
Abstract
Background
Recent increases in genomic studies of the developing human fetus and neonate have led to a need for widespread characterization of the functional roles of genes at different developmental stages. The Gene Ontology (GO), a valuable and widely-used resource for characterizing gene function, offers perhaps the most suitable functional annotation system for this purpose. However, due in part to the difficulty of studying molecular genetic effects in humans, even the current collection of comprehensive GO annotations for human genes and gene products often lacks adequate developmental context for scientists wishing to study gene function in the human fetus.
Description
The Developmental FunctionaL Annotation at Tufts (DFLAT) project aims to improve the quality of analyses of fetal gene expression and regulation by curating human fetal gene functions using both manual and semi-automated GO procedures. Eligible annotations are then contributed to the GO database and included in GO releases of human data. DFLAT has produced a considerable body of functional annotation that we demonstrate provides valuable information about developmental genomics. A collection of gene sets (genes implicated in the same function or biological process), made by combining existing GO annotations with the 13,344 new DFLAT annotations, is available for use in novel analyses. Gene set analyses of expression in several data sets, including amniotic fluid RNA from fetuses with trisomies 21 and 18, umbilical cord blood, and blood from newborns with bronchopulmonary dysplasia, were conducted both with and without the DFLAT annotation.
Conclusions
Functional analysis of expression data using the DFLAT annotation increases the number of implicated gene sets, reflecting the DFLAT’s improved representation of current knowledge. Blinded literature review supports the validity of newly significant findings obtained with the DFLAT annotations. Newly implicated significant gene sets also suggest specific hypotheses for future research. Overall, the DFLAT project contributes new functional annotation and gene sets likely to enhance our ability to interpret genomic studies of human fetal and neonatal development.
Similar content being viewed by others


Background
A growing awareness of developmental impacts on lifelong health [1, 2], as well as the need to understand the functional implications of prenatally diagnosed sonographic and chromosomal abnormalities, has motivated expanded interest in studying molecular processes that affect the fetus and neonate. In the past, biologists relied on analyses of cultured cells or animal models to understand human development. Now, gene expression data sets derived from the human placenta [3], cell-free RNA in amniotic fluid [4, 5], or the blood of pregnant women [6] are increasingly being made available. Correctly interpreting such data is important, because it is only a matter of time before sequence, expression, and clinical data are integrated to provide a comprehensive view of development that influences clinical decisions.
Our ability to understand and interpret such high-throughput molecular data, however, depends heavily on our knowledge of gene function [7]. This knowledge has long influenced the analysis of gene expression data [8, 9] and is relevant to the interpretation of sequence variation as well [10]. Most commonly, functional annotation databases, in which genes or gene products are linked to particular molecular, biological, or other functional processes, are scanned to find annotation categories that are statistically overrepresented in the set of implicated genes. In addition to post hoc analyses of functional categories overrepresented in lists of individually-implicated genes, it has become commonplace to use pre-defined gene sets as a group to identify the implicated pathways [11–15]. For example, a gene set implicated in the process of “single strand break repair” might consist of the genes APLF, APTX, LIG4, SIRT1, TDP1, and TNP1. Even if none of these genes is itself significantly upregulated in a set of phenotypically related samples, if all of the genes are moderately upregulated, the consistency of those changes might indicate that the process is indeed upregulated in the phenotype. Such gene-set analysis methods can be highly effective, but only if the functional annotation used to create the gene sets is informative about the specific conditions being studied [16].
There are several sources of functional pathway annotation used for this purpose. The most frequently referenced annotation source is the Gene Ontology (GO) [17], a collaborative effort to standardize the functional annotation of genes and gene products using a controlled vocabulary of terms connected by relationships that result in directed, acyclic graphs. The application of this vocabulary allows broad inferences to be made based on the grouping of many isolated annotations. Community participation and shared standards encourage consistent annotation across a wide range of species. GO annotations, linked to their supporting evidence in the primary literature, are publicly available and broadly relevant to a range of fields. Although not initially designed explicitly for this purpose, annotation from the GO is often used for gene set analysis [11, 18–20].
The Gene Ontology’s framework for representing developmental processes is quite detailed [21, 22]. However, necessarily, much of the human genetic information in the GO database is derived from research conducted on adult subjects or in cultured cells. Other annotations linking genes to human developmental processes are derived from studies of embryonic development in invertebrate model organisms such as C. elegans or D. melanogaster. Many of these genes do indeed have human orthologs with similar functions, especially in the realms of cell polarity, neurological development, and immunity [23]. However, other human developmental processes, particularly those crucial in later stages of development, are not as well modeled in these organisms as they are in vertebrates such as M. musculus[24, 25]. Molecular developmental annotation in mouse is substantial [22, 26, 27], but it has not yet systematically been leveraged to extend the human developmental annotation in GO. Thus, functional analyses in humans can lead to results that are insufficiently informative about the normal physiological changes occurring in the developing fetus.
Here, we describe our efforts to address this limitation through a project entitled “Developmental FunctionaL Annotation at Tufts” (DFLAT). The goal of the DFLAT project is to improve our understanding of human fetal development by adding appropriate human-specific, developmentally-relevant annotation to the Gene Ontology (GO) database and by maintaining a collection of gene sets tailored for use in studying human development. The next section of the manuscript, Construction and content, describes the methods used to derive the DFLAT annotation and gene sets.
