
Abstract
Infection with dengue virus (DENV) causes both mild dengue fever and severe dengue diseases, such as dengue hemorrhagic fever and dengue shock syndrome. The pathogenic mechanisms for DENV are complicated, involving viral cytotoxicity, immunopathogenesis, autoimmunity, and underlying host diseases. Viral load correlates with disease severity, while the antibody-dependent enhancement of infection largely determines the secondary effects of DENV infection. Epidemiological and experimental studies have revealed an association between the plasma levels of interleukin (IL)-10, which is the master anti-inflammatory cytokine, and disease severity in patients with DENV infection. Based on current knowledge of IL-10-mediated immune regulation during infection, researchers speculate an emerging role for IL-10 in clinical disease prognosis and dengue pathogenesis. However, the regulation of dengue pathogenesis has not been fully elucidated. This review article discusses the regulation and implications of IL-10 in DENV infection. For future strategies against DENV infection, manipulating IL-10 may be an effective antiviral treatment in addition to the development of a safe dengue vaccine.
Similar content being viewed by others


Review
Dengue virus infection
Infection with the four serotypes of dengue virus (DENV), a mosquito-borne virus belonging to the family Flaviviridae, causes a global burden of 50 million infections per year occurring across approximately 100 countries [1]. DENV infection results in a wide range of disorders, ranging from mild dengue fever (DF) to severe dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS), which can cause death in the absence of appropriate medication [1]. Dengue patients frequently present clinical symptoms ranging from a mild fever to an incapacitating high fever with severe headache, pain behind the eyes, muscle and joint pain, and rash. However, in patients with severe DHF/DSS, potentially lethal complications include plasma leakage, severe hemorrhage, and organ failure, and these complications can affect both children and adults [1, 2].
The enveloped single-stranded RNA virus of dengue virus (DENV) contains 3 structural proteins, including the envelope (E) protein, the precursor membrane (prM) protein, and the capsid protein, and 7 nonstructural (NS) proteins, including NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5 in endoplasmic reticulum (ER)‑derived membrane structures. The newly synthesized viral RNA is incorporated into viral proteins and assembled into immature virions within the ER lumen [3–5]. The life cycle of DENV starts with the entry of infectious virions into target cells through membrane fusion and the binding of surface receptors/co-receptors. Most DENV proteins play a crucial role in the biological functions and pathogenesis of DENV. The DENV E protein is a viral receptor for cell binding and fusion in monocytes/macrophages, dendritic cells, B cells, T cells, basophil/mast cells, endothelial cells, epithelial cells, and hepatocytes [6, 7]. Several surface molecules, including heparan sulfate [8], CD14 [9], dendritic cell-specific intracellular adhesion molecule 3 grabbing nonintegrin [10], GRP78 [11], laminin receptor [12], heat shock proteins [13], mannose receptor [14], C-type lectin domain family 5 member A [15], and integrins [16], are required for cell binding and entry during DENV infection. After receptor binding, DENV infects target cells through receptor-mediated endocytosis [7]. When the virions are internalized through endocytosis, the surface E protein is rearranged under environment acidification, leading to viral and vesicle membrane fusion and the subsequent release of viral RNA into the cytoplasm. Cytosolic NS1 and NS2A proteins control viral RNA replication complexes while NS4B modulates DENV replication via interactions with NS3 [17–20]. In addition, both soluble NS1 and membrane-bound NS1 proteins may play a role in complement activation following the binding of anti-NS antibodies [21–23]. The serum levels of soluble NS1 predict DHF progression [24]. The NS2B protein, which is a co-factor of NS3, forms a complex with NS2A/NS3 to regulate viral replication, post-translation modification, and virion assembly through multifaceted enzyme activities, including RNA helicase, RNA 5′-triphosphatase (RTPase), and RNA-stimulated nucleoside triphosphatase (NTPase) activity [25]. The NS5 protein, which is the largest and most highly conserved DENV protein, acts as an RNA-dependent RNA polymerase [26] and methyltransferase [25], and it interacts with the helicase domain of NS3, triggering its RTPase and NTPase activities [27, 28] during DENV replication.
Unfortunately, no safe dengue vaccine is available, even though considerable effort has been directed toward the development of several candidate vaccines [29–31]. The biggest challenge is the lack of a clear antiviral strategy, reflecting the multifaceted pathogenesis, including viral load; virulence; cytotoxicity; the nature of the immune response; autoimmunity [32, 33]; and the potential effects of underlying host diseases, such as allergies, diabetes, and hypertension [34, 35].
Dengue pathogenesis
The pathogenesis of DENV infection is classified into several types, including viral factors, cytokine storms, host genetic factors, autoimmunity, and antibody-dependent enhancement (ADE) [33, 36–39]. Many reports have suggested that the viral genotypic nucleotide variation is associated with disease severity [40, 41]. In addition, higher levels of plasma DENV RNA have been observed in DHF patients compared with DF patients [42, 43]. More data are needed to conclusively correlate viral load with disease severity. Interferons (IFNs) are central players in the innate immune system for defense against pathogen infection. However, DENV harbors a number of virulence proteins that interfere with the IFN signaling pathway [4, 44–48]. The NS2A, NS4A, and NS4B proteins contribute to immune invasion by disrupting type I IFN signaling [44, 46, 47]. Furthermore, NS5 inhibits IFN-α signaling by inhibiting signal transducer and activator of transcription (STAT) 2 phosphorylation [45].
In addition to viral factors, including viral load, serotype, and virulence, a number of proinflammatory and anti-inflammatory responses are generated in host cells that have been infected with DENV. While aberrant inflammatory responses have been identified in DENV-infected patients, a number of cytokines, including tumor necrosis factor (TNF) α, IFN-γ, granulocyte-macrophage colony-stimulating factor, interleukin (IL)-10, and soluble TNF-α receptors (sTNFR) I and sTNFRII, exhibit greater expression in DHF/DSS patients compared with DF patients [49–54]. The immunopathogenesis of DENV infection involves host-specific immune responses, including immune cell activation, the release of cytokines (IL-1β, IL-2, IL-6, IL-10, IL-13, IL-18, macrophage migration inhibitory factor, tumor growth factor-β, TNF, and IFNs) and chemokines (IL-8, monocyte chemoattractant protein-1, and regulated and normal T cell expressed and secreted), complement activation, the production of inflammatory mediators, and autoimmunity [6, 30, 32, 33, 38, 55, 56]. Recently, based on genome-wide association studies has determined that host genetic factors, including the human leukocyte antigens, antibody receptors, immune/inflammatory mediators, attachment molecules, cytokines, and other immunoregulatory factors, are associated with the pathogenesis of severe dengue [37].
During infection, antibodies against soluble NS1 may lead to the complement-mediated lysis of DENV-infected cells [23]. For DENV-induced autoimmunity, anti-DENV NS1 antibodies bind to human platelets and endothelial cells [57, 58]. Numerous studies [6, 59–62] have reported mechanisms of molecular mimicry in which antibodies directed against DENV NS1 cross-react with human platelets and endothelial cells and cause damage and dysfunction, which may also be associated with the clinical features of dengue disease. The C-terminus of NS1 may be responsible for cross-reactivity with endothelial cells and platelets, as demonstrated through experiments using a modified NS1 lacking cross-reactive epitopes [63]. In addition, the deletion of the C-terminus of DENV NS1 abolishes anti-NS1-mediated platelet dysfunction and associated bleeding [63]. In addition, antibodies against DENV E and prM proteins also have autoimmune potential. Monoclonal anti-E antibodies bind to coagulant factor, and anti-prM antibodies bind to host cells [64, 65]. Autoimmunity might therefore be involved in DENV pathogenesis; however, the timing of autoantibody generation and generated titers associated with clinical parameters need further clarification. Furthermore, the generation of autoantibodies may cause safety concerns for vaccine development.
