
Abstract
Macrophages (Mϕ) are the major source of inflammatory cytokines and aretarget cells for dengue virus (DV) replication. However, Mϕ areheterogeneous and their phenotypic and functional diversities are influenced bycytokines that regulate their differentiation, tissue distribution, and defenseagainst invading pathogens. In vitro, human primary macrophages arederived from peripheral blood CD14+ monocytes in the presence ofmacrophage colony-stimulating factor (M-CSF) or granulocyte macrophagecolony-stimulating factor (GM-CSF). These are essential for developingtissue/resting macrophages (M-Mϕ) and inflammatory macrophages(GM-Mϕ), respectively. While IFN production is similar between M-Mϕand GM-Mϕ, M-Mϕ cannot produce IL-1β after DV infection. Incontrast, GM-Mϕ is more susceptible to DV infection and DV triggers CLEC5Ain GM-Mϕ to activate NLRP3 inflammasomes, which in turn release IL-18 andIL-1β that are critical for Th17 activation and contribute to diseaseseverity. Thus, GM-Mϕ is more representative than M-Mϕ forinvestigating inflammasome activation in dengue infection, and is invaluable forrevealing the molecular mechanism of pathogen-induced inflammatory reaction.Distinct phenotypes of macrophage subsets under the influence of M-CSF andGM-CSF raise the question of optimal conditions for culturing primarymacrophages to study host-pathogen interaction.
Similar content being viewed by others


Review
Introduction
Dengue virus (DV) is a positive-sense, single-stranded RNA virus that belongs tothe flavivirus genus of the Flaviviridae family. It is transmittedamong humans by the Aedes mosquitoes and is prevalent in over 100tropical and sub-tropical countries, with about 2.5 billion people at risk [1]. Infection causes a spectrum of illness ranging from sub-clinical andmild febrile illness to classical dengue fever (DF) to severe and sometimesfatal hemorrhagic disease [1]. Classical DF is an acute febrile illness that usually occurs inolder children and adults and is often characterized by fever, frontal headache,myalgia, arthralgia, nausea, vomiting, and rash lasting 3–7 days [2]. While DF is self-limiting in most cases, it can progress into denguehemorrhagic fever (DHF) or dengue shock syndrome (DSS). The signaling pathwayleading to dengue infection had been unclear until the myeloid Syk-coupledC-type lectin CLEC5A was identified as the therapeutic target of DF/DHF [3].
Macrophages (Mϕ) are thought to originate from hematopoietic stem cells(HSCs) during development and reside in various tissues such as Kupffer cells inthe liver, microglia in the brain, alveolar macrophage in the lungs, osteoclastin the bone, and in lymph nodes and other tissues. Tissue macrophages play abroad role in maintaining tissue homeostasis via clearance of senescent cellsand tissue remodeling and repair. While macrophage colony-stimulating factor(M-CSF) has been applied to induce monocyte differentiation into macrophages forhost-pathogen interaction, recent studies reveal that granulocyte macrophagecolony-stimulating factor (GM-CSF) is influential in skewing macrophagedifferentiation into distinct phenotypes. Hamilton [4] proposed that a constant M-CSF level is necessary to keep the Mϕpopulation in a resting and homeostatic situation (M-Mϕ or restingmacrophage), while local GM-CSF elevation during infection triggers Mϕ intoan inflammatory condition (GM-Mϕ or inflammatory macrophage).
While GM-Mϕ has condensed nuclei and relatively abundant cytoplasm rich inmitochondria, M-Mϕ has relatively smaller nuclei and less cytoplasm filledwith lysosomes. Moreover, differential expression of Toll-like receptors (TLRs),C-type lectin receptors (CLRs), and cytosolic retinoid acid-inducible gene I(RIG-I)-like receptors (RLRs) have been observed [5]. While M-Mϕ is less sensitive to DV infection and do not produceinterleukin-1beta (IL-1β) and IL-18, GM-Mϕ is highly susceptible to DVinfection, release higher levels of tumor necrosis factor-alpha (TNF-α),and activate NLR family PYD-containing protein 3 (NLRP3) inflammasome to secreteIL-1β and IL-18 and become pyroptosis [5].