We then assess the impact of the DFLAT annotation. Functional annotation can be used in many ways, but one common application is for functional analyses of high-throughput molecular data. In the section Utility and discussion , we describe a case study in which we use Biological Process gene sets derived from DFLAT-augmented GO annotation to analyze data from several previously published gene expression microarray experiments. Comparison of the analytical results to those derived from existing annotation demonstrates that using the annotation and gene sets provided by DFLAT allows researchers to more accurately perform gene set and pathway enrichment analyses when studying human development.
Construction and content
Annotation: curation and inference
Annotation related to human development was manually collected from the literature by DFLAT curators using the Protein2GO curation tool [28] and the methodology of the Gene Ontology Consortium [29]. Annotation efforts were focused particularly on genes relating to developmental and biological processes that neonatologists considered likely to be detected in fetal expression data obtained during the second and third trimesters, including heart, lung, and brain development. Eligible annotations were submitted to the Gene Ontology directly through Protein2GO and included in subsequent data releases [30].
In examining the literature, curators often found that functional information as presented in some papers did not quite meet the current standards for human GO curation. For example, a paper might refer to a gene product in a vague way that could be mapped to two or more different UniProt IDs, or the relevant sentence might require a TAS (Traceable Author Statement) evidence code, which is not considered sufficiently strong evidence for human GO annotation unless the source of the assertion has been traced back to the original experimental data. (For example, GONE links the gene LGI1 and the GO term “hippocampus development” (GO:0021766) with a TAS evidence code. This annotation was derived from a sentence in one paper [31] that refers to another paper’s evidence about LG1’s role in synapse maturation in the hippocampus).
Such annotations can nonetheless be valuable in the aggregate for use by gene set analysis methods, which are designed to find significant patterns across multiple genes and are therefore easily able to handle a slightly higher noise rate in gene set membership. These annotations were stored in a separate collection, described as “Gene Ontology Non-Eligible” (GONE). DFLAT curators have so far submitted 613 manually-curated annotations to GO and 664 to GONE. Both sets of annotation are available on the DFLAT website (http://dflat.cs.tufts.edu).
Because of the slow pace of manual curation, we were interested in determining whether the judicious use of mammalian orthology might provide a fast yet accurate way to augment the human developmental annotation. Automated orthology-mapping of annotation was performed using one-to-one mouse-to-human orthologs taken from the Mouse Genome Informatics (MGI) database (orthology assertion as defined by Homologene [32]). Only annotations within the developmental branch of the GO were used to transfer annotations between species. Mapping was limited to annotations with experimental evidence codes (EXP, IDA, IPI, IMP, IGI, and IEP). Evidence codes indicating annotation derived from computational analyses, including ISS and its children, were removed to avoid generalizing annotations that might have originally been derived through orthology. Automatically assigned, author statements, and curator statements were also excluded. A similar approach is standard in the Mouse Genome Informatics’ Gene Ontology annotation pipeline.
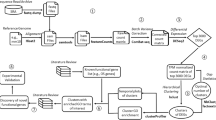
In total, 12,798 unique orthologous annotations were derived from this approach. An overview of the different sources of data in DFLAT appears in Figure 1.

Overview of sources of DFLAT annotation. On the left-hand side, neonatologists suggest keywords and developmental processes for manual curation of the literature, which proceeds according to the methodology of the Gene Ontology Consortium. Eligible annotations are submitted to the Gene Ontology and included in subsequent data releases. Others, valuable for our purposes, become part of the “GONE” collection. The right-hand side of the image depicts our procedure for deriving annotation from mouse orthologs. Mouse genes of interest are identified by having GO annotations in the “Developmental Process” subtree. For those genes for which MGI has identified unique human orthologs, all mouse annotations with the required evidence codes are mapped to the corresponding human gene.
We note that this use of orthologs affects our ability to focus on a specific developmental stage. Annotations to GO terms that are descendants of the term “Developmental Process” are not necessarily limited to fetal or even prenatal development. Furthermore, while there have been attempts to broadly align developmental stages in mouse and human (e.g., [33]), developmental processes in specific organs or systems may take place at very different times from those predicted by such alignment [34], with some processes that are completed in the human fetus continuing postnatally in the mouse (and vice-versa). (E.g., the human cortex is comparatively well developed at birth, and the development of some perceptual systems is much further along in neonatal humans than in mice [35]. For example, the onset of hearing in the mouse develops during the second postnatal week, with the maturation of the organ of Corti [36], while in the human this occurs prenatally by approximately the 24th week of gestation [37]). Thus, by including orthology-derived annotation, we are using a wide brush, potentially including information about not only fetal but also neonatal and perhaps subsequent developmental processes. Some implications of this decision are discussed in the section Utility and discussion, below.
Construction of DFLAT gene sets
Manually curated GO and GONE annotations, the ortholog-derived annotations described above, and human Gene Ontology Annotation (GOA) (downloaded 4/10/13) were collected in a single GAF 2.0 annotation file. Gene Symbols were derived from UniProt IDs (using the mapping at http://www.uniprot.org/). Annotations identified in more than one group (such as manually-curated and orthology-derived annotations) were consolidated. In total, 13,344 unique DFLAT annotations were combined with the existing Gene Ontology data.
