
Abstract
Background
Evidence for historical, demographic and selective factors affecting enzyme evolution can be obtained by examining nucleotide sequence variation in candidate genes such as Lactate dehydrogenase (Ldh). Two closely related Daphnia species can be distinguished by their electrophoretic Ldh genotype and habitat. Daphnia pulex populations are fixed for the S allele and inhabit temporary ponds, while D. pulicaria populations are fixed for the F allele and inhabit large stratified lakes. One locus is detected in most allozyme surveys, but genome sequencing has revealed two genes, LdhA and LdhB.
Results
We sequenced both Ldh genes from 70 isolates of these two species from North America to determine if the association between Ldh genotype and habitat shows evidence for selection, and to elucidate the evolutionary history of the two genes. We found that alleles in the pond-dwelling D. pulex and in the lake-dwelling D. pulicaria form distinct groups at both loci, and the substitution of Glutamine (S) for Glutamic acid (F) at amino acid 229 likely causes the electrophoretic mobility shift in the LDHA protein. Nucleotide diversity in both Ldh genes is much lower in D. pulicaria than in D. pulex. Moreover, the lack of spatial structuring of the variation in both genes over a wide geographic area is consistent with a recent demographic expansion of lake populations. Neutrality tests indicate that both genes are under purifying selection, but the intensity is much stronger on LdhA.
Conclusions
Although lake-dwelling D. pulicaria hybridizes with the other lineages in the pulex species complex, it remains distinct ecologically and genetically. This ecological divergence, coupled with the intensity of purifying selection on LdhA and the strong association between its genotype and habitat, suggests that experimental studies would be useful to determine if variation in molecular function provides evidence that LDHA variants are adaptive.
Similar content being viewed by others

Background
Understanding the evolutionary factors that affect genetic variation in natural populations remains the primary focus of population genetics. Early surveys of allozyme variation using protein electrophoresis revealed abundant polymorphism, but only about one third of the possible amino acid changes, and none of the variation at silent and non-coding sites, could be detected with this technique [1]. Analyses of genetic variation now involve nucleotide data, which can be used in a variety of statistical tests to discriminate between patterns of variation due to neutral processes (drift, population expansion, bottlenecks) and various types of selection (positive, purifying, balancing). Most studies have shown that levels of variation at silent and non-coding sites are substantially higher than levels of protein variation, suggesting that the majority of non-synonymous mutations are deleterious [1]. Even so, patterns of allozyme (protein) variation often appear to conform to neutral or nearly neutral expectations [2]. Conversely, detailed studies showing associations with different habitats, or clinal variation across wide geographic ranges have provided evidence that functional differences among allozyme variants can affect fitness (e.g. Alcohol dehydrogenase and Glucose-6-phosphate dehydrogenase in Drosophila melanogaster and Phosphoglucose isomerase in Colias butterflies; reviewed in [3]). Thus, a major challenge has been to determine the adaptive significance, if any, of polymorphism and interspecific divergence at allozyme loci [4].
L-Lactate dehydrogenase (L-LDH, EC 1.1.1.27) is involved in the interconversion of pyruvate and L(-)-lactate, which allows the aerobic metabolism of lactate accumulated by anaerobic glycolysis following periods of exposure to lowered environmental oxygen tension, or hypoxia [5]. There is evidence that selection is acting on LDH variation in well-studied species such as the killifish, Fundulus heteroclitus [6, 7]. For example, an amino acid substitution between two alleles that vary along a cline in temperature affects thermostability [8], and experimental studies have shown that this variation is involved in the adaptation of killifish to hypoxia [9, 10].
A strong association between Ldh genotype and habitat has been also observed in members of the Daphnia pulex species complex. Based on relative electrophoretic mobility, the pond species, D. pulex is characterized by homozygosity for the slow (S) Ldh allele and the closely related lake species, D. pulicaria by homozygosity for the fast (F) allele in North America [11–13]. In addition, the two species hybridize and hybrids are generally found in ponds or disturbed, intermediate habitats [13]. The genetic and habitat segregation is also associated with differences in physiological and life history traits [14, 15]. Surveys of allozyme variation in most Daphnia species are consistent with a one-locus model, but a recent analysis of the D. pulex genome sequence has identified two Ldh genes, LdhA and LdhB, that are approximately 26 cM apart [16, 17]. Preliminary gene expression work suggests that LdhA is expressed at a significantly higher level than LdhB [18], but the link between the allozyme locus and the genome sequences has not yet been made. Nevertheless, the fixation of different LDH variants in D. pulex and D. pulicaria in different aquatic habitats, despite the fact that they share polymorphisms at other allozyme loci [19–21], suggests the possibility that Ldh variation is adaptive. However, the hypothesis that selection has played a role in Ldh divergence in Daphnia has yet to be tested.
Genetic studies suggest that D. pulex and D. pulicaria are members of a relatively young species complex and appear to be undergoing ecological speciation [22]. Both species reproduce by cyclical parthenogenesis in which females produce offspring by apomictic parthenogenesis during favorable conditions and have a sexual phase that results in the production of diapausing eggs prior to the onset of unfavorable conditions. Natural hybrids between the two species reproduce by obligate parthenogenesis [23, 24] in which females produce broods of parthenogenetic offspring (like cyclic females), but also produce their diapausing eggs parthenogenetically. This results in a completely ameiotic life history and allows clonal genotypes to persist for many years.
The objectives of the present study are to analyze both LdhA and LdhB sequence variation to (1) determine which locus is detected in allozyme surveys, (2) elucidate the evolutionary history of the two genes in the context of speciation and hybridization, and (3) determine if there is evidence that selection is acting on the variation at each locus. To achieve these objectives, isolates of D. pulex and D. pulicaria from North America were chosen to maximize geographic coverage of the three Ldh genotypes (SS, SF and FF) and the two breeding systems.