Humoral immunity is commonly involved in DHF/DSS pathogenesis, particularly in patients with a secondary DENV infection. ADE, a phenomenon in which non-neutralizing antibodies cross-react with heterogeneous serotypes of DENV and facilitate their binding with Fcγ receptor-bearing cells, facilitates severe DHF/DSS during DENV infection [55, 66–68]. The generation of antibodies against the DENV E and prM proteins is fundamental for host defense; however, such immune responses may increase the risk of developing DHF/DSS upon re-infection, primarily due to the effects of ADE. In addition to the extrinsic ADE pathway, in which the Fcγ receptor directly facilitates DENV binding onto the cell surface for DENV infection/replication, an intrinsic ADE pathway induces IL-10-mediated immunosuppression [55, 69]. For the intrinsic pathway, the ADE of DENV infection triggers IL-10 production through an immune complex associated with the Fcγ receptor to enhance the infection severity. In the presence of ADE, the Fcγ receptor can facilitate viral entry and trigger intracellular signaling. Moreover, IL-10 overproduction can enhance downstream signaling protein suppressor of cytokine signaling (SOCS) 3 expression, followed by type I IFN signaling suppression in the human monocyte cell line THP-1 [69, 70]. However, the molecular mechanisms of host and viral regulation of IL-10 expression and the pathological role of IL-10 in DENV infection are mostly unknown. Therefore, the generation of autoimmunity and ADE may cause concerns for vaccine development against DENV infection. Both viral particles acting through the extrinsic pathway and Fcγ receptor signaling through the intrinsic pathway are important for IL-10 induction. To clarify the potential effects of these regulatory routes, determining the detailed molecular mechanisms underlying DENV-induced IL-10 production is an important target for research.
IL-10 expression and activation
The balance between inflammation and anti-inflammation is critical for infection control [71, 72]. IL-10, which was originally named cytokine synthesis inhibitory factor, is a cytokine that is produced by type 2 T-helper cells [73]. IL-10 exhibits anti-inflammatory properties, including the inhibition of immune mediator secretion, antigen presentation, and phagocytosis [74]. Currently, 6 IL-10-related cytokines, including IL-10, IL-19, IL-20, IL-22, IL-24, and IL-26, have been identified [75, 76]. All IL-10 family members utilize similar receptor complexes. Two transmembrane glycoproteins, IL-10 receptor (IL-10R) 1 and IL-10R2, form the complete IL-10R. There are 2 steps involved in the initiation of IL-10 signaling. IL-10 first binds to IL-10R1, and the interaction between IL-10/IL-10R1 changes the conformation of the IL-10/IL-10R1 complex to facilitate the interaction between IL-10/IL-10R1 and IL-10R2 [77]. The cross-reaction of IL-10Rs induces the Janus kinase (Jak) 1/Tyrosine kinase (Tyk) 2-mediated phosphorylation of IL-10R1 at tyrosine residue 446/496. Subsequently, STAT3 binding induces autophosphorylation [78, 79], followed by downstream gene transcription. A recent study showed that numerous immune cells, including dendritic cells, monocytes/macrophages, B cells, T cells, nature killer (NK) cells, mast cells, neutrophils, and eosinophils, produce IL-10 in vivo or in vitro[80].
Regulation of IL-10 production in DENV infection
In DENV-infected cells, a variety of immune mediators alter anti-viral responses and inflammatory activation [6, 38]; however, the mechanisms for such responses are in need of further investigation. Increased levels of serum IL-10 may be a useful prognostic hallmark in DHF/DSS patients, as discussed above. Aberrant IL-10 expression may also be involved in DENV pathogenesis, particularly for DENV infection/replication under ADE as demonstrated in vitro[55]. However, the significance of this in vivo IL-10 expression is not known.
IL-10 is a cytokine with pleiotropic effects in immunoregulation and inflammation. IL-10 may play a role in DENV pathogenesis, reflecting an immunosuppressive function that causes IFN resistance, followed by impaired immune clearance and a persistent infectious effect for acute viral infection. Duell and colleagues [81] summarized IL-10 induction in distinct pathogens. Microbes, including protozoa, nematodes, fungi, viruses, and bacteria, regulate host cell IL-10 expression to allow persistent infection [82–84]. In Table 1, we summarize a panel of epidemiological studies from the past decade that report a positive correlation between IL-10 levels and dengue disease severity [50, 85–92]. Overall, higher levels of IL-10 are detected in DHF/DSS patients compared with DF patients, and this trend is observed for infants, children, and adults. The time-kinetic analysis shows increased levels of IL-10 from the onset of fever to defervescence, and viremia primarily occurs during fever in dengue patients [50, 85, 93]. The relationship between IL-10 and viral replication is therefore speculated, and the possible pathogen effects may result from the IL-10-mediated inhibition of the antiviral IFN response [55]. Another study showed a late peak of IL-10 production after viremia at defervescence [90]. Maximal plasma IL-10 levels measured from the acute phase of infection correlated with the degree of plasma leakage, as determined by the pleural effusion index [50, 90]. Thus, IL-10 may cause lymphocyte dysfunction through the suppression of the T cell proliferative response to mitogens, which occurs in dengue patients during the early stages of infection [85, 94]. Furthermore, having a decreased number of platelets, called thrombocytopenia, has been associated with the presence of IL-10 [85, 90, 94]. Interestingly, serum IL-10 levels have been strongly associated with the serum levels of hepatic transaminases AST and ALT [91]. Moreover, the level of IL-10 is higher in secondary DENV-infected patients than in primary DENV-infected patients [95, 96]. IL-10 induction is associated with severe DENV infection and is a potential biomarker for acute DENV infection [93, 94]. Specifically, IL-10 expression acts as predictive marker of death for DHF patients [86].
Several possibilities have been proposed to explain DENV-induced regulation of IL-10. IL-10 is primarily produced by monocytes/macrophages, type 2 T-helper cells, and CD4+CD25+Foxp3+ regulatory T cells, which constitute a suppressive T cell population. An early report showed that increased frequencies of CD4+CD25high regulatory T cells are present in dengue patients with acute infection [97]. Based on these findings, the ratios of regulatory/effector T cells are also increased. Furthermore, the activation of this cell population and the generation of IL-10 are normal during infection. Activated regulatory T cells may be one of the IL-10-producing cell populations in circulation. Current studies have shown that cell type specificity and host genetic polymorphisms affect IL-10 production during ADE of DENV infection [98]. Specifically, in monocytes, as previously demonstrated [69, 70], IL-10 is induced only in ADE infection, but not in DENV infection alone. However, other Fcγ receptor-bearing cells, including dendritic cells, B cells, mast cells, and NK cells, may also produce IL-10 in an ADE-regulated manner. An analysis of the single nucleotide polymorphisms in the IL-10 promoter region revealed that the homozygous GCC haplotype is associated with an increased level of IL-10 [98]. However, another group showed that the IL-10 (−1082/-819/-592) ACC/ATA haplotype is associated with DHF even though this haplotype results in downregulated IL-10 [92]. Although host cell responses and genetic polymorphisms complicate IL-10 regulation, these studies do not support a strong role for IL-10 in ADE-facilitated DHF/DSS progression.
