340. 10.1101/gr.2821705.
Guindon S, Gascuel O: A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, 52 (5): 696-704. 10.1080/10635150390235520.
Ronquist F, Huelsenbeck JP: MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19 (12): 1572-1574. 10.1093/bioinformatics/btg180.
Adachi J, Hasegawa M: Model of amino acid substitution in proteins encoded by mitochondrial DNA. J Mol Evol. 1996, 42: 459-468. 10.1007/PL00013324.
Yang Z: Among-site rate variation and its impact on phylogenetics analysis. Trends Ecol Evol. 1996, 11 (9): 367-372. 10.1016/0169-5347(96)10041-0.
Cech TR, Damberger SH, Gutell RR: Representation of the secondary and tertiary structure of group I introns. Nat Struct Biol. 1994, 1 (5): 273-280. 10.1038/nsb0594-273.
Gonzalez P, Barroso G, Labarere J: Molecular analysis of the split cox1 gene from the Basidiomycota Agrocybe aegerita: relationship of its introns with homologous Ascomycota introns and divergence levels from common ancestral copies. Gene. 1998, 220 (1-2): 45-53. 10.1016/S0378-1119(98)00421-1.
Waring RB, Brown TA, Ray JA, Scazzocchio C, Davies RW: Three variant introns of the same general class in the mitochondrial gene for cytochrome oxidase subunit 1 in Aspergillus nidulans. Embo J. 1984, 3 (9): 2121-2128.
Lang BF: The mitochondrial genome of the fission yeast Schizosaccharomyces pombe: highly homologous introns are inserted at the same position of the otherwise less conserved cox1 genes in Schizosaccharomyces pombe and Aspergillus nidulans. Embo J. 1984, 3 (9): 2129-2136.
Woo PCY, Zhen HJ, Cai JJ, Yu J, Lau SKP, Wang J, Teng JLL, Wong SSY, Tse RH, Chen R, Yang HM, Liu B, Yuen KY: The mitochondrial genome of the thermal dimorphic fungus Penicillium marneffei is more closely related to those of molds than yeasts. FEBS Lett. 2003, 555 (3): 469-477. 10.1016/S0014-5793(03)01307-3.
Acknowledgements
We would like to thank Laura Steindler for her help with sponge DNA extraction, and Sara Kinamon for her help in the laboratory. Three anonymous reviewers provided valuable comments. Sponge tissue samples are part of the museum collections of Tel-Aviv University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
CR and IG extracted the sponge DNAs and sequenced the cox1 genes. MI initiated the study and was responsible for the collection and identification of the sponges studied. DH coordinated the study, performed the molecular analysis and wrote the paper. All authors contributed to the writing and the revision of the manuscript.
Chagai Rot, Itay Goldfarb contributed equally to this work.
Electronic supplementary material
12862_2006_256_MOESM1_ESM.fas
Additional File 1: Location of introns in the cox1 gene. DNA sequence alignment (in FASTA format) used to create Figure 1. The location of introns is indicated by X, gaps by -. (FAS 15 KB)
12862_2006_256_MOESM2_ESM.doc
Additional File 2: Sequencing primers. Name, sequence and direction of the primers used to sequence the cox1 gene. (DOC 31 KB)
12862_2006_256_MOESM3_ESM.fas
Additional File 3: COI protein alignment. Protein sequence alignment (in FASTA format) used to reconstruct the phylogenetic tree present in Figure 4. (FAS 10 KB)
12862_2006_256_MOESM4_ESM.fas
Additional File 4: LAGLIDADG protein alignment. Protein sequence alignment (in FASTA format) used to reconstruct the phylogenetic tree present in Figure 5. (FAS 26 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Rot, C., Goldfarb, I., Ilan, M. et al. Putative cross-kingdom horizontal gene transfer in sponge (Porifera) mitochondria. BMC Evol Biol 6, 71 (2006). https://doi.org/10.1186/1471-2148-6-71
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-6-71