Methods
Daphniasamples
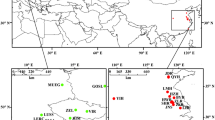
A total of 85 Daphnia isolates collected from 68 populations, mainly from North America, were included in this study (Figure 1, Additional file 1.1). These isolates include representatives of all the major lineages in the D. pulex species complex identified by Colbourne et al. [25] using phylogenetic analysis of mitochondrial DNA (Figure 2), as well as the C lineage of South American D. pulicaria identified by Mergeay et al. [26]. An isolate of Daphnia obtusa from Illinois [27] was also included as an outgroup for phylogenetic analyses.

Location of the North American Daphnia populations sampled for this study. Latitude and longitude for each site are available in Additional file 1.1. Populations are characterized by (a) species and (b) breeding system for D. pulex and D. pulicaria isolates.

Neighbor-joining tree of lineages in the Daphnia pulex species complex. Representative sequences of the mitochondrial ND5 gene were taken from Colbourne et al. [25]. The relationship of the SAPC-C lineage, first identified by Mergeay et al. [26], to the rest of the complex is based on the mitochondrial 12S rRNA gene. Attempts to amplify the ND5 fragment from members of this lineage have been unsuccessful. Taxa are indicated as follows: ARE = D. arenata, EPC = European D. pulicaria, EPX = European D. pulex, MEL = D. melanica, MID = D. middendorffiana, NAPC = North American D. pulicaria (e = eastern, w = western, p = polar), NAPX = North American D. pulex, SAPC = South America D. pulicaria, TEN = D. tenebrosa.
Genomic DNA was generally extracted from multiple parthenogenetically-produced offspring of a single progenitor female using the CTAB method [28]. Isolates from South America and Europe were extracted from single individuals preserved in 95% ethanol using the Mammalian genomic DNA GenElute kit (Sigma-Aldrich).
The breeding system (cyclic or obligate parthenogenesis) of most lab-reared isolates was diagnosed by examination of diapausing egg cases (ephippia) produced in the absence of males. While cyclical parthenogens often release empty ephippial cases unless the eggs have been fertilized, obligate parthenogens deposit eggs into ephippia even in the absence of males [29].
The Ldh genotype of all of the isolates from North America was determined by allozyme electrophoresis. In most cases, isolates were assigned to a species within the pulex complex [25] based on the sequence of the mitochondrial NAD dehydrogenase subunit 5 (ND5) gene generated by the lab from which the genomic DNA was obtained (Additional file 1.1). However, isolates that were collected from lakes and reproduce by cyclic parthenogenesis (i.e. sexually) were designated as D. pulicaria [13, 19] even though some of them have a D. pulex mitochondrial haplotype. Previous studies have shown that populations of D. pulicaria with either mitochondrial type are not distinguishable from one another based on nuclear allozyme and microsatellite variation, but are divergent from cyclically parthenogenetic populations of D. pulex [22, 30]. Of the 85 isolates included in this study, 70 were classified as either North American D. pulex or D. pulicaria.
Polymerase chain reaction, cloning and sequencing
PCR amplification of the two Ldh loci from Daphnia genomic DNA was carried out using two sets of primers: LDHA-u4F (5'-GAAAATGGCCACCAGCGTCG) and LDHA-1304R (5'-TTGAACTTCATTCAAAGTGGCAGC) for LdhA, and LDHB-u7F (5'-AATCAGAATGCAGACAAAGGCCTC) and LDHB-1477R (5'-TCAGAACACCAAGTTTGACTGAACTTC) for LdhB. Each 25 μl PCR reaction contained 1X Phusion HF buffer, 2 mM MgCl2, 2 mmol of each dNTP, 2 pmol of each primer, and 0.5 units of Phusion High Fidelity polymerase (Finnzymes), with 2 μl (20 to 50 ng) of the target genomic DNA. The thermocycling program for the LdhA primers was: 94°C for 2 min; 25 cycles of 94°C for 30 s, 54°C for 30 s, 72°C for 90 s; a further 10 cycles as above except that the 72°C extension time was increased to 2 min; and finally 72°C for 5 min. A similar program was used for the LdhB primers but with an annealing temperature of 50°C.
Amplicons of the expected size (~1.3 kb for LdhA, ~1.4 kb for LdhB) were cloned using the StrataClone Blunt PCR Cloning Kit (Stratagene) following the manufacturer's protocol. Colonies were grown on Luria Broth (LB) agar containing ampicillin (100 μg/ml) and X-gal (20 μg/ml). Glycerol stocks (LB + amp in 30% glycerol) were generated for 24 white colonies from each experiment and archived at -80°C.
Initially, eight plasmids of the cloned PCR product from each isolate were selected for sequencing. The cloned insert was amplified directly from bacterial cells by PCR using the M13F (5'-GTTGTAAAACGACGGCCAGTG) and M13R (5'-CAGGAAACAGCTATGACCATG) sequencing primers, and Platinum Taq polymerase (Invitrogen). If an amplicon of the expected size was not obtained from all eight colonies, the process was repeated until at least eight amplicons of the expected size were obtained. Cycle-sequencing of these amplicons in both directions was carried out using the BigDye 3.1 kit (Applied Biosystems) and the M13F and M13R primers. Each 10 μl sequencing reaction contained 0.25 μl of BigDye mix, 1 μl of sequencing buffer, 10 pmol of primer, 6.75 μl of ultrapure water, and 1 μl of PCR amplicon. Completed reactions were run on an ABI3730 DNA Analyzer.
Sequence diversity analyses
A reference sequence was created from the D. pulex genome sequence [18] for each of the Ldh genes indicating the location of exons, introns and the start and stop codons. Sequence electropherograms from each cloning experiment were aligned and edited in Sequencher v.4.5 (Gene Codes) using the reference sequences to orient the alignment and to trim the ends to the start and stop codons. The consensus sequence of each allele present within an isolate was assembled into a master alignment of all the Ldh alleles for a particular gene. Unique differences among the eight clones from an isolate were assumed to be PCR, cloning or sequencing errors and were excluded from the analysis.
An alignment containing alleles from all isolates was generated for each gene in BioEdit [31]. Separate alignments for the exon and intron sequences were also generated using the reference sequences as a guide.