Aberrant production of IL-10 could be the result of intrinsic regulation by ADE in DENV infection [70]. IL-10 activation followed by SOCS3 expression has been demonstrated during ADE in DENV infection and is also observed in patients with DHF/DSS [69]. Following DENV infection of monocytes, IL-10 expression is induced in a time-dependent manner; notably, ADE significantly facilitates this response. This study was the first report to show that DENV and ADE directly co-regulate IL-10, which is increased in severe DHF/DSS patients. To explain the effects of ADE on IL-10 upregulation, intrinsic signaling through Fcγ receptor-mediated sequential activation of splenic tyrosine kinases mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK) has been suggested [55]. This potential molecular mechanism needs further exploration in the near future, particularly at the level of the transcriptional and translational regulation of IL-10.
Various transcription factors are involved in the production of IL-10 by monocytes/macrophages, including activating transcription factor 1, CCAAT/enhancer binding protein-β, cAMP-responsive element-binding protein (CREB), nuclear factor-κB (NF-κB), pre-B-cell leukemia transcription factor 1, PBX-regulating protein 1, specific protein 1, and MAF [99, 100]. Notably, these transcription factors are commonly regulated by MAPKs, including p38 MAPK and ERK. Recent studies [101, 102] reported that inhibiting glycogen synthase kinase (GSK)-3, a multi-functional serine/threonine kinase that controls protein synthesis, cell proliferation, division, differentiation, motility, inflammation, and apoptosis, downregulates Toll-like receptor (TLR)-mediated inflammatory responses but increases IL-10 production. We recently showed that GSK-3 regulates inflammatory activation in lipopolysaccharide (LPS)-activated macrophages, partly through inhibiting IL-10 [103]. Mechanistically, GSK-3 negatively regulates CREB, a transcription factor that promotes IL-10 [101, 102, 104]. We recently showed the mechanisms through which IFN-γ upregulates LPS-induced nitric oxide (NO) biosynthesis in macrophages through GSK-3-mediated IL-10 inhibition [105]. In the presence of TLR signaling, inhibiting GSK-3 can increase the phosphorylation of the transcription factor CREB. CREB activation is positively mediated by protein kinase A (PKA)-, phosphatidylinositol 3-kinase/PKB-, and PKC-mediated phosphorylation [106]. In addition to PKA, PKB, and PKC, CREB is also regulated by GSK-3β, which decreases CREB stability by phosphorylating CREB at Ser129 [107, 108]. Both of these kinases act upstream of GSK-3 and inactivate GSK-3 through phosphorylation at serine residues [107, 109, 110]. Another study showed that the overexpression of IL-10 is mediated by GSK-3 inhibition-induced PKC and ERK activation [84]. In Leishmania infection, GSK-3 negatively regulates myeloid cell IL-10 production in a PI3K/PKB/CREB-dependent manner [111]. During ADE of DENV infection, Fcγ receptor may also trigger both ERK and PKC signaling [55]. Therefore, GSK-3 may be inactivated during DENV infection, which might be important for DENV-induced IL-10 production.
Implications of IL-10 in dengue pathogenesis
Consistent with many human viruses, such as human immunodeficiency virus, hepatitis C virus, and Epstein-Barr virus, DENV infection also induces IL-10 production [69, 112–114]. In ADE infections, very early IL-10 overproduction is correlated with the suppression of anti-viral responses, indicating that the timing of IL-10 expression is important for immunosurveillance. Extrinsic ADE infection contributes to a high rate of viral infection in Fcγ receptor-bearing cells, whereas the intrinsic ADE effect via IL-10 suppresses the activation of the IFN-mediated antiviral response. For modulating the immune response, SOCS3 plays a key role downstream of IL-10 signaling [115]. Interactions between IL-10 and IL-10 receptors activate the Jak/STAT pathway, leading to downstream gene transcription that promotes the anti-inflammatory response [80, 116–119]. Several reports have shown that IL-10 might suppress the immune response through negatively regulating MyD88 expression in mononuclear cells [116, 120]. ADE of DENV infection may be the principal cause of IL-10-mediated immunopathogenesis. Strategies to manipulate IL-10 regulation may facilitate the development of a safe DENV vaccine, perhaps by providing a way to protect against the effects of ADE caused by current candidate vaccines.
IL-10 can block NF-κB activity, and NF-κB is critical for TLR-mediated antiviral IFN responses; pro-inflammatory activation; production of IL-2, IL-12, TNF-α, and IFN-γ; and expression of MHC class II antigens and co-stimulatory molecules [71, 121]. In severe DHF/DSS patients, the levels of IL-2, IL-12, and IFN-γ are decreased [122]; however, the mechanisms underlying this decrease are still unknown. IL-10 is released to inhibit the action of antiviral NK cells during the immune response to viral infection [123, 124]. This release may prolong viral infection, and inhibiting IL-10 might facilitate the antiviral response. High titers of viremia, caused by the ADE of DENV infection, determine the frequency of DHF/DSS progression [39, 94, 125]. In addition to the involvement of extrinsic ADE-mediated viral infection, delayed viral clearance mediated through IL-10 immunosuppression may be involved in DENV pathogenesis.
The type II T-helper cell-derived cytokine IL-10 typically attenuates the type I T-helper cell-derived IFN-γ-activated Jak/STAT signaling pathway [80, 115, 117–119]. IL-10-induced SOCS3 can block the interaction of STAT1 and the IFN-γ receptor to inhibit the activation of IFN-γ. IFN-γ activity is important for preventing DENV-induced mortality, as demonstrated in an experimental murine model [126]. An antiviral axis of IFN-γ/inducible NO synthase/NO-mediated control of viral replication is exhibited in host cells that have been infected with DENV. Consistent with the findings that Bordetella parapertusis-induced IL-10 limits host cytoprotective IFN-γ responses [127], aberrant IL-10 production may also be required for IFN-γ resistance during ADE of DENV infection. Notably, the ADE of DENV infection causes aberrant production of IL-10, followed by aberrant SOCS3 expression and IFN resistance [69, 70]. An intrinsic pathway involving the Fcγ receptor may facilitate DENV infection/replication following IL-10-mediated blockade of antiviral IFN responses. During microbial infection, the generation of such infectious immune complexes may also cause similar IL-10-mediated immunopathogenesis [55].
Molecular mimicry between DENV proteins and host proteins may cause autoimmunopathogenesis in DENV infection [33]. However, the mechanisms through which B cells are activated and immunotolerance is compromised remain unclear. IL-10 can enhance B cell survival, proliferation, maturation, and antibody production [128, 129], implying a possible role of IL-10 for autoimmunity during ADE of DENV infection. However, it is still controversial whether IL-10 attenuates autoimmunity by blocking IFN-mediated autoimmune-associated inflammation in lupus [130]. The implication of IL-10 in DENV-induced autoimmunity needs further study.
Conclusions
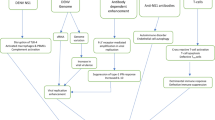
IL-10 has immunomodulatory effects and is generally considered anti-inflammatory. Excessive or poorly timed IL-10 production may allow viruses to escape from immune surveillance during DENV pathogenesis. DENV-induced IL-10 production, which may be exacerbated by ADE through Fcγ receptor-mediated extrinsic and intrinsic pathways, leads to IL-10/SOCS3-mediated immunosuppression and enhanced viral replication (Figure 1). The molecular basis for IL-10 induction should be investigated in cells during DENV infection and during the ADE of DENV infection. After DENV infection, the major IL-10-producing cells in the host should be identified, and the pathogenic roles of IL-10 must be clarified. In addition, the involvement of viral receptor- and Fcγ receptor-mediated signaling is key for exploring the regulation of IL-10. Targeting IL-10 regulation and signaling pharmacologically using neutralizing antibodies, antagonists, and inhibitors may represent a viable therapeutic strategy for combating the progression of severe dengue diseases.