We then used this full annotation collection to create GMT-formatted gene sets (http://www.broadinstitute.org/cancer/software/gsea/wiki/index.php/Data_formats) for use in Gene Set Enrichment Analysis. This simple text file format is readily convertible for use with other gene set analysis methods.
In an effort to maximize consistency with the GO-derived collection of gene sets (known as “C5”) in GSEA’s Molecular Signature database [38], we used only GO and GONE annotations with the evidence codes IDA, IPI, IMP, IGI, IEP, ISS, or TAS in the formation of our gene sets (http://www.broadinstitute.org/gsea/msigdb/collection_details.jsp). All orthologous annotations were included as well. Annotations with the NOT qualifier were excluded. Our gene set collection and the C5 collection both consist of gene sets corresponding to GO terms, with each gene set containing the human genes annotated with that particular term.
To reflect the propagation of annotation terms through the Gene Ontology, and for further consistency with the C5 collection, gene annotations were propagated to all ancestors of the original annotated GO term (Figure 2). For this purpose, ancestors were determined by is_a and part_of relationships, as defined in the gene_ontology_ext.obo file (downloaded 4/10/13). DFLAT’s developmentally-focused human gene sets, as well as the scripts used to create them, are freely available on the DFLAT website (http://dflat.cs.tufts.edu).

Propagation of Gene Ontology annotation. Genes or gene products (letters A-P) are propagated up the GO graph from the original GO annotation term to all of its parents via is_a (solid arrows) and part_of (dashed arrows) relationships. Bold text letters indicate genes or gene products directly annotated to the GO term; the remaining letters indicate propagated annotations.
Despite our efforts to match the methods used in the construction of the C5 collection, unavoidable differences in the GOA source data (based on date of download) and manual review steps in the C5 gene set construction process made it impossible to evaluate the impact of the new annotation by direct comparison to C5. Accordingly, for the purpose of comparison, we also created a collection of gene sets using the same methods used to create the DFLAT gene sets described above, but containing only the downloaded GOA data. These “GOA-only” gene sets are also available on our web site. Gene sets with fewer than 7 genes are generally considered too small to be used in gene set analysis [11], although we included the smaller gene sets in our data release for completeness. Compared to these “GOA-only” gene sets, the Biological Process gene set collection created with the additional DFLAT annotation included 381 additional GO terms whose corresponding gene sets contain at least 7 genes (and a total of 476 additional GO terms of any size).
Utility and discussion
DFLAT’s primary purpose is to enable the study of human development through gene-set or pathway-based analyses of high throughput expression data. We hypothesized that the collection of additional annotation tailored to fetal and neonatal developmental stages would result in more biologically relevant and accurate results in the analysis of expression data from patients in these age groups than would be obtained using existing functional annotation. To demonstrate DFLAT’s impact, we therefore used gene set enrichment analysis (GSEA) to analyze five gene expression data sets with the GOA-only gene set collection and, separately, with the DFLAT-augmented gene set collection. The results of this comparative analysis demonstrate that DFLAT is serving its intended role by improving our functional analyses of fetal, neonatal, and even later developmental expression data.
Data and methods for comparison of DFLAT and GOA-only analyses
The data sets include two studies of cell-free mRNA gene expression in amniotic fluid of aneuploid fetuses compared to euploid controls. The first study identified patterns of oxidative stress and its consequences in second-trimester trisomy 21 fetuses compared to age- and sex-matched controls [39]. The second characterized the differences between second-trimester amniotic fluid in trisomy 18 fetuses and euploid controls [40], implicating ion transport, immunity, glycosylation, and G-protein mediated signaling pathways. The third data set comes from a paper demonstrating the possibility of identifying cell-free fetal RNA in maternal blood taken shortly before full-term elective cesarean deliveries [6]. The fourth data set characterizes complications of prematurity in expression patterns of blood samples drawn from neonates born below 1500 g and before 32 weeks’ gestational age [41]. From this complex data set, we focused on the long-term respiratory complication of prematurity, bronchopulmonary dysplasia (BPD). We selected just the samples taken on approximately the 14th postnatal day (time point B, corresponding to subjects still below 34 weeks’ gestational age) and categorized them as either having BPD (including mild, moderate, and severe cases as indicated by the authors) or not. The final data set compared acute megakaryoblastic leukemia in patients with and without Down syndrome [42]. Because the DFLAT annotation is focused on fetal development, we hypothesized that its impact would be greatest on gene expression data from very young subjects. Yet few of the samples are truly neonatal. Accordingly, we selected from the leukemia data set only those samples from subjects younger than 24 months. We refer to these five data sets in the tables below as “trisomy 21”, “trisomy 18”, “maternal/fetal blood”, “BPD”, and “leukemia”, respectively. Further details about the data sets, including their Gene Expression Omnibus (GEO) IDs, appear in Additional file 1: Table S1. Each data set was normalized using the method described in the associated GEO record.
For all analyses, we wrote scripts to run the java implementation of GSEA (version 2–2.07) [11] in batch mode. In all cases but one, the analysis was a straightforward comparison of two classes, and the normalized data sets were provided as input to GSEA. The exception was the maternal/fetal blood data set. Because this analysis results from a more complex comparison that is not easily expressed as a simple binary classification problem, we instead ran a GSEA “pre-ranked” analysis. For this analysis, microarray qualifiers were ranked by the log of the maximum p-value from one of two comparisons: antepartum maternal blood compared to postpartum maternal blood from the same mother, and antepartum maternal blood compared to umbilical cord blood from each mother and her baby [6]. The genes with the most extreme negative ranks are those that are most significantly up-regulated in antepartum maternal blood compared to both postpartum maternal blood and cord blood, and are thus potentially fetal in origin.