Inflammasome is composed of three components: the nucleotide-binding domain andleucine-rich repeat containing (NLR) proteins or the pyrin and HIN domaincontaining family member (PYHIN), apoptosis-associated speck-like proteincontaining a CARD (Asc), and pro-caspase-1. Activation of TLRs and CLRs resultsin the assembling of inflammasome to activate caspase-1, which further processespro-IL-1β and IL-18 into mature forms and induces pyroptosis. Sinceinflammasomes play critical roles in Th17 activation and tissue damage duringacute and chronic inflammation, GM-Mϕ may be an ideal in vitromodel system to investigate the regulation of inflammasome activation bypathogens [6].
In addition to DV, the influenza virus elicits different responses frommacrophage subsets, which is attributed to the distinct culture conditionsin vitro. Cheung et al. demonstrated that H5N1 virus induced higherlevels of TNF-α and interferon beta (IFNβ) than H1N1 and H3N2 in humanmacrophages differentiated by heat-inactivated autologous plasma [7]. However, Friesenhagen et al. suggested that induction ofpro-inflammatory cytokines and type I IFNs were significantly abolished inH5N1-infected macrophages differentiated by cultivating monocytes in Teflon bagswith RPMI-1640 medium, supplemented by 10% human AB serum than in H1N1-infectedcells [8]. Thus, contradicting results seem come from distinct differentiationmethods for macrophage subsets used in the study of host-pathogeninteraction.
To address this important issue, cytokines optimal for macrophage differentiationis discussed and the current strategy of using human M-CSF to drive monocytedifferentiation in vitro models to study host-pathogen interaction isre-visited.
Signals for macrophage differentiation and activation
Macrophages can differentiate from either hematopoietic progenitor cells (HPC) orcirculating monocytes, and display distinct phenotypes in host-pathogeninteraction and the resolution of inflammatory reactions. Various cytokines andstimulatory signals are involved in the process. Monocytes differentiate intoresting or inflammatory macrophages under the influence of M-CSF and GM-CSF,respectively [9], or into M1 and M2 by interferon-gamma (IFN-γ) and IL-4 priming,respectively [10–12]. Stimulation of macrophage subsets by pathogen-associated molecularpatterns (PAMPs), damage-associated molecular patterns (DAMPs), or distinctresolution signal like IL-10, TGF-β and glucocorticoids, determine theconsequence of host immune responses [12, 13].
The M1 macrophages are responsible for the high levels of pro-inflammatorycytokines (i.e., TNF-α, IL-1β, and IL-6), IL-12 and IL-23, chemokines(Chemokine [C-C motif] ligand 5, CCL5, and C-X-C motif chemokine, CXCL10), andlow levels of IL-10. As a result, M1 macrophages express strong anti-microbialactivity and contribute to Th1 response. In contrast, M2 macrophages can befurther classified into three major groups: M2a (induced by IL-4 or IL-13), M2b(induced by immune complexes and agonists of IL-1 receptors or TLRs), and M2c(induced by glucocorticoids or IL-10 or transforming growth factor beta,TGF-β). The M2 macrophages are characterized by low IL-12 and high IL-10production. They are also responsible for resolving Th1 response and modulatingtissue repair and remodeling [10, 11, 13].
Role of M-CSF and GM-CSF in macrophage differentiation
The M-CSF controls the primary regulator of mononuclear phagocyte productionin vivo and plays an essential role in the survival, proliferation,differentiation, and maturation of the macrophage myeloid lineage [14]. Mutation of M-CSF results in profound macrophage deficiency [15, 16] similar to that observed in M-CSF receptor knock-out mice [17]. In contrast, disturbed hematopoiesis and deficient macrophages arenot observed in GM-CSF knockout mice, even though GM-CSF-deficient mice developabnormal lungs, including peri-brochovascular lymphocyte infiltration andsurfactant accumulation in the alveoli. Moreover, opportunistic bacterial andfungal infections in lung tissue are the significant features ofGM-CSF-deficient mice [18]. The GM-CSF−/− mice is less able to controlinfluenza virus infection than WT mice, and GM-CSF over-expression in lungepithelial cells in GM-CSF−/− mice enhance mice survivalafter influenza virus infection [19]. This suggests that GM-CSF is necessary for host defense againstpathogen invasion, while M-CSF is essential for driving monocyte differentiationinto macrophage in vivo.