Theoretical model for IL-10 production and its pathogenic role during DENV infection. IL-10 is produced in T cells (TH2 and Treg) through an unknown mechanism and in Fcγ receptor-bearing cells directly through viral receptors and indirectly through an ADE-facilitated manner. The extrinsic ADE pathway occurs through Fcγ receptor-facilitated virus contact and entry following viral receptor-mediated endocytosis. An alternative intrinsic ADE pathway may trigger Fcγ receptor-mediated signaling to activate Syk/PKC/ERK-regulated IL-10 expression. However, the molecular mechanisms for IL-10 production remain unclear. Crosstalk between PKC/PKA/PKB-regulated GSK-3 for the transcriptional regulation of IL-10 and the involvement of genetic factors are also summarized. IL-10-regulated SOCS3 expression may benefit DENV replication by facilitating IFN resistance.
Abbreviations
- ADE:
-
Antibody-dependent enhancement
- CREB:
-
cAMP-responsive element-binding protein
- DF:
-
Dengue fever
- DHF:
-
Dengue hemorrhagic fever
- DSS:
-
Dengue shock syndrome
- DENV:
-
Dengue virus
- E:
-
Envelope protein
- ERK:
-
Extracellular signal-regulated kinase
- GSK:
-
Glycogen synthase kinase
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- IFN:
-
Interferon
- IL:
-
Interleukin
- LPS:
-
Lipopolysaccharide
- MAPK:
-
Mitogen-activated protein kinase
- NO:
-
Nitric oxide
- NS:
-
Nonstructural
- NF-κB:
-
Nuclear factor-κB
- NTPase:
-
Nucleoside triphosphatases
- prM:
-
Precursor membrane
- PKA:
-
Protein kinase A
- RTPase:
-
RNA 5′-triphosphatase
- STAT:
-
Signal transducer and activator of transcription
- sTNFR:
-
Soluble TNF-α receptor
- SOCS:
-
Suppressor of cytokine signaling
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor.
References
Diaz-Quijano FA: Dengue. N Engl J Med. 2012, 367: 180-author reply 181
Alexander N, Balmaseda A, Coelho IC, Dimaano E, Hien TT, Hung NT, Janisch T, Kroeger A, Lum LC, Martinez E: Multicentre prospective study on dengue classification in four South-east Asian and three Latin American countries. Trop med int health TM IH. 2011, 16: 936-948. 10.1111/j.1365-3156.2011.02793.x.
Guzman MG, Halstead SB, Artsob H, Buchy P, Farrar J, Gubler DJ, Hunsperger E, Kroeger A, Margolis HS, Martinez E: Dengue: a continuing global threat. Nat Rev Microbiol. 2010, 8: S7-S16. 10.1038/nrmicro2460.
Morrison J, Aguirre S, Fernandez-Sesma A: Innate immunity evasion by Dengue virus. Viruses. 2012, 4: 397-413. 10.3390/v4030397.
Perera R, Kuhn RJ: Structural proteomics of dengue virus. Curr Opin Microbiol. 2008, 11: 369-377. 10.1016/j.mib.2008.06.004.
Clyde K, Kyle JL, Harris E: Recent advances in deciphering viral and host determinants of dengue virus replication and pathogenesis. J Virol. 2006, 80: 11418-11431. 10.1128/JVI.01257-06.
Rodenhuis-Zybert IA, Wilschut J, Smit JM: Dengue virus life cycle: viral and host factors modulating infectivity. Cell mol life sci CMLS. 2010, 67: 2773-2786. 10.1007/s00018-010-0357-z.
Chen Y, Maguire T, Hileman RE, Fromm JR, Esko JD, Linhardt RJ, Marks RM: Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nature med. 1997, 3: 866-871. 10.1038/nm0897-866.
Chen YC, Wang SY, King CC: Bacterial lipopolysaccharide inhibits dengue virus infection of primary human monocytes/macrophages by blockade of virus entry via a CD14-dependent mechanism. J Virol. 1999, 73: 2650-2657.
Tassaneetrithep B, Burgess TH, Granelli-Piperno A, Trumpfheller C, Finke J, Sun W, Eller MA, Pattanapanyasat K, Sarasombath S, Birx DL: DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J exper med. 2003, 197: 823-829. 10.1084/jem.20021840.
Jindadamrongwech S, Thepparit C, Smith DR: Identification of GRP 78 (BiP) as a liver cell expressed receptor element for dengue virus serotype 2. Arch Virol. 2004, 149: 915-927. 10.1007/s00705-003-0263-x.
Thepparit C, Smith DR: Serotype-specific entry of dengue virus into liver cells: identification of the 37-kilodalton/67-kilodalton high-affinity laminin receptor as a dengue virus serotype 1 receptor. J Virol. 2004, 78: 12647-12656. 10.1128/JVI.78.22.12647-12656.2004.
Reyes-Del Valle J, Chavez-Salinas S, Medina F, Del Angel RM: Heat shock protein 90 and heat shock protein 70 are components of dengue virus receptor complex in human cells. J Virol. 2005, 79: 4557-4567. 10.1128/JVI.79.8.4557-4567.2005.
Miller JL, de Wet BJ, Martinez-Pomares L, Radcliffe CM, Dwek RA, Rudd PM, Gordon S: The mannose receptor mediates dengue virus infection of macrophages. PLoS pathogens. 2008, 4: e17-10.1371/journal.ppat.0040017.
Chen ST, Lin YL, Huang MT, Wu MF, Cheng SC, Lei HY, Lee CK, Chiou TW, Wong CH, Hsieh SL: CLEC5A is critical for dengue-virus-induced lethal disease. Nature. 2008, 453: 672-676. 10.1038/nature07013.
Wan SW, Lin CF, Lu YT, Lei HY, Anderson R, Lin YS: Endothelial cell surface expression of protein disulfide isomerase activates beta1 and beta3 integrins and facilitates dengue virus infection. J Cell Biochem. 2012, 113: 1681-1691.
Falgout B, Chanock R, Lai CJ: Proper processing of dengue virus nonstructural glycoprotein NS1 requires the N-terminal hydrophobic signal sequence and the downstream nonstructural protein NS2a. J Virol. 1989, 63: 1852-1860.
Kanlaya R, Pattanakitsakul SN, Sinchaikul S, Chen ST, Thongboonkerd V: Vimentin interacts with heterogeneous nuclear ribonucleoproteins and dengue nonstructural protein 1 and is important for viral replication and release. Mol Biosyst. 2010, 6: 795-806. 10.1039/b923864f.
Lindenbach BD, Rice CM: Genetic interaction of flavivirus nonstructural proteins NS1 and NS4A as a determinant of replicase function. J Virol. 1999, 73: 4611-4621.
Umareddy I, Chao A, Sampath A, Gu F, Vasudevan SG: Dengue virus NS4B interacts with NS3 and dissociates it from single-stranded RNA. J Gen Virol. 2006, 87: 2605-2614. 10.1099/vir.0.81844-0.
Falkler WA, Diwan AR, Halstead SB: Human antibody to dengue soluble complement-fixing (SCF) antigens. J Immunol. 1973, 111: 1804-1809.
Avirutnan P, Punyadee N, Noisakran S, Komoltri C, Thiemmeca S, Auethavornanan K, Jairungsri A, Kanlaya R, Tangthawornchaikul N, Puttikhunt C: Vascular leakage in severe dengue virus infections: a potential role for the nonstructural viral protein NS1 and complement. J Infect Dis. 2006, 193: 1078-1088. 10.1086/500949.