The most meaningful measurement of gene set enrichment is often the false discovery rate (FDR) q-value, which GSEA derives through permutation. However, for several reasons, we will discuss only the raw p-values here. First, it is difficult to compare false-discovery rates fairly between gene set collections containing different numbers of gene sets. In addition, some of the data sets, particularly the trisomy 18 study, contain relatively few samples in each class. Most gene sets in such experiments have q-values of 1.0 for both gene set collections, but their p-values still provide evidence about the differences between the two gene set collections. Finally, adjusting for multiple testing between highly-overlapping gene sets such as those derived from the Gene Ontology is a fraught process to begin with, and such adjustment methods ought to take the overlap structure into account [43, 44]. Yet this overlap structure also differs between the collections. For the purposes of evaluating the differences between the two collections, therefore, we have chosen to focus exclusively on the unadjusted p-values reported by GSEA.
Gene set availability increases with DFLAT
In gene set enrichment analysis (GSEA), the gene sets considered for each analysis are typically selected based on the gene set sizes (i.e., the number of genes in each gene set that are included in the input gene expression data). We chose to require a minimum of 7 genes and a maximum of 150 genes for all gene sets in the analyses described here, because with fewer than about 7 genes the permutation analyses to assess significance lack sufficient granularity [11], and because gene sets larger than approximately 150 genes tend to be high-level, broad categories that are not very informative. DFLAT contains more annotations than GOA, increasing the number of genes in many of the gene sets. As a result, using the DFLAT collection allows some gene sets to grow large enough to meet the minimum size threshold in GSEA. We call these newly-included gene sets “unique to DFLAT”. However, other, larger gene sets may grow in size enough to be disqualified; we call these “unique to GOA”.
One important question is therefore to evaluate the tradeoff between the gene sets gained and those lost in the DFLAT analysis. In all five studies, we find more gene sets unique to DFLAT than unique to GOA (Table 1). The total number of gene sets meeting these size criteria, for DFLAT/GOA respectively, is 3,700/3,358 for the three data sets on Affymetrix U133 Plus 2.0 arrays (Trisomy 21, Trisomy 18, and BPD); 3,563/3,245 for the Maternal/Fetal Blood data set on U133A arrays, and 3,649/3,332 for the Leukemia data set (where the published data in GEO mapped the qualifiers on the U133A arrays to gene symbols). Even if we restrict our attention to the most significantly-changing gene sets, those with p-values below 0.05 (Table 1, center and right-hand columns), we see that DFLAT is contributing more significantly-implicated gene sets to the results than it is taking away.
The annotation we have added is intentionally functionally-biased, augmenting some categories more than others. Previous work has demonstrated that despite this bias, such functionally-focused annotation efforts can have a positive impact on the outcome of downstream analyses [45]. We contend that having larger numbers of gene sets available for analysis is likely to have a similar positive effect, because functionally-coherent differential expression can only be detected if the relevant gene sets are considered [16]. The primary lesson from Table 1 is that by adding focused annotation, we have increased the number of available gene sets. The resulting analyses indeed show a corresponding increase in the number of gene sets significantly implicated in the differential expression analyses.
Table 1 does not, however, suggest that the gene sets unique to DFLAT are more likely to be significantly enriched than their unique-to-GOA counterparts. By a one-sided Fisher’s exact test, there is no evidence that the fraction of unique DFLAT gene sets that are significant at the .05 level is larger than the fraction of GOA gene sets that are significant at the .05 level, except in the BPD data set (p-value < 0.038). And in fact, the leukemia data set shows a significant difference (p-value < 0.01) in the other direction. Possibly this is due to the fact that it is the only one of these data sets including subjects beyond the fetal and neonatal developmental stages targeted in the DFLAT curation process. But even if the number of significant gene sets is roughly proportional to the number of gene sets considered, as long as the user’s false discovery rate cutoff remains the same, an increase in the number of gene sets will likely translate to a larger number of correct discoveries.
The addition of DFLAT annotation caused widespread changes in gene set membership. Therefore, the results of GSEA differential expression analyses across all five studies change substantially. We can compare these results further by looking at the gene sets that meet the size criteria for inclusion under both gene set collections (i.e., those that are not “unique to” either collection); we call these the “common” gene sets. Among common gene sets, the use of the DFLAT collection changed the reported raw p-values in nearly all of them. If we again focus just on those gene sets that are most enriched (those with p-values < 0.05 in either study), we still find that nearly all of them (91.4-99.5%) change in significance (data not shown). In most of the studies, about half of the gene sets common to both collections showed increased significance using DFLAT, and about half showed increased significance using the GOA-only collection.
When we consider all gene sets together (both common and unique), we find that by incorporating the DFLAT annotation, we typically gain more gene sets meeting a particular significance cutoff (e.g., in Table 2, p < 0.05) than we lose. Of course, drawing a hard line to indicate significance is somewhat arbitrary and problematic, but such cutoffs at least indicate that the additional annotation is expanding the number of enriched GO terms detected at a particular threshold.