The M-CSF circulates at detectable levels in a steady state(<60 μg/animal) in normal healthy individuals. It is constitutivelyproduced in vitro by several cell types, including fibroblasts,endothelial cells, stromal cells, macrophages, smooth muscle cells, andosteoblasts [20]. On the other hand, GM-CSF expression is spatially regulated anddramatically up-regulated at inflammation or infection sites [4], suggesting that macrophage differentiation during inflammatoryreactions is under the influence of GM-CSF, which causes a massive increase inthe macrophage population of the spleen and liver to induce hepato-splenomegaly [21]. Thus, the basal level of M-CSF is required to maintain thehomeostasis of tissue macrophage through M-CSF signaling, while the local andtemporal increase in GM-CSF, which inhibits M-CSF signaling during inflammation,polarizes monocytes to differentiate into inflammatory Mϕ during theinflammatory reaction and shift back to resting macrophages after theinfection-induced inflammation is removed [4].
At present, inflammatory macrophages are considered to contribute to pathogenclearance by releasing many mediators like cytotoxic/pro-inflammatory/chemokinemolecules, to eliminate pathogen infection and regulate other cell types whileresting macrophages inhibit inflammation and initiate wound repair. Furthermore,excessive activation without resolution may result in tissue injury and evenmultisystem organ failure and death. The persistence of pro-inflammatorymediators may lead to the development of chronic inflammation. Therefore, thefinal outcome of the response of tissue injury or repair depends on the balancebetween two opposing forces affecting macrophages [4, 13, 22].
Differential response of murine “inflammatory Mϕ” and“resting Mϕ” to lipopolysaccharide (LPS)
Recently, Fleetwood et al. compared the different responses of murine bonemarrow-derived macrophages subsets GM-BMϕ and M-BMϕ to LPSstimulation. After LPS stimulation, GM-BMϕ preferentially producedTNF-α, IL-6, IL-12p70, and IL-23 whereas, while M-BMϕ generated moreIL-10 and CCL2 under similar conditions. Interestingly, phenotypes ofGM-BMϕ and M-BMϕ adopt the phenotype of other populations ifpre-treated with M-CSF and GM-CSF, respectively. This indicates the plasticityof GM-BMϕ and M-BMϕ by M-CSF and GM-CSF, and further supports theargument that GM-BMϕ may be the dominant macrophage subset during theinflammatory reaction [23].
In addition to GM-CSF, type I IFN has crucial regulatory function in M-BMϕand GM-BMϕ. Compared to GM-BMϕ, M-BMϕ constitutively expresshigher levels of IFN-β to enhance type I IFN signaling-dependent geneexpression, including Ccl5, Ccl12, Irf7,Stat1, Stat2 and Cxcl10. The autocrine type I IFNsignaling in GM-BMϕ and M-BMϕ differentially regulates the productionof M1 and M2 cytokines after LPS stimulation. These results indicate thatendogenous and LPS-induced type I IFNs participate in regulating the phenotypeand functions of M-BMϕ and GM-BMϕ [24].
Lacey et al. also revealed that IL-10−/− M-BMϕproduced higher amounts of TNF, IL-6, IL-12p70, and IL-23p19 after LPSstimulation. Unlike M-BMϕ, GM-BMϕ has a similar response to LPSregardless of whether they are derived from wild type orIL-10−/− mice [25]. Since the phenotype of M-BMϕ reflects steady-state macrophages,the selective influence of IFN-β and IL-10 is consistent with the conceptthat the micro-environment can influence the polarization of macrophagedifferentiation at the start of immune response, while GM-BMϕ in theinflammatory sites are resistant to the influence of exogenous cytokines such asIFN-β and IL-10. This partly explains the failure of IL-10 to suppressinflammatory reaction in vivo, where most activated macrophages behaveas GM-BMϕ resistant to IL-10-mediated immuno-suppression in mousemodels.
Inflammasome activation and viral infections
Unlike other pro-inflammatory cytokines, the production of IL-1β and IL-18is tightly controlled by the activation of inflammasome. External signals inducethe assembling of inflammasome to activate caspase-1, which further processespro-IL-1β and pro-IL-18 into mature cytokines and induce pyroptosis [6]. Three inflammasomes of the NLR family (NLRP1, NLR familyCARD-containing protein [NLRC4], and NLRP3) and one PYHIN family member (absentin melanoma 2 [AIM2]) have been clearly identified to regulate IL-1β andIL-18 secretion in macrophages. The NLRP1 inflammasome senses anthrax lethaltoxin while NLRC4 recognizes flagellin delivered through bacterial type III(T3SS) or type IV secretion systems (T4SS). The AIM2 inflammasome responds tocytosolic double-stranded DNA contributed by bacteria or virus. To date, theNLRP3 inflammasome is the well-characterized inflammasome that can sense manystimuli, including microbial stimuli (i.e., microbial lipopeptide, bacterialRNA, dsRNA) and particular molecules (e.g., amyloid deposit, silica, andaluminum salts) [26].