Avirutnan P, Hauhart RE, Somnuke P, Blom AM, Diamond MS, Atkinson JP: Binding of flavivirus nonstructural protein NS1 to C4b binding protein modulates complement activation. J Immunol. 2011, 187: 424-433. 10.4049/jimmunol.1100750.
Pawitan JA: Dengue virus infection: predictors for severe dengue. Acta medica Indonesiana. 2011, 43: 129-135.
Benarroch D, Egloff MP, Mulard L, Guerreiro C, Romette JL, Canard B: A structural basis for the inhibition of the NS5 dengue virus mRNA 2′-O-methyltransferase domain by ribavirin 5′-triphosphate. J Biol Chem. 2004, 279: 35638-35643. 10.1074/jbc.M400460200.
Tan BH, Fu J, Sugrue RJ, Yap EH, Chan YC, Tan YH: Recombinant dengue type 1 virus NS5 protein expressed in Escherichia coli exhibits RNA-dependent RNA polymerase activity. Virology. 1996, 216: 317-325. 10.1006/viro.1996.0067.
Kapoor M, Zhang L, Ramachandra M, Kusukawa J, Ebner KE, Padmanabhan R: Association between NS3 and NS5 proteins of dengue virus type 2 in the putative RNA replicase is linked to differential phosphorylation of NS5. J Biol Chem. 1995, 270: 19100-19106. 10.1074/jbc.270.32.19100.
Pryor MJ, Rawlinson SM, Butcher RE, Barton CL, Waterhouse TA, Vasudevan SG, Bardin PG, Wright PJ, Jans DA, Davidson AD: Nuclear localization of dengue virus nonstructural protein 5 through its importin alpha/beta-recognized nuclear localization sequences is integral to viral infection. Traffic. 2007, 8: 795-807. 10.1111/j.1600-0854.2007.00579.x.
Murphy BR, Whitehead SS: Immune response to dengue virus and prospects for a vaccine. Annu Rev Immunol. 2011, 29: 587-619. 10.1146/annurev-immunol-031210-101315.
Simmons CP, Farrar JJ, Nguyen V, Wills B: Dengue. N Engl J Med. 2012, 366: 1423-1432. 10.1056/NEJMra1110265.
Thomas SJ, Endy TP: Critical issues in dengue vaccine development. Curr Opin Infect Dis. 2011, 24: 442-450. 10.1097/QCO.0b013e32834a1b0b.
Halstead SB: Controversies in dengue pathogenesis. Paediatrics inter child health. 2012, 32 (Suppl 1): 5-9.
Wan SW, Lin CF, Yeh TM, Liu CC, Liu HS, Wang S, Ling P, Anderson R, Lei HY, Lin YS: Autoimmunity in dengue pathogenesis. J Formosan Med Assoc = Taiwan yi zhi. 2013, 112: 3-11. 10.1016/j.jfma.2012.11.006.
Figueiredo MA, Rodrigues LC, Barreto ML, Lima JW, Costa MC, Morato V, Blanton R, Vasconcelos PF, Nunes MR, Teixeira MG: Allergies and diabetes as risk factors for dengue hemorrhagic fever: results of a case control study. PLoS Negl Trop Dis. 2010, 4: e699-10.1371/journal.pntd.0000699.
Pang J, Salim A, Lee VJ, Hibberd ML, Chia KS, Leo YS, Lye DC: Diabetes with hypertension as risk factors for adult dengue hemorrhagic fever in a predominantly dengue serotype 2 epidemic: a case control study. PLoS Negl Trop Dis. 2012, 6: e1641-10.1371/journal.pntd.0001641.
Guilarde AO, Turchi MD, Siqueira JB, Feres VC, Rocha B, Levi JE, Souza VA, Boas LS, Pannuti CS, Martelli CM: Dengue and dengue hemorrhagic fever among adults: clinical outcomes related to viremia, serotypes, and antibody response. J Infect Dis. 2008, 197: 817-824. 10.1086/528805.
Lan NT, Hirayama K: Host genetic susceptibility to severe dengue infection. Tropical med health. 2011, 39: 73-81. 10.2149/tmh.2011-02.
Rothman AL: Immunity to dengue virus: a tale of original antigenic sin and tropical cytokine storms. Nat Rev Immunol. 2011, 11: 532-543. 10.1038/nri3014.
Vaughn DW, Green S, Kalayanarooj S, Innis BL, Nimmannitya S, Suntayakorn S, Endy TP, Raengsakulrach B, Rothman AL, Ennis FA, Nisalak A: Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J Infect Dis. 2000, 181: 2-9. 10.1086/315215.
Cologna R, Rico-Hesse R: American genotype structures decrease dengue virus output from human monocytes and dendritic cells. J Virol. 2003, 77: 3929-3938. 10.1128/JVI.77.7.3929-3938.2003.
Tuiskunen A, Monteil V, Plumet S, Boubis L, Wahlstrom M, Duong V, Buchy P, Lundkvist A, Tolou H, Leparc-Goffart I: Phenotypic and genotypic characterization of dengue virus isolates differentiates dengue fever and dengue hemorrhagic fever from dengue shock syndrome. Arch Virol. 2011, 156: 2023-2032. 10.1007/s00705-011-1100-2.
Sudiro TM, Zivny J, Ishiko H, Green S, Vaughn DW, Kalayanarooj S, Nisalak A, Norman JE, Ennis FA, Rothman AL: Analysis of plasma viral RNA levels during acute dengue virus infection using quantitative competitor reverse transcription-polymerase chain reaction. J Med Virol. 2001, 63: 29-34. 10.1002/1096-9071(200101)63:1<29::AID-JMV1004>3.0.CO;2-S.
Wang WK, Chao DY, Kao CL, Wu HC, Liu YC, Li CM, Lin SC, Ho ST, Huang JH, King CC: High levels of plasma dengue viral load during defervescence in patients with dengue hemorrhagic fever: implications for pathogenesis. Virology. 2003, 305: 330-338. 10.1006/viro.2002.1704.
Jones M, Davidson A, Hibbert L, Gruenwald P, Schlaak J, Ball S, Foster GR, Jacobs M: Dengue virus inhibits alpha interferon signaling by reducing STAT2 expression. J Virol. 2005, 79: 5414-5420. 10.1128/JVI.79.9.5414-5420.2005.
Mazzon M, Jones M, Davidson A, Chain B, Jacobs M: Dengue virus NS5 inhibits interferon-alpha signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J Infect Dis. 2009, 200: 1261-1270. 10.1086/605847.
Munoz-Jordan JL, Laurent-Rolle M, Ashour J, Martinez-Sobrido L, Ashok M, Lipkin WI, Garcia-Sastre A: Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J Virol. 2005, 79: 8004-8013. 10.1128/JVI.79.13.8004-8013.2005.
Munoz-Jordan JL, Sanchez-Burgos GG, Laurent-Rolle M, Garcia-Sastre A: Inhibition of interferon signaling by dengue virus. Proc Natl Acad Sci USA. 2003, 100: 14333-14338. 10.1073/pnas.2335168100.
Pagni S, Fernandez-Sesma A: Evasion of the human innate immune system by dengue virus. Immunol Res. 2012, 54 (1-3): 152-159. 10.1007/s12026-012-8334-2.
Green S, Vaughn DW, Kalayanarooj S, Nimmannitya S, Suntayakorn S, Nisalak A, Lew R, Innis BL, Kurane I, Rothman AL, Ennis FA: Early immune activation in acute dengue illness is related to development of plasma leakage and disease severity. J Infect Dis. 1999, 179: 755-762. 10.1086/314680.