Analyses of nucleotide diversity included only alleles obtained from isolates of North American D. pulex and D. pulicaria and their hybrids. Alleles at both loci were divided into pulex and pulicaria groups based on their phylogenetic relationship to alleles obtained from cyclically parthenogenetic isolates from lakes (D. pulicaria) and ponds (D. pulex). Alleles from isolates that are likely to be hybrids with a species other than North American D. pulex or D. pulicaria (one isolate of D. middendorffiana, three isolates of South American D. pulicaria) were excluded from the diversity analyses.
Standard measures of nucleotide diversity including the number of haplotypes (K), the number of segregating sites (S), haplotype diversity (H), the average number of pairwise differences per site between sequences (π) and diversity per site based on the number of segregating sites (θ) were calculated using the program DNASPv5.10 [32]. Both π and θ were estimated for all sites (πT, θT), as well as introns (πi, θi), synonymous coding sites (πs, θs), and nonsynonymous coding sites (πn, θn). In addition, DNASP was used to perform tests of neutrality (Tajima's D; Fu and Li's D and D*; Fu and Li's F and F*, Fay and Wu's H). Patterns of diversity between the two loci in each of the pulex and pulicaria allele groups were analysed using an HKA test [33] in DNASP using the D. obtusa sequence as an outgroup. We used the algorithm of Betrán et al. [34] in DNASP to detect gene conversion between the pulex and pulicaria allele groups in each locus.
Separate genetic distance matrices were calculated for each allele group (pulex and pulicaria) and each locus based on the number of nucleotide differences between pairs of alleles in Arlequin 3.11 [35]. The geographic distance matrix was constructed by calculating the great circle distance between each pair of sites based on the decimal degrees of latitude and longitude using the function "rdist.earth" with earth radius = 6371 km in the Fields 5.02 package in R 2.9.2 [36]. A Mantel test with 1000 permutations was then conducted between the genetic and geographic distance matrices for each of the four allele groups using Arlequin.
Phylogenetic analysis
We performed Bayesian phylogenetic analyses on the unique nt sequences (total and introns only, indels excluded) of each Ldh gene using Mr. Bayes v3.1.2 [37] with default prior settings and a GTR model. All trees were rooted through a sequence from D. obtusa, which is not a member of the pulex complex but a member of its sister species complex [38]. We ran two independent and simultaneous Markov Chain Monte Carlo (MCMC) analyses of 15 heated and one cold chain for six million generations sampling from the chain every 100 generations. The initial 30% of trees were discarded as a burn-in. A 50% majority-rule consensus topology with posterior probability (PP) values for each node was constructed from the post-burn-in trees. We also constructed Neighbor-joining trees [39] of the amino acid sequences using MEGA4 [40] and Poisson-corrected estimates of sequence divergence.
Results
Characterization of sequence variation
We obtained a total of 143 LdhA sequences from the 85 isolates included in the study, of which 76 (53.1%) are unique. Our sequences begin at the start codon and end 15 nt upstream of the stop codon. This gene consists of six exons and five introns [18]. The total length of the sequence alignment is 1394 nt with individual sequences varying from 1299 to 1352 nt and all length variation occurring in introns. The coding region is 981 nt resulting in 327 codons for a protein length of 332 amino acids when the five missing from our PCR amplicon are included. The total length of intron sequences in LdhA varies from 318 to 371 nt. The amino acid substitution of Glutamine (uncharged) to Glutamic acid (negative) at amino acid 229 changes the overall charge of the protein, which would cause an electrophoretic mobility shift. Thus LdhA is most likely the locus that is scored in allozyme analyses. The "C" to "G" transversion (CAA to GAA) that causes this substitution corresponds to nt 912 in the alignment (nt 685 in the coding sequence).
We obtained a total of 157 sequences for LdhB of which 115 (73.2%) are unique. This gene consists of eight exons and seven introns [18]. Our sequences begin with the start codon and end with the stop codon resulting in a total alignment length of 1545 nt with individual sequences varying from 1446 nt to 1488 nt. With the exception of a 2-nt insertion in exon 8 of a single sequence, all other length variation occurs in introns. The coding region of 972 nt encodes a protein of 324 amino acids. The total length of intron sequences varies from 471 to 513 nt. The alignments of all the LdhA and LdhB nucleotide and protein sequences are provided as supplementary information (Additional files 2, 3, 4, 5). All unique nucleotide sequences have been submitted to GenBank (accession numbers JN117725-JN117915).
Phylogenetic analysis
The Bayesian phylogenetic analysis based on complete, unique LdhA sequences shows that all of the fast LdhA alleles from North American (NA) D. pulicaria (Additional file 1.2) and hybrids involving this species are monophyletic (Figure 3a). In addition, alleles from NA D. pulex form a monophyletic group with the exception of 15 recombinant alleles (described below). Because one allele from this group was isolated from a cyclically parthenogenetic pond population, these alleles were assigned to the pulex allele group. The two alleles from the European D. pulex isolate are the sister group to all the other members of the pulex species complex (Figure 3a), which is concordant with the phylogeny based on mitochondrial (mt)DNA ([25], Figure 2). However, the sequences from all the other lineages in the complex cluster with the NA D. pulicaria alleles, which is not consistent with the mtDNA phylogeny. For example, D. tenebrosa and European D. pulicaria form a separate cluster (the tenebrosa group) from species in the pulicaria group in the mtDNA phylogeny (Figure 2). The tree based on LdhA intron sequences (Additional file 6a) is similar to the full-sequence tree but relationships within the cluster of alleles from NA D. pulicaria, which also contains alleles from all the other species, are not well resolved.