A recent study indicates that inflammasome activation plays critical roles invirus infections. Compared to the wild-type mice,IL-1RI−/− mice have higher mortality after influenzavirus infection [27]. IL-1β−/− mice also express decreasedimmune response and increased viral load compared to wild-type mice after herpessimplex virus 1 (HSV-1) infection [28]. Like IL-1RI−/− mice,IL-18−/− mice have increased viral load and mortalityafter influenza virus infection compared to the wild-type mice [29]. In other study, administration of IL-18 before HSV-1 infectionraises the survival rates of HSV-1-infected mice [30]. These indicate that IL-1β and IL-18 supports immune controlagainst the influenza virus and protects against HSV-1-induced encephalitis.
To escape from host immunity, viruses also evolve distinct mechanisms to evadeinflammasome activation, including 1) inhibiting inflammasome assembly (i.e.,Kaposi's sarcoma-associated herpes virus and measles virus [MV]), 2) blockingcaspase-1 function (e.g., orthopoxviruses and influenza virus), and 3)neutralizing IL-1β and IL-18 (e.g., vaccinia virus and cytoplasmicpolyhedrosis virus) [31]. Thus, understanding of different activations of inflammasome inmacrophage subsets may help illustrate the pathogenesis of dengue fever anddengue virus-induced lethal diseases.
Distinct regulation of inflammasome activation by DV in human“inflammatory Mϕ” and “resting Mϕ”
Human M-Mϕ and dendritic cells (DCs) are the primary targets of DVinfections [32–35]. Unlike DCs, which undergo apoptosis upon DV infection [35], human M-Mϕ survive for at least 45 days after DVinfection, suggesting that M-Mϕ may be regarded as major sources ofpro-inflammatory cytokines in vivo[32]. Chen et al. further demonstrated that DV activates M-Mϕ tosecrete pro-inflammatory cytokines via CLEC5A, a DNAX-activating protein(DAP12)-associated C-type lectin, that is expressed on human M-Mϕ.Furthermore, antagonistic mAb against murine CLEC5A can prevent DV-inducedpro-inflammatory cytokine release and lethal diseases in vivo[3]. This demonstrates that CLEC5A is crucial for the onset of DF andDHF/DSS, and M-Mϕ may be the most important cell subset in dengueinfection.
However, whether or not human inflammatory macrophage subsets display distinctreactions to dengue virus infection has not been systemically addressed. In thestudy by Wu et al. [5], GM-Mϕ is more susceptible to DV infection than M-Mϕ(100-fold difference) and supernatant from DV-infected GM-Mϕ is more potentin increasing the permeability of endothelia cells, HMEC-1. While both celltypes produce similar amounts of IFN-α, both IL-1β and IL-18 areundetectable in DV-infected M-Mϕ. In contrast, GM-Mϕ produces muchhigher amounts of TNF-α, IL-1β, and IL-18, and less IL-10.Furthermore, DV-infected GM-Mϕ can become pyroptosis due to caspase-1activation. It is interesting to note that DV up-regulates NLRP3 expressionwithout affecting NLRC4 and NLRP1, whereas NLRP3 siRNA inhibits DV-inducedIL-1β and IL-18 secretion specifically in GM-Mϕ.
Since LPS-priming reportedly induce IL-1β transcription and enhanceIL-1β production, Wu et al. further compared LPS-primed M-Mϕ andGM-Mϕ to DV infection. While LPS-priming dramatically increased productionof IL-1β (25-fold), LPS-primed M-Mϕ still failed to produce detectableIL-1β and IL-18. Since IL-1β and IL-18 production is under the controlof inflammasomes, this clearly shows the distinct regulation of inflammasomes inM-Mϕ and GM-Mϕ. Wu et al. further demonstrated that DV can triggerCLEC5A on GM-Mϕ to activate NLRP3 inflammasome, leading to the secretion ofIL-1β and IL-18 [5]. This observation further indicates CLEC5A may play a critical rolein DV-induced inflammasome activation.