Green S, Vaughn DW, Kalayanarooj S, Nimmannitya S, Suntayakorn S, Nisalak A, Rothman AL, Ennis FA: Elevated plasma interleukin-10 levels in acute dengue correlate with disease severity. J Med Virol. 1999, 59: 329-334. 10.1002/(SICI)1096-9071(199911)59:3<329::AID-JMV12>3.0.CO;2-G.
Huang YH, Lei HY, Liu HS, Lin YS, Liu CC, Yeh TM: Dengue virus infects human endothelial cells and induces IL-6 and IL-8 production. Am J Trop Med Hyg. 2000, 63: 71-75.
Kurane I, Innis BL, Nimmannitya S, Nisalak A, Meager A, Ennis FA: High levels of interferon alpha in the sera of children with dengue virus infection. Am J Trop Med Hyg. 1993, 48: 222-229.
Kurane I, Innis BL, Nimmannitya S, Nisalak A, Meager A, Janus J, Ennis FA: Activation of T lymphocytes in dengue virus infections. High levels of soluble interleukin 2 receptor, soluble CD4, soluble CD8, interleukin 2, and interferon-gamma in sera of children with dengue. J Clin Invest. 1991, 88: 1473-1480. 10.1172/JCI115457.
Kurane I, Janus J, Ennis FA: Dengue virus infection of human skin fibroblasts in vitro production of IFN-beta, IL-6 and GM-CSF. Arch Virol. 1992, 124: 21-30. 10.1007/BF01314622.
Halstead SB, Mahalingam S, Marovich MA, Ubol S, Mosser DM: Intrinsic antibody-dependent enhancement of microbial infection in macrophages: disease regulation by immune complexes. Lancet Infect Dis. 2010, 10: 712-722. 10.1016/S1473-3099(10)70166-3.
Srikiatkhachorn A, Green S: Markers of dengue disease severity. Curr Top Microbiol Immunol. 2010, 338: 67-82. 10.1007/978-3-642-02215-9_6.
Lin CF, Lei HY, Liu CC, Liu HS, Yeh TM, Wang ST, Yang TI, Sheu FC, Kuo CF, Lin YS: Generation of IgM anti-platelet autoantibody in dengue patients. J Med Virol. 2001, 63: 143-149. 10.1002/1096-9071(20000201)63:2<143::AID-JMV1009>3.0.CO;2-L.
Lin CF, Lei HY, Shiau AL, Liu CC, Liu HS, Yeh TM, Chen SH, Lin YS: Antibodies from dengue patient sera cross-react with endothelial cells and induce damage. J Med Virol. 2003, 69: 82-90. 10.1002/jmv.10261.
Green S, Rothman A: Immunopathological mechanisms in dengue and dengue hemorrhagic fever. Curr Opin Infect Dis. 2006, 19: 429-436. 10.1097/01.qco.0000244047.31135.fa.
Lin CF, Wan SW, Cheng HJ, Lei HY, Lin YS: Autoimmune pathogenesis in dengue virus infection. Viral immunology. 2006, 19: 127-132. 10.1089/vim.2006.19.127.
Lin YS, Yeh TM, Lin CF, Wan SW, Chuang YC, Hsu TK, Liu HS, Liu CC, Anderson R, Lei HY: Molecular mimicry between virus and host and its implications for dengue disease pathogenesis. Exp Biol Med (Maywood). 2011, 236: 515-523. 10.1258/ebm.2011.010339.
Pang T, Cardosa MJ, Guzman MG: Of cascades and perfect storms: the immunopathogenesis of dengue haemorrhagic fever-dengue shock syndrome (DHF/DSS). Immunol Cell Biol. 2007, 85: 43-45. 10.1038/sj.icb.7100008.
Chen MC, Lin CF, Lei HY, Lin SC, Liu HS, Yeh TM, Anderson R, Lin YS: Deletion of the C-terminal region of dengue virus nonstructural protein 1 (NS1) abolishes anti-NS1-mediated platelet dysfunction and bleeding tendency. J Immunol. 2009, 183: 1797-1803. 10.4049/jimmunol.0800672.
Huang YH, Chang BI, Lei HY, Liu HS, Liu CC, Wu HL, Yeh TM: Antibodies against dengue virus E protein peptide bind to human plasminogen and inhibit plasmin activity. Clin Exp Immunol. 1997, 110: 35-40. 10.1111/j.1365-2249.1997.499-ce1398.x.
Huang KJ, Yang YC, Lin YS, Huang JH, Liu HS, Yeh TM, Chen SH, Liu CC, Lei HY: The dual-specific binding of dengue virus and target cells for the antibody-dependent enhancement of dengue virus infection. J Immunol. 2006, 176: 2825-2832.
Boonnak K, Slike BM, Burgess TH, Mason RM, Wu SJ, Sun P, Porter K, Rudiman IF, Yuwono D, Puthavathana P, Marovich MA: Role of dendritic cells in antibody-dependent enhancement of dengue virus infection. J Virol. 2008, 82: 3939-3951. 10.1128/JVI.02484-07.
Goncalvez AP, Engle RE, St Claire M, Purcell RH, Lai CJ: Monoclonal antibody-mediated enhancement of dengue virus infection in vitro and in vivo and strategies for prevention. Proc Nat Acad Sci United States Am. 2007, 104: 9422-9427. 10.1073/pnas.0703498104.
Halstead SB: Neutralization and antibody-dependent enhancement of dengue viruses. Adv virus res. 2003, 60: 421-467.
Ubol S, Phuklia W, Kalayanarooj S, Modhiran N: Mechanisms of immune evasion induced by a complex of dengue virus and preexisting enhancing antibodies. J Infect Dis. 2010, 201: 923-935. 10.1086/651018.
Chareonsirisuthigul T, Kalayanarooj S, Ubol S: Dengue virus (DENV) antibody-dependent enhancement of infection upregulates the production of anti-inflammatory cytokines, but suppresses anti-DENV free radical and pro-inflammatory cytokine production, in THP-1 cells. J Gen Virol. 2007, 88: 365-375. 10.1099/vir.0.82537-0.
Couper KN, Blount DG, Riley EM: IL-10: the master regulator of immunity to infection. J Immunol. 2008, 180: 5771-5777.
Wilson EB, Brooks DG: The role of IL-10 in regulating immunity to persistent viral infections. Curr Top Microbiol Immunol. 2011, 350: 39-65.
Fiorentino DF, Bond MW, Mosmann TR: Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J exper med. 1989, 170: 2081-2095. 10.1084/jem.170.6.2081.
Jung M, Sabat R, Kratzschmar J, Seidel H, Wolk K, Schonbein C, Schutt S, Friedrich M, Docke WD, Asadullah K: Expression profiling of IL-10-regulated genes in human monocytes and peripheral blood mononuclear cells from psoriatic patients during IL-10 therapy. Eur J Immunol. 2004, 34: 481-493. 10.1002/eji.200324323.
Fickenscher H, Hor S, Kupers H, Knappe A, Wittmann S, Sticht H: The interleukin-10 family of cytokines. Trends in immunology. 2002, 23: 89-96. 10.1016/S1471-4906(01)02149-4.
Kotenko SV: The family of IL-10-related cytokines and their receptors: related, but to what extent?. Cytokine growth factor rev. 2002, 13: 223-240. 10.1016/S1359-6101(02)00012-6.
Yoon SI, Logsdon NJ, Sheikh F, Donnelly RP, Walter MR: Conformational changes mediate interleukin-10 receptor 2 (IL-10R2) binding to IL-10 and assembly of the signaling complex. J Biol Chem. 2006, 281: 35088-35096. 10.1074/jbc.M606791200.