These results appear encouraging. However, it is difficult to evaluate them properly without knowing which gene sets should be enriched in each analysis. This is a much more difficult problem, which we now address.
Literature verification supports DFLAT’s new contributions
The best representation of the current state of biomedical knowledge on a particular topic is the collection of relevant published literature. Indeed, this is the source of the Gene Ontology annotation in the first place; it is just that the labor-intensive annotation effort required and the accelerating pace of publication means that there is a substantial amount of information in the literature that has not yet been incorporated into GO.
We therefore attempted to partly assess the accuracy of annotation terms that have changed in “status” (i.e., crossed some significance cutoff) between the DFLAT and GOA-only analyses by reviewing the literature to find corroborative experimental evidence for such terms. Because this validation process is also labor-intensive, we performed this analysis only for the trisomy 21 and trisomy 18 studies (chosen because the phenotypes under investigation are relatively clear and well represented in the literature). Published evidence was sought linking the gene sets significantly enriched under only one annotation collection, tallied in Table 2, to the indicated aneuploidy. In order to eliminate bias, the team members performing the literature review were blinded to whether each listed gene set was significantly implicated in the DFLAT or the GOA-only analysis.
Each term was considered in the context of the indicated trisomy, and was assigned to one of two categories: either the indicated function was supported by a report in the literature, or no evidence about that function was found. A full list of the terms considered and the supporting references identified is available in Additional file 2: Table S2.
Our results, shown in Tables 3 (trisomy 21) and 4 (trisomy 18), indicate that GSEA analysis with DFLAT produces more newly-significant biological process terms whose relevance to the indicated trisomy is supported by publications than GSEA analysis of the same data using the GOA-only collection. The tables show the percentage (and numbers, in parentheses) of terms identified as significant with the indicated gene set collection that had literature support. Although we cannot easily determine the significance of the differences reported in Tables 3 and 4, both the trend and the science behind the implicated gene sets are consistent with the hypothesis that the additional DFLAT annotation provides new and valuable information about the data sets being analyzed.
A closer look at specific gene sets identified in the trisomy 21 analyses gives insight into what new information DFLAT is providing or culling. Several gene sets that reach the 0.05 significance cut off in the GOA-only analysis are vague umbrella terms that provide little specific insight into trisomy 21, such as Axis specification, Determination of left/right symmetry, and Modified amino acid transport. None of these broad processes was directly linked to trisomy 21 in the literature. Instead, the enriched terms enriched from the DFLAT collection implicate a diverse spectrum of organ systems affected by trisomy 21, including the nervous system (Cranial nerve morphogenesis, Myelin, Cerebellum development, Midbrain development, Regulation of action potential of neuron), eye (Lens fiber cell differentiation, Photoreceptor cell development), urogenital tract (Renal tubule development, Ureter development, Prostate gland growth, Collecting duct development), and muscle (Regulation of striated muscle development, Myotube development, Muscle adaptation). All of these were supported by literature. The results of the GOA-only analysis, while implicating some of the same organ systems, tended to highlight broader gene sets or earlier stages of development (e.g., Embryonic camera-type eye development, Heart looping, Mesonephros development). That said, there were some informative and probably correct terms that were significant in the GOA-only analysis but not the DFLAT analysis, including Glial cell differentiation, Spermatogenesis, and Positive regulation of biomineral tissue development.
There is less literature describing the trisomy 18 phenotype, so it is not surprising that fewer Gene Ontology terms from either collection garnered literature support in the trisomy 18 analysis. However, many of those that were identified seem plausible. For example, Regulation of cardiac muscle cell differentiation and Atrioventricular valve development were identified in the DFLAT analysis, consistent with the observation that heart defects are common in trisomy 18 [46], while no significant terms related to heart development were unique to GOA.
Other molecular processes implicated by these analyses might form the basis for future studies. Particularly, in several cases, significantly enriched gene sets represent processes that seem very likely to be affected in trisomy 18, but direct research would be needed to confirm them. For example, neural tube defects including spina bifida are common in patients with trisomy 18 [46, 47]. Thus, while no current research directly links trisomy 18 explicitly with changes in Spinal cord dorsal/ventral patterning (a term that appeared significant with DFLAT), future studies could likely confirm this connection. Similarly, it would not be unexpected for Axon extension, Schwann cell development, and Schwann cell differentiation to be relevant. Several cases of malformed kidneys have also been described in trisomy 18, and it has been noted that renal dysplasia deserves further study in this context. Several kidney-related gene set terms (such as Glomerular basement membrane and Mesonephric tubule development) were identified as significant only with DFLAT. Though these terms do not have literature support, they provide specific testable hypotheses about the trisomy 18 phenotype.
Conclusions
A major contribution of the DFLAT project has been to capture critical information about developmental context to further characterize literature-derived functional annotation. The demonstrated impact of this effort on the interpretation of high-throughput molecular data suggests that ongoing Gene Ontology Consortium efforts to better model functional contexts will prove valuable.