Supernatants from DV-infected GM-Mϕ are more potent than that fromDV-infected M-Mϕ in increasing the permeability change of endothelialcells. As such, GM-Mϕ seems more critical than M-Mϕ in thepathogenesis of dengue fever, dengue hemorrhagic fever, and dengue shocksyndrome. The DV-induced inflammasome activation pathways in GM-Mϕ andM-Mϕ are summarized in Figures 1 and 2[5].

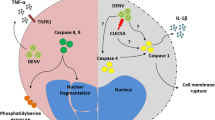
Activation of NLRP3 inflammasome in DV-infected GM-Mϕ.GM-Mϕ is infected with DV directly (A), or after LPS priming(B). DV binding to CLEC5A recruits DAP12, which isphosphorylated by Src, and then activates Syk. Activated Syk induces thetranscription of IL-1β, IL-18, and NLRP3 to activate inflammasomeand caspase-1, leading to cell death (pyroptosis) and cleavage ofpro-IL-1β and pro-IL-18. Secondary signaling, potassium efflux, andlysosome cathepsin B are also involved in NLRP3 inflammasome activationand the release of IL-1β and-IL-18 from DV–infectedGM-Mϕ. LPS priming further enhances the transcription of IL-1β(significantly), IL-18 (slightly), and NLRP3 (slightly), and furtherincreases the secretion of IL-1β. DV, dengue virus; NLRP, NLRfamily PYD-containing protein; LPS, lipopolysaccharides; IL-1β,interleukin -1beta.

Activation of NLRP3 inflammasome in DV–infected M-Mϕ.M-Mϕ is incubated with DV directly (A), or after LPS priming(B). DV activates Syk via CLEC5A to up-regulate thetranscription of IL-1β (slightly) and IL-18 (significantly), but isunable to induce NLRP3 transcription. LPS priming further up-regulatesthe transcription of IL-1β, but down-regulates IL-18 transcription.LPS priming cannot enhance the transcription of NLRP3. NLRP3inflammasome is not activated in DV-infected M-Mϕ, and thus, isunable to activate caspase-1 to process pro- IL-1β and pro-IL-18.NLRP, NLR family PYD-containing protein; DV, dengue virus; LPS,lipopolysaccharides; IL-1β, interleukin -1beta.
In addition to different responses of human M-Mϕ and GM-Mϕ to DV,Verreck et al. [36] cultured human GM-Mϕ and M-Mϕ to study their distinct rolesin mycobacteria. They found that GM-Mϕ secreted high levels of IL-23(p40/p19) but not IL-12 (p40/p35) after mycobacterial infection, while asecondary signal, IFN-γ, induced IL-12p35 transcription and IL-12production. In contrast to GM-Mϕ, M-Mϕ predominantly produced IL-10,but not IL-12 and IL-23. Also, only GM-Mϕ, not M-Mϕ, supported Th1response after mycobacterial infection. These results indicate that IL-23, butnot IL-12, is the major type 1 cytokine produced by mycobacteria-stimulatedGM-Mϕ, and that GM-Mϕ and M-Mϕ also play essential roles inanti-mycobacterial immunity.
Differential expression of innate immunity receptors and inflammasomes inhuman macrophage subsets
Recently, a comprehensive study was conducted to shed light on the expression ofTLRs, CLRs, and inflammasome components involved in recognizing DV in humanM-Mϕ and GM-Mϕ. Before incubation with DV, baseline levels of TLRs(TLR 3, 7, and 8), Dendritic Cell-Specific Intercellular adhesionmolecule-3-Grabbing Non-integrin (DC-SIGN), and most of the inflammasomesreceptors (except AIM-2 and NLRP12) were higher in M-Mϕ. Higher expressionlevels of CLEC5A and MR were found in GM-Mϕ (Figure 3).

Relative expression levels of TLRs, CLRs, and inflammasome componentsbetween GM-Mϕ and M-Mϕ. Expression levels of thesegenes are determined by real time PCR and the differential expressionlevels between GM-Mϕ and M-Mϕ are indicated in color: blue(<2 fold), green (2–5 fold), blue (6–20 fold), brown(21–50 fold), and red (>50 fold). *Indicates the proteinexpression level for the indicated receptor as confirmed by flowcytometry. TLR, Toll-like receptor; CLR, C-type lectin receptor.
Infection with DV up-regulated the expression levels of pro-inflammatorycytokines, chemokines, TLRs, and most members of NLRs. In contrast, theexpressions of CLEC5A, MR, ASC, mitochondrial antiviral signaling protein(MAVS), and members of NLRs (including NLRP1, NLRP12, NLRC4, and the NLR familymember X1 [NLRX1]) were down-regulated in both M-Mϕ and GM-Mϕ(Figures 4 and 5).