Finbloom DS, Winestock KD: IL-10 induces the tyrosine phosphorylation of tyk2 and Jak1 and the differential assembly of STAT1 alpha and STAT3 complexes in human T cells and monocytes. J Immunol. 1995, 155: 1079-1090.
Weber-Nordt RM, Riley JK, Greenlund AC, Moore KW, Darnell JE, Schreiber RD: Stat3 recruitment by two distinct ligand-induced, tyrosine-phosphorylated docking sites in the interleukin-10 receptor intracellular domain. J Biol Chem. 1996, 271: 27954-27961. 10.1074/jbc.271.44.27954.
Sabat R, Grutz G, Warszawska K, Kirsch S, Witte E, Wolk K, Geginat J: Biology of interleukin-10. Cytokine growth factor rev. 2010, 21: 331-344. 10.1016/j.cytogfr.2010.09.002.
Duell BL, Tan CK, Carey AJ, Wu F, Cripps AW, Ulett GC: Recent insights into microbial triggers of interleukin-10 production in the host and the impact on infectious disease pathogenesis. FEMS Immunol Med Microbiol. 2012, 64: 295-313. 10.1111/j.1574-695X.2012.00931.x.
Cheng YL, Wang CY, Huang WC, Tsai CC, Chen CL, Shen CF, Chi CY, Lin CF: Staphylococcus aureus induces microglial inflammation via a glycogen synthase kinase 3beta-regulated pathway. Infect Immun. 2009, 77: 4002-4008. 10.1128/IAI.00176-09.
Gee K, Angel JB, Mishra S, Blahoianu MA, Kumar A: IL-10 regulation by HIV-Tat in primary human monocytic cells: involvement of calmodulin/calmodulin-dependent protein kinase-activated p38 MAPK and Sp-1 and CREB-1 transcription factors. J Immunol. 2007, 178: 798-807.
Noh KT, Son KH, Jung ID, Kang HK, Hwang SA, Lee WS, You JC, Park YM: Protein kinase C delta (PKCdelta)-extracellular signal-regulated kinase 1/2 (ERK1/2) signaling cascade regulates glycogen synthase kinase-3 (GSK-3) inhibition-mediated interleukin-10 (IL-10) expression in lipopolysaccharide (LPS)-induced endotoxemia. J Biol Chem. 2012, 287: 14226-14233. 10.1074/jbc.M111.308841.
Azeredo EL, Zagne SM, Santiago MA, Gouvea AS, Santana AA, Neves-Souza PC, Nogueira RM, Miagostovich MP, Kubelka CF: Characterisation of lymphocyte response and cytokine patterns in patients with dengue fever. Immunobiology. 2001, 204: 494-507. 10.1078/0171-2985-00058.
Chen LC, Lei HY, Liu CC, Shiesh SC, Chen SH, Liu HS, Lin YS, Wang ST, Shyu HW, Yeh TM: Correlation of serum levels of macrophage migration inhibitory factor with disease severity and clinical outcome in dengue patients. Am J Trop Med Hyg. 2006, 74: 142-147.
Chen RF, Liu JW, Yeh WT, Wang L, Chang JC, Yu HR, Cheng JT, Yang KD: Altered T helper 1 reaction but not increase of virus load in patients with dengue hemorrhagic fever. FEMS Immunol Med Microbiol. 2005, 44: 43-50. 10.1016/j.femsim.2004.11.012.
Guerrero CD, Arrieta AF, Ramirez ND, Rodriguez LS, Vega R, Bosch I, Rodriguez JA, Narvaez CF, Salgado DM: High plasma levels of soluble ST2 but not its ligand IL-33 is associated with severe forms of pediatric dengue. Cytokine. 2013, 61: 766-771. 10.1016/j.cyto.2012.12.024.
Houghton-Trivino N, Salgado DM, Rodriguez JA, Bosch I, Castellanos JE: Levels of soluble ST2 in serum associated with severity of dengue due to tumour necrosis factor alpha stimulation. J Gen Virol. 2010, 91: 697-706. 10.1099/vir.0.012971-0.
Libraty DH, Endy TP, Houng HS, Green S, Kalayanarooj S, Suntayakorn S, Chansiriwongs W, Vaughn DW, Nisalak A, Ennis FA, Rothman AL: Differing influences of virus burden and immune activation on disease severity in secondary dengue-3 virus infections. J Infect Dis. 2002, 185: 1213-1221. 10.1086/340365.
Nguyen TH, Lei HY, Nguyen TL, Lin YS, Huang KJ, Le BL, Lin CF, Yeh TM, Do QH, Vu TQ: Dengue hemorrhagic fever in infants: a study of clinical and cytokine profiles. J Infect Dis. 2004, 189: 221-232. 10.1086/380762.
Perez AB, Garcia G, Sierra B, Alvarez M, Vazquez S, Cabrera MV, Rodriguez R, Rosario D, Martinez E, Denny T, Guzman MG: IL-10 levels in Dengue patients: some findings from the exceptional epidemiological conditions in Cuba. J Med Virol. 2004, 73: 230-234. 10.1002/jmv.20080.
Butthep P, Chunhakan S, Yoksan S, Tangnararatchakit K, Chuansumrit A: Alteration of cytokines and chemokines during febrile episodes associated with endothelial cell damage and plasma leakage in dengue hemorrhagic fever. Pediatr Infect Dis J. 2012, 31 (12): e232-e238. 10.1097/INF.0b013e31826fd456.
Brasier AR, Ju H, Garcia J, Spratt HM, Victor SS, Forshey BM, Halsey ES, Comach G, Sierra G, Blair PJ: A three-component biomarker panel for prediction of dengue hemorrhagic fever. Am J Trop Med Hyg. 2012, 86: 341-348. 10.4269/ajtmh.2012.11-0469.
Tang Y, Kou Z, Zhang F, Yao X, Liu S, Ma J, Zhou Y, Zhao W, Tang X, Jin X: Both viremia and cytokine levels associate with the lack of severe disease in secondary dengue 1 infection among adult Chinese patients. PLoS One. 2010, 5: e15631-10.1371/journal.pone.0015631.
Yeh WT, Chen RF, Wang L, Liu JW, Shaio MF, Yang KD: Implications of previous subclinical dengue infection but not virus load in dengue hemorrhagic fever. FEMS Immunol Med Microbiol. 2006, 48: 84-90. 10.1111/j.1574-695X.2006.00127.x.
Luhn K, Simmons CP, Moran E, Dung NT, Chau TN, Quyen NT, le Thao TT, Van Ngoc T, Dung NM, Wills B: Increased frequencies of CD4+ CD25(high) regulatory T cells in acute dengue infection. J exper med. 2007, 204: 979-985. 10.1084/jem.20061381.
Boonnak K, Dambach KM, Donofrio GC, Tassaneetrithep B, Marovich MA: Cell type specificity and host genetic polymorphisms influence antibody-dependent enhancement of dengue virus infection. J Virol. 2011, 85: 1671-1683. 10.1128/JVI.00220-10.
Mosser DM, Zhang X: Interleukin-10: new perspectives on an old cytokine. Immunol Rev. 2008, 226: 205-218. 10.1111/j.1600-065X.2008.00706.x.
Saraiva M, O’Garra A: The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010, 10: 170-181. 10.1038/nri2711.
Martin M, Rehani K, Jope RS, Michalek SM: Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005, 6: 777-784.
Woodgett JR, Ohashi PS: GSK3: an in-Toll-erant protein kinase?. Nat Immunol. 2005, 6: 751-752.