Bayesian phylogeny of (a) Ldh A and (b) Ldh B gene sequences from 85 isolates in the Daphnia pulex species complex. The trees are rooted through Daphnia obtusa. Species codes are given in Figure 2 except that North American D. pulicaria isolates have been shortened to "C" and North American D. pulex isolates have been shortened to "X". Hybrids between these two species are designated "CX". Isolates within groups are numbered and alleles within heterozygotes are designated "a" and "b". The 2-letter province or state code is appended to C, X and CX isolates. Allele groups in D. pulicaria (C) and D. pulex (X) that were used in the nucleotide diversity analyses are boxed. The R box indicates recombinant LdhA alleles (see text). Isolates that reproduce by obligate parthenogenesis are indicated with asterisks (*)
As is the case for LdhA, LdhB alleles from NA D. pulex and D. pulicaria form two distinct groups (Figure 3b, Additional file 1.2). In contrast to the LdhA tree, LdhB alleles from D. arenata do not belong to either of these major groups and their relationship with them is unresolved. In addition, European D. pulex alleles are not the sister group to alleles from all the other species, as is the case for the LdhA and mtDNA trees. The topology based on intron sequences (Additional file 6b) is similar except that the D. arenata sequences are the sister group to alleles from NA D. pulex and all other sequences cluster with the alleles from NA D. pulicaria.
Recombination between pulex and pulicaria alleles
Analysis of recombination identified gene conversion tracts between the pulex and pulicaria alleles in both genes (Additional file 1.3, 1.4). In most cases these are short (<100 nt) but there are two larger tracts (127 nt in LdhA and 501 nt in LdhB) which both occur in only one allele. With one exception for each gene, conversion tracts consist of pulicaria variants in pulex alleles. A pair of tracts in LdhA; 28 nt from position 812-839 and 74 nt from position1161-1280 (indels are excluded from the tracts), occur in 15 pulex alleles from isolates as far east as Quebec and as far west as the Northwest Territories with most of the alleles occurring in isolates from Manitoba and Saskatchewan. Indeed, these 15 alleles form a cluster that groups more closely with the pulicaria than the pulex LdhA alleles in the phylogenetic tree (Figure 3a).
As most of the LdhA heterozygotes are obligately parthenogenetic (Additional file 1.1), and are thought to be F1 hybrids [11, 19], we would expect them to be heterozygous for pulex and pulicaria alleles at LdhB as well. This is indeed the case with nine exceptions, six of which come from Manitoba and Saskatchewan (Table 1). In all but one case, the isolate is heterozygous at one locus but homozygous at the other. The exception is an isolate from Saskatchewan that is homozygous for a pulex allele at LdhA but homozygous for a pulicaria allele at LdhB. There is no bias with respect to locus or species in the eight isolates; three are homozygous for the pulicaria allele at one locus, while five are homozygous for the pulex allele. Similarly, LdhA is the homozygous locus in four isolates, while LdhB is the homozygous locus in the other four.
Amino acid variation
Amino acid variation in LDHA is very limited. Indeed, the outgroup, D. obtusa, and most of the lineages in the pulex complex have the same amino acid sequence, which is slow electrophoretically. Fast alleles are only found in D. melanica, NA D. pulicaria and hybrids, which also include the three SA D. pulicaria isolates and one isolate of D. middendorfficana. Overall, the 143 nucleotide sequences encode 11 amino acid sequences (Figure 4). Of these, seven are unique and four ("slow", "fast", D. arenata and European D. pulex) occur more than once. There are nine (2.8%) variable amino acid positions and of these, five variants (55.5%) only occur in one sequence.

Neighbor-joining tree of LDHA protein sequences from 85 isolates in the Daphnia pulex species complex. The number of sequences shared by multiple isolates of a particular species is given in brackets. "Slow" and "Fast" refer to electrophoretic mobility in allozyme analyses. C = North American D. pulicaria, X = North American D. pulex and CX = hybrid isolates. Other taxon codes are defined in Figure 3.
The primary differences between pulex (slow) and pulicaria (fast) LDHA are amino acid substitutions at two sites; the substitution of Aspartic acid in pulex for Glutamic acid in pulicaria at position 6, and the substitution of Glutamine in pulex for Glutamic acid in pulicaria at position 229, which likely causes the slow/fast mobility shift. In addition, the D. arenata and European D. pulex proteins have Asparagine at position 314 while all others have Serine. Finally, the protein encoded by both European D. pulex alleles has Isoleucine at position 132 while all other proteins have Leucine.
There is considerably more amino acid variation among LDHB proteins, with little differentiation in any particular lineage (Figure 5). The 157 nucleotide sequences encode a total of 37 protein sequences of which only 14 (37.8%) occur more than once. There are 36 (11.1%) variable amino acid positions and of these, 17 variants (47.2%) only occur in one sequence.

Neighbor-joining tree of LDHB protein sequences from 85 isolates in the Daphnia pulex species complex. The number of sequences shared by multiple isolates of a particular species is given in brackets. C = North American D. pulicaria, X = North American D. pulex and CX = hybrid isolates. Other taxon codes are defined in Figure 3.
Sequence diversity and neutrality tests of pulex and pulicaria alleles
In general, nucleotide diversity (π and θ) is substantially higher in alleles from NA D. pulex (pulex alleles) than in alleles from NA D. pulicaria (pulicaria alleles) for both genes, and higher in LdhB than LdhA, with the exception of πs in pulicaria alleles and πi and θi in pulex alleles (Table 2, Figure 6). In addition, diversity tends to be higher at synonymous sites than in introns, except for pulicaria LdhB alleles where diversity is higher in the introns. As expected, nucleotide diversity at nonsynonymous sites is much lower than that at synonymous sites and introns (Table 2). The dN/dS ratios for all four groups of alleles are less than one (Table 3). Pulex LdhA alleles have the lowest dN/dS ratio (0.032) while pulicaria LdhB alleles have the highest ratio (0.348).

Nucleotide diversity in Ldh A and Ldh B alleles from Daphnia pulex and Daphnia pulicaria. The thin vertical lines represent 1 standard deviation. T = total variation, i = variation in introns, s = variation at synonymous sites in exons and n = variation at non-synonymous sites in exons.