The five data sets we analyzed here cover a range of developmental stages, including second-trimester fetuses, fetuses near term, premature and full-term neonates, and even infants and toddlers. Yet the DFLAT annotation appears to implicate additional pathways in the analysis of all of these data sets. DFLAT’s impact on the analyses of many different developmental stages is likely due to the introduction of the orthology-derived annotation, but it may also reflect the fact that some genes with known roles in fetal development may continue to play similar roles after birth. Future work will assess DFLAT’s impact on analyses of additional pediatric studies. Given the growing interest in molecular analyses of such pediatric disorders as autism, ADHD, and asthma, the potential availability of tools to improve the interpretation of such data would be quite valuable.
The orthology data used here is based heavily on mouse models. Recent controversial work on mouse models of human inflammatory disease has raised questions about when it is appropriate to rely on data from such models [48–52]. While anatomical comparisons of developmental landmarks are well established and generally consistent between the two organisms [53], there are still some species-specific differences, as mentioned above in the Construction and content section. These may very well increase the rate of false positive results. Future work might mitigate the impact of these differences by incorporating developmental data from additional vertebrate models such as rat and zebrafish. In addition, the EMAGE database [27] provides detailed temporospatial information characterizing expression patterns in the developing mouse. The possibilities for leveraging these and related data to further augment the DFLAT collection are intriguing. Although the risks of inferring function from expression are considerable, it might be possible to combine multiple data sources to do so reliably, potentially yielding a valuable resource combining molecular and anatomical data. Ultimately, of course, while functional analyses of expression data can be greatly helpful in generating or refining hypotheses, the biological significance of results obtained through the use of DFLAT – or any functional enrichment analysis – must be confirmed through further laboratory experiments.
We have focused here on DFLAT’s implications for gene expression data analysis. However, we emphasize that the interpretation of sequence variation also often relies on pre-defined gene sets, and that the integration of expression and sequence data is a powerful tool for analyzing the functional interactions of genetic and environmental factors [54]. The fraction of fetal DNA in maternal circulation is relatively low [55], but with the availability of low-cost deep sequencing technologies, it has become possible to detect fetal aneuploidy reliably and non-invasively, by comparing the chromosomal distribution of sequences from maternal blood to the expected distribution [56]. The recent sequencing of an entire fetal genome from amniocytes [57], and even from the cell-free fetal DNA that circulates within maternal plasma [58] has opened the door to a much larger range of personalized prenatal diagnostics [57]. It will thus be valuable to assess the impact of the DFLAT annotation on the interpretation of fetal genomic sequence as whole genome DNA sequencing becomes integrated into prenatal clinical care.
Overall, DFLAT has added more developmentally focused human annotation to GOA, providing a useful tool for researchers studying development in humans. In particular, DFLAT is well suited for gene set and pathway analysis on human fetal and neonatal expression data. Using DFLAT-derived gene sets in gene set enrichment analyses not only provides a larger number of enriched functions, but provides results that are more likely to be confirmed in the literature. Even those that have not yet been confirmed tend to implicate more specific molecular processes, leading to the development of novel, focused hypotheses about the molecular mechanisms behind specific pathologies.
Although there is growing evidence of a relationship between fetal, pediatric, and adult health, little is currently known about the molecular connections between neonatal health and later-onset disorders. The DFLAT annotation augments the infrastructure needed to analyze developmental gene function. We therefore expect that future applications of DFLAT annotation will facilitate the identification of previously-obscured developmental roles from genomic and clinical data sets, engendering novel insights into developmental impacts on life-long health.
Availability and requirements
All DFLAT annotations and gene sets are freely available from http://dflat.cs.tufts.edu. The code used to generate the automated annotation and gene sets can also be downloaded from this site. Those annotations meeting Gene Ontology curation standards are also available through the standard GO releases.
References
Barker DJ: The developmental origins of adult disease. Eur J Epidemiol. 2003, 18 (8): 733-736.
Calkins K, Devaskar SU: Fetal origins of adult disease. Curr Probl Pediatr Adolesc Health Care. 2011, 41 (6): 158-176. 10.1016/j.cppeds.2011.01.001.
Sood R, et al: Gene expression patterns in human placenta. Proc Natl Acad Sci USA. 2006, 103 (14): 5478-5483. 10.1073/pnas.0508035103.
Hui L, et al: The amniotic fluid transcriptome: a source of novel information about human fetal development. Obstet Gynecol. 2012, 119 (1): 111-118. 10.1097/AOG.0b013e31823d4150.
Larrabee PB, et al: Global gene expression analysis of the living human fetus using cell-free messenger RNA in amniotic fluid. JAMA. 2005, 293 (7): 836-842. 10.1001/jama.293.7.836.
Maron JL, et al: Gene expression analysis in pregnant women and their infants identifies unique fetal biomarkers that circulate in maternal blood. J Clin Invest. 2007, 117 (10): 3007-3019. 10.1172/JCI29959.
Slonim DK, Yanai I: Getting started in gene expression microarray analysis. PLoS Comput Biol. 2009, 5 (10): e1000543-10.1371/journal.pcbi.1000543.
Curtis RK, Oresic M, Vidal-Puig A: Pathways to the analysis of microarray data. Trends Biotechnol. 2005, 23 (8): 429-435. 10.1016/j.tibtech.2005.05.011.
Khatri P, Draghici S: Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 2005, 21 (18): 3587-3595. 10.1093/bioinformatics/bti565.
Rebbeck TR, Spitz M, Wu X: Assessing the function of genetic variants in candidate gene association studies. Nat Rev Genet. 2004, 5 (8): 589-597. 10.1038/nrg1403.