Expression levels of TLRs, CLRs, and inflammasome components inGM-Mϕ after DV infection. After incubation with DV for24 hours, the expression levels of each gene were determined byreal-time PCR. The difference in expression levels between mock and DVis indicated in color: blue (<2 fold), green (2–5 fold), blue(6–20 fold), brown (21–50 fold), and red (>50 fold). TLR,Toll-like receptor; CLR, C-type lectin receptor; DV, dengue virus.

Expression levels of TLRs, CLRs, and inflammasome components inM-Mϕ after DV infection. After incubation with DV for24 hours, the expression levels of each gene were determined byreal-time PCR. The difference in expression levels between mock and DVare indicated in color: blue (<2 fold), green (2–5 fold), blue(6–20 fold), brown (21–50 fold), and red (>50 fold). TLR,Toll-like receptor; CLR, C-type lectin receptor; DV, dengue virus.
Although the expression profiling of CLRs, TLRs, inflammasomes, pro-inflammatorycytokines, and chemokines modulated by DV is similar, the relative expressionlevels of genes between M-Mϕ and GM-Mϕ are enormous after DV infection(Figure 6). The expressions of CLEC5A, MR, NLRP1,and NLRC4 in GM-Mϕ are also higher (2- to 20-fold) than in M-Mϕ. It issurprising that NLRP12 expression is much higher (30-fold) in GM-Mϕ than inM-Mϕ. In addition, although NLRP3 gene expression in DV-infected GM-Mϕis slightly higher than in M-Mϕ (Figure 6), DVinfection induces NLRP3 gene up-regulation (Figure 4). This effect is not observed in M-Mϕ (Figure 5).

Relative expression levels of TLRs, CLRs, inflammasome components,cytokines, and chemokines between GM-Mϕ and M-Mϕ after DVinfection. The difference in gene expression levels betweenGM-Mϕ and M-Mϕ are indicated in color: blue (<2 fold),green (2–5 fold), blue (6–20 fold), brown (20–50fold), and red (>50 fold). TLR, Toll-like receptor; CLR, C-type lectinreceptor; DV, dengue virus.
In contrast, expressions of TLR8, melanoma differentiation-associated antigen 5(MDA5), NLPRP6, NLRP10, IL-18, IL-25, and CXCL10 (Interferon gamma-inducedprotein 10, IP-10) is higher (2- to 20-fold) in M-Mϕ than in GM-Mϕ. Itis interesting to note that expressions of TLR7, DC-SIGN, IL-6, and CCL2(monocyte chemotactic protein-1, MCP-1) are up-regulated by more than 30-fold inDV-infected M-Mϕ (Figure 6). The IL-18 mRNAexpression is higher in DV-infected M-Mϕ, but IL-18 is still not detectablein DV-infected M-Mϕ supernatant. This suggests the presence of a negativeregulator controlling NLRP3 activation in DV-infected M-Mϕ. The differentexpression profiling of cytokines and innate immunity receptors/sensors betweenM-Mϕ and GM-Mϕ further supports the notion that these two subsets havedistinct functions in DV infection.
Aside from mediating immune response to pathogen infection, IL-1β and IL-18play an important role in driving adaptive immunity during infection. Thecollaboration of IL-1β, IL-18, and IL-23 triggers the secretion of IL-17from Th17 cells and IL-17-secreting γδ T cells. As a result,regulation for the synthesis and production of IL-1β and IL-18 is the keypoint for modulating IL-17-associated diseases. Recent studies have shown thatIL-1β can induce the expression of IL-23 and the secretion of IL-6, whichis essential for Th17 cells differentiation [37–39]. The potential role of IL-1β and IL-18 released from DV-infectedGM-Mϕ in the Th17 differentiation is shown in Figure 7.

The combination of NLRP3 inflammasome-processed cytokines and IL-23during DV infection induced the production of IL-17 fromTh17/γδ Tcells. Stimulation of TLRs in GM-Mϕ withDV can induce the activation of NF-κB and MAPK, which promotes thetranscription of a range of pro-inflammatory cytokines. NLRP3inflammasome-activated caspase-1 further processes the pro-IL-1βand pro-IL-18 into their mature cytokine form, IL-1β and IL-18.IL-1β can also enhance the production of IL-23 and IL-6. Thereleased IL-1β, IL-18, and IL-23 induce Th17/γδ T cellsto produce pro-inflammatory cytokines which are responsible for hostimmune responses against DV infection.