The results of the Mantel test indicate that there is no significant correlation between sequence divergence and geographic distance for the pulicaria alleles at either locus (Figure 7a and 7c), but there is a significant positive correlation for the pulex alleles at both loci (Figure 7b and 7d).

Mantel tests of the association between geographic distance and nucleotide diversity in Ldh sequences from Daphnia pulex (X) and Daphnia pulicaria (C). (a, b) LdhA. (c, d) LdhB.
All the neutrality tests, except for Fay and Wu's H, are significant for pulicaria LdhA alleles, and the Fu and Li tests are also significant for pulicaria LdhB alleles (Table 2). These test statistics are all negative indicating an excess of rare polymorphisms. The negative values of Fay and Wu's H tests indicate an excess of high frequency variants, but they are not significant. The HKA tests are not significant for either the pulex or pulicaria allele groups (Chi-square (pulex) = 0.215, p = 0.64; Chi-square (pulicaria) = 2.26, p = 0.13), indicating that the ratio of intraspecific polymorphism to interspecific divergence is not significantly different between the two loci.
Discussion
Analysis of sequence variation in protein coding genes can be used to determine the selective and non-selective processes that have contributed to current patterns of genetic diversity. It has been shown that Ldh variation is associated with differentiation between two closely related Daphnia species that occupy contrasting habitats, and may be a consequence of ecological speciation. In this study, we found that patterns of variation at two Ldh loci are likely a consequence of recent demographic expansion and purifying selection in D. pulicaria.
Phylogeny
It is clear that alleles in lake-dwelling NA D. pulicaria and pond-dwelling NA D. pulex form distinct groups at both Ldh loci. Moreover, alleles from D. pulex are divergent from alleles from every other lineage in the pulex species complex, including those that share the same amino acid sequence. In addition, relationships among species in the complex differ substantially in trees based on the nuclear Ldh and the mitochondrial ND5 (Figure 2) genes. For example, alleles from species in the tenebrosa group cluster with those from NA D. pulicaria in trees based on both Ldh loci. Moreover, D. arenata is the sister taxon to the other members of the complex in the LdhB tree, although European D. pulex occupies this position in the LdhA tree, which is consistent with the mtDNA phylogeny (Figure 2). Vergilino et al. [30] analyzed sequence variation in the nuclear Rab4 gene from lineages in the pulex species complex and also found that tenebrosa group alleles cluster with NA D. pulicaria. However, they found that D. arenata clusters with NA D. pulex, which is consistent with the mtDNA phylogeny. The only other phylogenetic analysis of this species complex based on nuclear genes is that of Omilian and Lynch [22], who analyzed variation at six nuclear protein-coding genes (one of which was Rab4) in D. arenata, NA D. pulex and NA D. pulicaria. They also found that the relationship between D. arenata alleles and alleles from the other two species varies across the six loci, which could be a consequence of hybridization and/or incomplete sorting of ancestral polymorphism [30].
The fact that Daphnia species within the pulex complex have hybridized in the past, and continue to do so, is well-known [38, 41] and this undoubtedly contributes to the inconsistency between phylogenies based on mitochondrial and nuclear genes. Despite the lack of congruence across different nuclear loci, alleles from each species do tend to form monophyletic groups suggesting that gene pools in this species complex have been isolated long enough to diverge from one another at the nucleotide level. One explanation for this pattern is that the nuclear genomes of some species in this complex are mosaics of genes derived from multiple species generated by a complex history of recurrent introgressive hybridization followed by isolation and divergence [42]. Phylogenetic analysis of additional nuclear loci will be required to test this hypothesis.
Hybridization and recombination
Ongoing hybridization between D. pulex and D. pulicaria should provide opportunities for recombination between the pulex and pulicaria allele groups at both Ldh loci, and some instances were observed in this study. Although the hybrids observed in nature appear to reproduce by obligate parthenogenesis, Heier & Dudycha [43] found that crosses between cyclically parthenogenetic D. pulex females and D. pulicaria males produce offspring that are themselves cyclically parthenogenetic, and are able to successfully backcross with both parental species and each other, with no apparent loss of sexual function. Indeed, we identified a group of 15 recombinant pulex LdhA alleles with two tracts of sequence from pulicaria alleles that appear to have arisen in populations from the Canadian prairies, and are now geographically widespread. In addition, six of the nine isolates with "recombinant" LdhA/LdhB genotypes (Table 1) came from Saskatchewan and Manitoba, including the isolate that is a pulex LdhA and pulicaria LdhB homozygote. Although, the breeding system of these isolates is unknown, the pattern of Ldh variation we observed suggests that substantial introgressive hybridization may be occurring between D. pulex and D. pulicaria in this region, where bodies of water vary substantially in depth, surface area and the presence of fish, and thus provide a continuum of habitats between "ponds" and "lakes". This is also consistent with the occurrence of the Ldh-S allele at low frequency in lake populations from central and western U.S.A [11] and Oregon [20], and with the results of Omilian and Lynch [22] whose analysis indicated that gene flow between these two species is on the order of 1.7 gene migrants per generation.
Sequence diversity in D. pulex and D. pulicaria
Nucleotide diversity at synonymous sites in pulex LdhA (πs = 0.027) and LdhB (πs = 0.031) alleles is similar to the average estimate of nuclear protein-coding genes in other invertebrates (πs = 0.027, [44]), and well within the range of values estimated by Omilian & Lynch [22] for six nuclear genes in NA D. pulex (mean πs = 0.019, range = 0.000 - 0.050) and NA D. pulicaria (πs = 0.015, range 0.000 to 0.037). In four of the six genes, estimates of πs are higher in D. pulicaria than in D. pulex. In contrast, synonymous nucleotide diversity in both pulicaria LdhA (πs = 0.006) and LdhB (πs = 0.007) alleles is substantially lower than that in D. pulex (πs = 0.027 for LdhA, πs = 0.031 for LdhB).