Subramanian A, et al: Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005, 102 (43): 15545-15550. 10.1073/pnas.0506580102.
Tian L, et al: Discovering statistically significant pathways in expression profiling studies. Proc Natl Acad Sci USA. 2005, 102 (38): 13544-13549. 10.1073/pnas.0506577102.
Efron B, Tibshirani R: On testing the significance of sets of genes. Annals of Applied Statistics. 2007, 1 (1): 107-129. 10.1214/07-AOAS101.
Torkamani A, Topol EJ, Schork NJ: Pathway analysis of seven common diseases assessed by genome-wide association. Genomics. 2008, 92 (5): 265-272. 10.1016/j.ygeno.2008.07.011.
Tarca AL, et al: Down-weighting overlapping genes improves gene set analysis. BMC Bioinformatics. 2012, 13: 136-10.1186/1471-2105-13-136.
Turcan S, et al: Mining functionally relevant gene sets for analyzing physiologically novel clinical expression data. Pac Symp Biocomput. 2011, 50: 61-
Gene Ontology Consortium: Gene ontology: tool for the unification of biology. Nat Genet. 2000, 25 (1): 25-29. 10.1038/75556.
Falcon S, Gentleman R: Using GOstats to test gene lists for GO term association. Bioinformatics. 2007, 23 (2): 257-258. 10.1093/bioinformatics/btl567.
Khatri P, et al: Profiling gene expression using onto-express. Genomics. 2002, 79 (2): 266-270. 10.1006/geno.2002.6698.
Dennis G, et al: DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4 (5): P3-10.1186/gb-2003-4-5-p3.
Gene Ontology Consortium: The Gene Ontology in 2010: extensions and refinements. Nucleic Acids Res. 2010, 38 (Database issue): D331-D335.
Khodiyar VK, et al: The representation of heart development in the gene ontology. Dev Biol. 2011, 354 (1): 9-17. 10.1016/j.ydbio.2011.03.011.
Anderson KV, Ingham PW: The transformation of the model organism: a decade of developmental genetics. Nat Genet. 2003, 33 (Suppl): 285-293.
Huelsken J, Birchmeier W: New aspects of Wnt signaling pathways in higher vertebrates. Curr Opin Genet Dev. 2001, 11 (5): 547-553. 10.1016/S0959-437X(00)00231-8.
Taylor MV: Comparison of Muscle Development in Drosophila and Vertebrates., in Madame Curie Bioscience Database [Internet]. 2000, Landes Bioscience: Austin (TX)
Armit C, et al: eMouseAtlas, EMAGE, and the spatial dimension of the transcriptome. Mamm Genome. 2012, 23 (9–10): 514-524.
Richardson L, et al: EMAGE: Electronic Mouse Atlas of Gene Expression. Methods Mol Biol. 2014, 1092: 61-79. 10.1007/978-1-60327-292-6_5.
Activities at the Universal Protein Resource (UniProt): UniProt Consortium. Nucleic Acids Res. 2014, 42 (1): D191-D198.
Gene Ontology Consortium: GO Annotation Policies and Guidelines. 1999, Available from: http://www.geneontology.org/GO.annotation.shtml see also http://www.geneontology.org/GO.annotation.SOP.shtml?all - literature, –2013
Gene Ontology Consortium: The Gene Ontology: enhancements for 2011. Nucleic Acids Res. 2012, 40 (Database issue): D559-D564.
Thomas R, et al: LGI1 is a Nogo receptor 1 ligand that antagonizes myelin-based growth inhibition. J Neurosci. 2010, 30 (19): 6607-6612. 10.1523/JNEUROSCI.5147-09.2010.
NCBI Resource Coordinators: Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2013, 41 (Database issue): D8-D20.
O’Rahilly R: Early human development and the chief sources of information on staged human embryos. Eur J Obstet Gynecol Reprod Biol. 1979, 9 (4): 273-280. 10.1016/0028-2243(79)90068-6.
Wessels A, Sedmera D: Developmental anatomy of the heart: a tale of mice and man. Physiol Genomics. 2003, 15 (3): 165-176.
Clancy B, et al: Extrapolating brain development from experimental species to humans. Neurotoxicology. 2007, 28 (5): 931-937. 10.1016/j.neuro.2007.01.014.
Theiler K: Stage 27: Newborn Mouse, in The House Mouse: Atlas of Mouse Development. 1989, New York: Springer-Verlag
Igarashi Y, Ishii T: Embryonic development of the human organ of Corti: electron microscopic study. Int J Pediatr Otorhinolaryngol. 1980, 2 (1): 51-62. 10.1016/0165-5876(80)90028-2.
Subramanian A, et al: GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics. 2007, 23 (23): 3251-3253. 10.1093/bioinformatics/btm369.
Slonim DK, et al: Functional genomic analysis of amniotic fluid cell-free mRNA suggests that oxidative stress is significant in Down syndrome fetuses. Proc Natl Acad Sci USA. 2009, 106 (23): 9425-9429. 10.1073/pnas.0903909106.
Koide K, et al: Transcriptomic analysis of cell-free fetal RNA suggests a specific molecular phenotype in trisomy 18. Hum Genet. 2011, 129 (3): 295-305. 10.1007/s00439-010-0923-3.