Huang WC, Lin YS, Chen CL, Wang CY, Chiu WH, Lin CF: Glycogen synthase kinase-3beta mediates endoplasmic reticulum stress-induced lysosomal apoptosis in leukemia. J Pharmacol Exp Ther. 2009, 329: 524-531. 10.1124/jpet.108.148122.
Hu X, Paik PK, Chen J, Yarilina A, Kockeritz L, Lu TT, Woodgett JR, Ivashkiv LB: IFN-gamma suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006, 24: 563-574. 10.1016/j.immuni.2006.02.014.
Lin CF, Tsai CC, Huang WC, Wang CY, Tseng HC, Wang Y, Kai JI, Wang SW, Cheng YL: IFN-gamma synergizes with LPS to induce nitric oxide biosynthesis through glycogen synthase kinase-3-inhibited IL-10. J Cell Biochem. 2008, 105: 746-755. 10.1002/jcb.21868.
Mayr B, Montminy M: Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001, 2: 599-609. 10.1038/35085068.
Chiu CT, Chuang DM: Molecular actions and therapeutic potential of lithium in preclinical and clinical studies of CNS disorders. Pharmacol Ther. 2010, 128: 281-304. 10.1016/j.pharmthera.2010.07.006.
Fiol CJ, Williams JS, Chou CH, Wang QM, Roach PJ, Andrisani OM: A secondary phosphorylation of CREB341 at Ser129 is required for the cAMP-mediated control of gene expression. A role for glycogen synthase kinase-3 in the control of gene expression. J Biol Chem. 1994, 269: 32187-32193.
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA: Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995, 378: 785-789. 10.1038/378785a0.
Fang X, Yu SX, Lu Y, Bast RC, Woodgett JR, Mills GB: Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc Natl Acad Sci U S A. 2000, 97: 11960-11965. 10.1073/pnas.220413597.
Nandan D, Camargo De Oliveira C, Moeenrezakhanlou A, Lopez M, Silverman JM, Subek J, Reiner NE: Myeloid cell IL-10 production in response to leishmania involves inactivation of glycogen synthase kinase-3beta downstream of phosphatidylinositol-3 kinase. J Immunol. 2012, 188: 367-378. 10.4049/jimmunol.1100076.
Lambert SL, Martinez OM: Latent membrane protein 1 of EBV activates phosphatidylinositol 3-kinase to induce production of IL-10. J Immunol. 2007, 179: 8225-8234.
Ostrowski MA, Gu JX, Kovacs C, Freedman J, Luscher MA, MacDonald KS: Quantitative and qualitative assessment of human immunodeficiency virus type 1 (HIV-1)-specific CD4+ T cell immunity to gag in HIV-1-infected individuals with differential disease progression: reciprocal interferon-gamma and interleukin-10 responses. J Infect Dis. 2001, 184: 1268-1278. 10.1086/324005.
Woitas RP, Petersen U, Moshage D, Brackmann HH, Matz B, Sauerbruch T, Spengler U: HCV-specific cytokine induction in monocytes of patients with different outcomes of hepatitis C. World j gastroenterol WJG. 2002, 8: 562-566.
Yoshimura A, Naka T, Kubo M: SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007, 7: 454-465. 10.1038/nri2093.
Chang J, Kunkel SL, Chang CH: Negative regulation of MyD88-dependent signaling by IL-10 in dendritic cells. Proc Natl Acad Sci USA. 2009, 106: 18327-18332. 10.1073/pnas.0905815106.
El Kasmi KC, Holst J, Coffre M, Mielke L, de Pauw A, Lhocine N, Smith AM, Rutschman R, Kaushal D, Shen Y: General nature of the STAT3-activated anti-inflammatory response. J Immunol. 2006, 177: 7880-7888.
Moore KW, de Waal MR, Coffman RL, O’Garra A: Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001, 19: 683-765. 10.1146/annurev.immunol.19.1.683.
Murray PJ: STAT3-mediated anti-inflammatory signalling. Biochem Soc Trans. 2006, 34: 1028-1031.
Dagvadorj J, Naiki Y, Tumurkhuu G, Hassan F, Islam S, Koide N, Mori I, Yoshida T, Yokochi T: Interleukin-10 inhibits tumor necrosis factor-alpha production in lipopolysaccharide-stimulated RAW 264.7 cells through reduced MyD88 expression. Innate immunity. 2008, 14: 109-115. 10.1177/1753425908089618.
Vicari AP, Trinchieri G: Interleukin-10 in viral diseases and cancer: exiting the labyrinth?. Immunol Rev. 2004, 202: 223-236. 10.1111/j.0105-2896.2004.00216.x.
Chaturvedi UC, Agarwal R, Elbishbishi EA, Mustafa AS: Cytokine cascade in dengue hemorrhagic fever: implications for pathogenesis. FEMS Immunol Med Microbiol. 2000, 28: 183-188. 10.1111/j.1574-695X.2000.tb01474.x.
Blackburn SD, Wherry EJ: IL-10, T cell exhaustion and viral persistence. Trends Microbiol. 2007, 15: 143-146. 10.1016/j.tim.2007.02.006.
Brooks DG, Lee AM, Elsaesser H, McGavern DB, Oldstone MB: IL-10 blockade facilitates DNA vaccine-induced T cell responses and enhances clearance of persistent virus infection. J exper med. 2008, 205: 533-541. 10.1084/jem.20071948.
Endy TP, Nisalak A, Chunsuttitwat S, Vaughn DW, Green S, Ennis FA, Rothman AL, Libraty DH: Relationship of preexisting dengue virus (DV) neutralizing antibody levels to viremia and severity of disease in a prospective cohort study of DV infection in Thailand. J Infect Dis. 2004, 189: 990-1000. 10.1086/382280.
Costa VV, Fagundes CT, Valadao DF, Cisalpino D, Dias AC, Silveira KD, Kangussu LM, Avila TV, Bonfim MR, Bonaventura D: A model of DENV-3 infection that recapitulates severe disease and highlights the importance of IFN-gamma in host resistance to infection. PLoS Negl Trop Dis. 2012, 6: e1663-10.1371/journal.pntd.0001663.
Wolfe DN, Karanikas AT, Hester SE, Kennett MJ, Harvill ET: IL-10 induction by Bordetella parapertussis limits a protective IFN-gamma response. J Immunol. 2010, 184: 1392-1400. 10.4049/jimmunol.0803045.
Groux H, Cottrez F: The complex role of interleukin-10 in autoimmunity. J Autoimmun. 2003, 20: 281-285. 10.1016/S0896-8411(03)00044-1.
Wakkach A, Cottrez F, Groux H: Can interleukin-10 be used as a true immunoregulatory cytokine?. Eur Cytokine Netw. 2000, 11: 153-160.
Hill N, Sarvetnick N: Cytokines: promoters and dampeners of autoimmunity. Curr Opin Immunol. 2002, 14: 791-797. 10.1016/S0952-7915(02)00403-X.
Acknowledgements
We apologize to colleagues whose relevant contributions were not cited due to space limitations. This work was supported by grants from the National Health Research Institutes (NHRI-EX101-9917NC) and the National Science Council (NSC101-2321-B-006-029), Taiwan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TTT, YJC, YSL, SWW, CLC, and CFL designed the concept, collected information, and prepared the manuscript and figures. TTT, YJC, and CFL wrote the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Tsai, TT., Chuang, YJ., Lin, YS. et al. An emerging role for the anti-inflammatory cytokine interleukin-10 in dengue virus infection. J Biomed Sci 20, 40 (2013). https://doi.org/10.1186/1423-0127-20-40
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1423-0127-20-40