The neutrality tests are negative in all cases but only significant for pulicaria alleles (Table 3), which is consistent with the operation of purifying selection, a selective sweep and/or demographic expansion [45]. The latter two alternatives are also consistent with the lack of correlation between sequence divergence and geographic distance for the pulicaria alleles at both loci in the Mantel tests (Figure 7). However, Fay and Wu's H statistic is not significant for either gene, which does not support a selective sweep. Taken together, the lower level of genetic variation in both Ldh genes in D. pulicaria, and the lack of geographic structuring of this variation suggest a recent demographic expansion of lake populations over a very wide geographic area after the evolution of the fast allele at LdhA. Indeed, isolates from SA D. pulicaria are heterozygous for a pulicaria-group allele at both LdhA and LdhB. The other alleles in these isolates form a distinct South American cluster. Recent expansion of D. pulicaria is also consistent with the results of Omilian and Lynch [22], which suggested that effective population size (Ne) is much lower in D. pulicaria than D. pulex, and that the nuclear genomes of these two species diverged from a common ancestor on the order of 80,000 years ago.
D. pulicaria and D. pulex are relatively young species and retain the ability to hybridize, so it is not surprising that they share alleles at many allozyme loci [19, 20]. Despite the occurrence of gene flow between them [22], they show divergence in physiology and life history traits [14, 15] associated with adaptation to pond and lake habitats. Previous allozyme surveys have identified two loci in addition to Ldh, Hexokinase (Hk) and a Peptidase that are fixed or nearly so for alternate alleles in lake and pond populations [19]. It has been suggested that selection, directly on these loci or on loci to which they are closely linked, may be involved in this divergence. For example, Pfrender et al. [21] analyzed variation at 13 allozyme loci, 11 microsatellite loci and mtDNA in Daphnia populations from western Oregon. They identified two pond populations that are genetically distinct from the D. arenata that typically inhabit ponds in this region [20, 21]. Instead, these two populations are very similar to Oregon lake D. pulicaria at all loci except Ldh and Hk, where they are fixed, or nearly so for the alleles Ldh-S and Hk-M, which invariably predominate in pond populations.
It is extremely difficult to detect positive selection on individual amino acid sites by applying standard statistical tests to sequences from one species or closely related species [46, 47]. However, it has been shown that one or a few amino acid substitutions can cause adaptive changes in protein function [48–50]. LDH is known to be involved in response to hypoxia, and variation in levels and temporal patterns of dissolved oxygen are very different in ponds and lakes. For example, oxygen tension in shallow temporary ponds decreases while temperature increases throughout the spring and summer and Daphnia populations generally die by mid-summer due to anoxia or desiccation [14]. Conversely, deep lakes are generally stratified during the summer months and Daphnia take refuge in the cold hypolimnion to avoid fish predation [14, 51]. Members of the pulex species complex tend to occupy temporary ponds, although species in the tenebrosa group also occur in permanent ponds and small lakes. However, D. pulicaria is the only species in the complex that has invaded large, stratified lakes. Moreover, the amino acid sequence of the slow LDHA protein is highly conserved despite substantial genetic variation at synonymous sites and introns indicating that this protein is generally under strong purifying selection. The slow and fast LDHA proteins only differ at two amino acid sites and neither change is involved in substrate or NAD binding. Even so, these changes could affect the catalytic properties of the enzyme, or other properties such as thermostability. For example, an amino acid substitution between LDHB alleles in the fish, F. heteroclitus increases thermostability of the proteins found in southern populations [8]. Overall, the strong association between LdhA genotype and habitat, and different patterns of protein variation in the two Ldh loci, suggest that experimental studies to determine the adaptive significance of amino acid variation in LDHA are warranted.
Conclusions
The two L-Ldh loci in the Daphnia pulex species complex show substantial variation at the nucleotide level, but the amino acid sequence of LDHA is much more conserved than that of LDHB suggesting that the intensity of purifying selection on LdhA is much stronger. Patterns of nucleotide variation also indicate that the fast LdhA allele arose very recently and spread rapidly over a very wide geographic area, possibly during a recent demographic expansion of D. pulicaria lake populations. Although lake-dwelling D. pulicaria hybridizes with the other lineages in the pulex complex, it remains distinct ecologically and genetically. This, coupled with the strong association between LdhA genotype and habitat, suggests that experimental studies would be useful to determine if there is a link between the protein variants and differences in enzymatic function, which could then be evaluated for their effects on fitness to test the hypothesis that LDHA variation is adaptive.
References
Charlesworth B: Molecular population genomics: a short history. Genet Res Camb. 2010, 92: 397-411. 10.1017/S0016672310000522.
Avise J: Molecular Markers, Natural History, and Evolution. 2004, Sunderland, Sinauer, Second
Eanes WF: Analysis of selection on enzyme polymorphisms. Annu Rev Ecol Syst. 1999, 30: 301-326. 10.1146/annurev.ecolsys.30.1.301.
Storz JF, Wheat CW: Integrating evolutionary and functional approaches to infer adaptation at specific loci. Evolution. 2010, 64: 2489-2509. 10.1111/j.1558-5646.2010.01044.x.
Dalziel AC, Rogers SM, Schulte PM: Linking genotypes to phenotypes and fitness: how mechanistic biology can inform molecular ecology. Mol Ecol. 2009, 18: 4997-5017. 10.1111/j.1365-294X.2009.04427.x.
Powers DA, Schulte PM: Evolutionary adaptations of gene structure and expression in natural populations in relation to a changing environment: A multidisciplinary approach to address the million-year saga of a small fish. J Exp Zool. 1998, 282: 71-94. 10.1002/(SICI)1097-010X(199809/10)282:1/2<71::AID-JEZ11>3.0.CO;2-J.
Schulte PM: Environmental adaptations as windows on molecular evolution. Comp Biochem Physiol B: Biochem Mol Biol. 2001, 128: 597-611. 10.1016/S1096-4959(00)00357-2.
Bernardi G, Sordino P, Powers DA: Concordant mitochondrial and nuclear-DNA phylogenies for populations of the teleost fish Fundulus heteroclitus. Proc Natl Acad Sci USA. 1993, 90: 9271-9274. 10.1073/pnas.90.20.9271.