Pietrzyk JJ, et al: The use of microarrays for gene expression analysis in premature children--new strategy of searching for genetic basis of late complications of prematurity--preliminary research. Przegl Lek. 2011, 68 (1): 44-46.
Bourquin JP, et al: Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene expression profiling. Proc Natl Acad Sci USA. 2006, 103 (9): 3339-3344. 10.1073/pnas.0511150103.
Alexa A, Rahnenfuhrer J, Lengauer T: Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006, 22 (13): 1600-1607. 10.1093/bioinformatics/btl140.
Goeman JJ, Mansmann U: Multiple testing on the directed acyclic graph of gene ontology. Bioinformatics. 2008, 24 (4): 537-544. 10.1093/bioinformatics/btm628.
Alam-Faruque Y, et al: The impact of focused Gene Ontology curation of specific mammalian systems. PLoS One. 2011, 6 (12): e27541-10.1371/journal.pone.0027541.
Nyberg DA, Souter VL: Sonographic markers of fetal trisomies: second trimester. J Ultrasound Med. 2001, 20 (6): 655-674.
Ferreira AF, et al: Posterior brain in fetuses with trisomy 18, trisomy 13 and triploidy at 11 to 13 weeks’ gestation. Prenat Diagn. 2012, 32 (9): 854-858.
Seok J, et al: Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013, 110 (9): 3507-3512. 10.1073/pnas.1222878110.
Cauwels A, Vandendriessche B, Brouckaert P: Of mice, men, and inflammation. Proc Natl Acad Sci USA. 2013, 110 (34): E3150-10.1073/pnas.1308333110.
Osterburg AR, et al: Concerns over interspecies transcriptional comparisons in mice and humans after trauma. Proc Natl Acad Sci USA. 2013, 110 (36): E3370-10.1073/pnas.1306033110.
Tompkins RG, et al: Reply to Osterburg et al: To study human inflammatory diseases in humans. Proc Natl Acad Sci USA. 2013, 110 (36): E3371-10.1073/pnas.1307452110.
Warren HS, et al: Reply to Cauwels et al.: Of men, not mice, and inflammation. Proc Natl Acad Sci USA. 2013, 110 (34): E3151-10.1073/pnas.1308943110.
Sissman NJ: Developmental landmarks in cardiac morphogenesis: comparative chronology. Am J Cardiol. 1970, 25 (2): 141-148. 10.1016/0002-9149(70)90575-8.
Idaghdour Y, Awadalla P: Exploiting gene expression variation to capture gene-environment interactions for disease. Front Genet. 2012, 3: 228-
Ashoor G, et al: Fetal fraction in maternal plasma cell-free DNA at 11–13 weeks’ gestation: relation to maternal and fetal characteristics. Ultrasound Obstet Gynecol. 2013, 41 (1): 26-32. 10.1002/uog.12331.
Bianchi DW, et al: Genome-wide fetal aneuploidy detection by maternal plasma DNA sequencing. Obstet Gynecol. 2012, 119 (5): 890-901. 10.1097/AOG.0b013e31824fb482.
Talkowski ME, et al: Clinical diagnosis by whole-genome sequencing of a prenatal sample. N Engl J Med. 2012, 367 (23): 2226-2232. 10.1056/NEJMoa1208594.
Snyder MW, et al: Noninvasive fetal genome sequencing: a primer. Prenat Diagn. 2013, 33 (6): 547-554. 10.1002/pd.4097.
Acknowledgements
We thank David Hill of the Jackson Laboratory for his help with the developmental Gene Ontology and mouse orthology data. We thank members of the GOA team, including Rolf Apweiler, Emily Dimmer, Rachel Huntley, and Amelia Ireland, for their support and for helpful discussions and advice. We are also grateful to Arthur Lieberzon, Aravind Subrmanian, and Jill Mesirov of the Broad Institute for their willingness to share detailed information about the construction of the Molecular Signature Database. The work reported in this publication was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development under award number R01HD058880 and R01HD076140. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DS and DB conceived of the study; with HW they participated in its design. DS provided general oversight of the project; DB provided oversight regarding medical practice and neonatal genetics; JB provided oversight regarding the Gene Ontology and functional annotation standards. HD trained HW and CF in GO literature curation; HW, CF, and HD contributed curated annotation to DFLAT and GO. HN, MS, and HW wrote and tested computer programs for ortholog mapping and gene set construction. Literature validation of the trisomy 21 and 18 results was performed by JH and HW. The manuscript was drafted by HW and DS and edited by DB, JB, and HD. All authors read and approved the final manuscript.
Electronic supplementary material
12859_2013_6309_MOESM1_ESM.docx
Additional file 1: Table S1: Details of the five gene expression data sets described in the Utility and discussion section. (DOCX 84 KB)
12859_2013_6309_MOESM2_ESM.docx
Additional file 2: Table S2: a. Uniquely significant gene sets in trisomy 21 and Pubmed IDs of papers providing supporting evidence. b. Uniquely significant gene sets in trisomy 18 and Pubmed IDs of papers providing supporting evidence. (DOCX 154 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wick, H.C., Drabkin, H., Ngu, H. et al. DFLAT: functional annotation for human development. BMC Bioinformatics 15, 45 (2014). https://doi.org/10.1186/1471-2105-15-45
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-15-45