Rees BB, Bowman JAL, Schulte PM: Structure and sequence conservation of a putative hypoxia response element in the lactate dehydrogenase-B gene of Fundulus. Biol Bull. 2001, 200: 247-251. 10.2307/1543505.
Rees BB, Figueroa YG, Wiese TE, Beckman BS, Schulte PM: A novel hypoxia-response element in the lactate dehydrogenase-B gene of the killifish Fundulus heteroclitus. Comp Biochem Physiol A: Mol Integr Physiol. 2009, 154: 70-77. 10.1016/j.cbpa.2009.05.001.
Černý M, Hebert PDN: Genetic diversity and breeding system variation in Daphnia pulicaria from North American lakes. Heredity. 1993, 71: 497-507. 10.1038/hdy.1993.168.
Hebert PDN: Clonal diversity in cladoceran populations. Population Biology: Retrospect and Prospect. Edited by: King C, Dawson P. 1983, New York, Columbia University Press, 37-60.
Hebert PDN, Beaton MJ, Schwartz SS, Stanton DJ: Polyphyletic origins of asexuality in Daphnia pulex. 1. Breeding-system variation and levels of clonal diversity. Evolution. 1989, 43: 1004-1015. 10.2307/2409581.
Dudycha JL, Tessier AJ: Natural genetic variation of life span, reproduction, and juvenile growth in Daphnia. Evolution. 1999, 53: 1744-1756. 10.2307/2640437.
Dudycha JL: Mortality dynamics of Daphnia in contrasting habitats and their role in ecological divergence. Freshwater Biology. 2004, 49: 505-514. 10.1111/j.1365-2427.2004.01201.x.
Cristescu ME, Colbourne JK, Radivojc J, Lynch M: A micro satellite-based genetic linkage map of the waterflea, Daphnia pulex: On the prospect of crustacean genomics. Genomics. 2006, 88: 415-430. 10.1016/j.ygeno.2006.03.007.
Cristescu ME, Innes DJ, Stillman JH, Crease TJ: D- and L-lactate dehydrogenases during invertebrate evolution. BMC Evol Biol. 2008, 8: 268-10.1186/1471-2148-8-268.
Cristescu ME, Egbosimba EE: Evolutionary history of d-lactate dehydrogenases: A phylogenomic perspective on functional diversity in the FAD binding oxidoreductase/transferase type 4 family. J Mol Evol. 2009, 69: 276-287. 10.1007/s00239-009-9274-x.
Hebert PDN, Schwartz SS, Ward RD, Finston TL: Macrogeographic patterns of breeding system diversity in the Daphnia pulex group. 1. Breeding systems of Canadian populations. Heredity. 1993, 70: 148-161. 10.1038/hdy.1993.24.
Crease TJ, Lee SK, Yu SL, Spitze K, Lehman N, Lynch M: Allozyme and mtDNA variation in populations of the Daphnia pulex complex from both sides of the Rocky Mountains. Heredity. 1997, 79: 242-251. 10.1038/hdy.1997.151.
Pfrender ME, Spitze K, Lehman N: Multi-locus genetic evidence for rapid ecologically based specation in Daphnia. Mol Ecol. 2000, 9: 1717-1735. 10.1046/j.1365-294x.2000.01062.x.
Omilian AR, Lynch M: Patterns of intraspecific DNA variation in the Daphnia nuclear genome. Genetics. 2009, 182: 325-336. 10.1534/genetics.108.099549.
Hebert PDN, Crease TJ: Clonal diversity in populations of Daphnia pulex reproducing by obligate parthenogenesis. Heredity. 1983, 51: 353-369. 10.1038/hdy.1983.40.
Weider LJ: Life-history variation among low arctic clones of obligately parthenogenetic Daphnia pulex - a diploid-polyploid complex. Oecologia. 1987, 73: 251-256. 10.1007/BF00377515.
Colbourne JK, Crease TJ, Weider LJ, Hebert PDN, Dufresne F, Hobæk A: Phylogenetics and evolution of a circumarctic species complex (Cladocera: Daphnia pulex). Biol J Linn Soc. 1998, 65: 347-365.
Mergeay J, Aguilera X, Declerck S, Petrusek A, Huyse T, De Meester L: The genetic legacy of polyploid Bolivian Daphnia: the tropical Andes as a source for the North and South American D. pulicaria complex. Mol Ecol. 2008, 17: 1789-1800. 10.1111/j.1365-294X.2007.03679.x.
McTaggart S, Dudycha JL, Omilian A, Crease TJ: Rates of recombination in the ribosomal DNA of apomictically propagated Daphnia obtusa lines. Genetics. 2007, 175: 311-320.
Doyle JJ, Doyle JL: A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987, 19: 11-15.
Innes DJ, Schwartz SS, Hebert PDN: Genotypic diversity and variation in mode of reproduction among populations in the Daphnia pulex group. Heredity. 1986, 57: 345-355. 10.1038/hdy.1986.134.
Vergilino R, Markova S, Ventura M, Manca M, Dufresne F: Reticulate evolution of the Daphnia pulex complex as revealed by nuclear markers. Mol Ecol. 2011, 20: 1191-1207. 10.1111/j.1365-294X.2011.05004.x.
Librado P, Rozas J: DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009, 25: 1451-1452. 10.1093/bioinformatics/btp187.
Hudson RR, Kreitman M, Aguade M: A test of neutral molecular evolution based on nucleotide data. Genetics. 1987, 116: 153-159.
Betrán EJ, Rozas J, Navarro A, Barbadilla A: The estimation of the number and the length distribution of gene conversion tracts from population DNA sequence data. Genetics. 1997, 146: 89-99.
Excoffier L, Laval G, Schneider S: Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evol Bioinf Online. 2005, 1: 47-50.
R Development Core Team: R: A Language and Environment for Statistical Computing. Vienna, R Foundation for Statistical Computing
Mr. Bayes v3.1.2. [http://mrbayes.csit.fsu.edu]
Colbourne JK, Hebert PDN: The systematics of North American Daphnia (Crustacea: Anomopoda): A molecular phylogenetic approach. Philos Trans R Soc London, Ser B. 1996, 351: 349-360. 10.1098/rstb.1996.0028.
Saitou N, Nei M: The neighbor-joining method - a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987, 4: 406-425.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007, 24: 1596-1599. 10.1093/molbev/msm092.
Dufresne F, Hebert PDN: Pleistocene glaciations and polyphyletic origins of polyploidy in an arctic cladoceran. Proc R Soc London, Ser B. 1997, 264: 201-206. 10.1098/rspb.1997.0028.
Wang-Sattler R, Blandin S, Ning Y, Blass C, Dolo G, et al: Mosaic genome architecture of the Anopheles gambiae species complex. PLoS ONE. 2007, 2: e1249-10.1371/journal.pone.0001249.
Heier CR, Dudycha JL: Ecological speciation in a cyclic parthenogen: Sexual capability of experimental hybrids between Daphnia pulex and Daphnia pulicaria. Limnol Oceanogr. 2009, 54: 492-502. 10.4319/lo.2009.54.2.0492.
Lynch M: The origins of eukaryotic gene structure. Mol Biol Evol. 2006, 23: 450-468.
Simonsen KL, Churchill GA, Aquadro CF: Properties of statistical tests of neutrality for DNA polymorphism data. Genetics. 1995, 141: 413-429.
Anisimova M, Bielawski JP, Yang Z: Accuracy and power of bayes prediction of amino acid sites under positive selection. Mol Biol Evol. 2002, 19: 950-958.
Kryazhimskiy S, Plotkin JB: The population genetics of dN/dS. PLoS Genetics. 2008, 4: e1000304-10.1371/journal.pgen.1000304.
Hoekstra HE, Hirschmann RJ, Bundey RA, Insel PA, Crossland JP: A single amino acid mutation contributes to adaptive beach mouse color pattern. Science. 2006, 313: 101-104. 10.1126/science.1126121.
Wheat CW, Haag C, Marden JH, Hanski I, Frilander M: Nucleotide polymorphism at a gene (Pgi) under balancing selection in a butterfly metapopulation. Mol Biol Evol. 2010, 27: 267-281. 10.1093/molbev/msp227.
Scott GR, Schulte PM, Egginton S, Scott ALM, Richards JG, Milsom WK: Molecular evolution of cytochrome c oxidase underlies high-altitude adaptation in the bar-headed goose. Mol Biol Evol. 2011, 28: 351-363. 10.1093/molbev/msq205.
Tessier AJ, Welser J: Cladoceran assemblages, seasonal succession and the importance of a hypolimnetic refuge. Freshwater Biology. 1991, 25: 85-93. 10.1111/j.1365-2427.1991.tb00475.x.
Zeng K, Fu YX, Shi S, Wu CI: Statistical tests for detecting positive selection by utilizing high-frequency variants. Genetics. 2006, 174: 1431-1439. 10.1534/genetics.106.061432.
Acknowledgements
This work was funded by Discovery Grants from the Natural Sciences and Engineering Research Council to TJC, MEC and DI. We thank E. Egbosimba and B. Demiri for assistance with cloning and sequencing. S. Adamowicz, C. Cáceres, A. Constantin, F. Dufresne, M. Lynch, A. Petrusek, and R. Vergilino provided Daphnia or DNA samples. We thank the Genomics Facility at the University of Guelph for processing the sequencing reactions. Comments from several anonymous reviewers improved earlier versions of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
RF cloned the Ldh genes and collected the sequence data. TJC performed the diversity analyses and constructed the NJ trees. MEC constructed the Bayesian trees. DI performed the Mantel tests. TJC, MEC and DI conceived the study and participated in its design. All authors helped to draft the manuscript and approved the final version.
Electronic supplementary material
12862_2011_1805_MOESM1_ESM.PDF
Additional file 1: Supplementary tables. This PDF file contains 4 tables as follows. 1.1 - Isolates from the Daphnia pulex species complex analyzed for this study. 1.2 - List of LdhA and LdhB allele groups used in the nt diversity analysis of D. pulex and D. pulicaria. 1.3 - Results of the recombination analysis among alleles of LdhA in D. pulicaria and D. pulex. 1.4 - Results of the recombination analysis among alleles of LdhB in D. pulicaria and D. pulex. (PDF 76 KB)
12862_2011_1805_MOESM2_ESM.TXT
Additional file 2: Ldh A nucleotide sequence alignment. A sequential alignment of LdhA sequences from members of the Daphnia pulex species complex in FastA and Nexus formats. (TXT 403 KB)
12862_2011_1805_MOESM3_ESM.TXT
Additional file 3: Ldh B nucleotide sequence alignment. A sequential alignment of LdhB sequences from members of the Daphnia pulex species complex in FastA and Nexus formats. (TXT 490 KB)
12862_2011_1805_MOESM4_ESM.TXT
Additional file 4: LDHA amino acid alignment. Sequential alignment of LDHA sequences from members of the Daphnia pulex species complex in FastA and Nexus formats. (TXT 106 KB)
12862_2011_1805_MOESM5_ESM.TXT
Additional file 5: LHDB amino acid alignment. Sequential alignment of LDHB sequences from members of the Daphnia pulex species complex in FastA and Nexus formats. (TXT 116 KB)
12862_2011_1805_MOESM6_ESM.PDF
Additional file 6: Intron phylogenies. Bayesian phylogeny of (a) LdhA and (b) LdhB intron sequences from members of the Daphnia pulex species complex. Estimates of posterior probability larger than 50 are indicated on the nodes of the tree. The trees are rooted through the Daphnia obtusa sequence. (PDF 497 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Crease, T.J., Floyd, R., Cristescu, M.E. et al. Evolutionary factors affecting Lactate dehydrogenase A and B variation in the Daphnia pulexspecies complex. BMC Evol Biol 11, 212 (2011). https://doi.org/10.1186/1471-2148-11-212
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-11-212