Conclusions
IL-1β is the most potent endogenous pyrogen [40, 41] and is essential for the differentiation of Th17 and other cell subsetsto fight pathogens [42]. IL-18 serum level correlates with thrombocytopenia and dengue hemorrhage [43]. Recent studies further demonstrates that serum IL-1β and IL-18levels correlate with susceptibility to dengue [44, 45]. Thus, GM-Mϕ seems to be crucial in understanding the pathogenesisof DV-induced lethal diseases. In our recent study [5], we observed the differential responses of M-Mϕ and GM-Mϕ toDV, such as infection rate, and the potential ability for IL-1β and IL-18production. The differential expression level of MR may determine the differentialinfection rate because of its strong binding to DV, while the distinct regulation ofinflammasome activation in M-Mϕ and GM-Mϕ contribute to the differentialproduction of IL-1β and IL-18. In GM-Mϕ, activation of Syk-coupled CLEC5Ainduces the transcription of pro-IL-1β and NLRP3 as well as the activation ofcaspase-1 during DV infection. Moreover, LPS priming further enhances IL-1βproduction by increasing pro-IL-1β transcription and translation(Figure 1). In contrast, transcription ofpro-IL-1β, NLRP3 and caspase-1 activation are not observed in M-Mϕ, thusfails to produce mature IL-1β/IL-18 even with LPS priming (Figure 2).
In contrast, avian influenza virus (HPAIV)-infected macrophages can escapeinflammasome activation and IL-1β production because of the lack of viral M2protein required for NLRP3 inflammasome activation in other influenza virus stains [8, 46]. This escape mechanism for HPAIV may affect the immune response of humanmacrophages and enhance the possibility for HPAIV causing systemic infection and acytokine storm in the later stage of infection.
Aside from DV and the influenza virus, intracellular bacteria (likeMycobacterium, Salmonella, and Listeria monocytogenes) andfungi (such as Candida albicans and Aspergillus fumigatus) alsoinvade and replicate in macrophages [26]. However, most studies incubate pathogens with M-Mϕ invitro and do not compare the different responses of GM-Mϕ andM-Mϕ. Whether the phenomenon observed in vitro reflects event invivo need to be re-evaluated.
Inflammasome activation is crucial for starting innate immunity and controlling hostimmune response to PAMPs and DAMPs [47]. GM-Mϕ is invaluable for the identification of novel genes involvedin regulating inflammasome activation. By comparing the gene expression profiling inGM-Mϕ and M-Mϕ using microarray, it becomes possible to find positive andnegative regulators to control inflammasome activation and inhibition, and helpidentify novel therapeutic targets for treating human diseases due to exaggeratedactivation or inhibition in the future.
Abbreviations
- M-CSF:
-
Macrophage colony-stimulating factor
- GM-CSF:
-
Granulocyte macrophagecolony-stimulating factor
- DF:
-
Dengue fever
- DHF:
-
Dengue hemorrhagic fever
- DSS:
-
Dengue shock syndrome
- HSCs:
-
Hematopoietic stem cells
- TLR:
-
Toll-like receptor
- RLR:
-
Retinoid acid-inducible gene I (RIG-I)-like receptors
- CLR:
-
C-type lectin receptor
- MR:
-
Mannose receptor
- DC-SIGN:
-
Dendritic Cell-Specific Intercellular adhesionmolecule-3-Grabbing Non-integrin
- RIG-I:
-
Retinoic acid inducible gene-I
- MDA5:
-
Melanoma differentiation-associated antigen 5
- AIM2:
-
Absent in melanoma 2
- MAVS:
-
Mitochondrial anti-viral signaling protein
- ASC:
-
Apoptosis-associated speck-likeprotein containing a CARD
- NLR:
-
Nucleotide-binding domain leucine-rich repeat
- PYHIN:
-
Pyrin and HIN domain containing
- NLRP:
-
NLR family PYD-containing protein
- NLRC:
-
NLR family CARD-containing protein
- NLRX1:
-
NLR family member X1
- DAP12:
-
DNAX-activating protein 12
- TNF-α:
-
Tumor necrosis factor
- IL-1β:
-
Interleukin-1 beta
- IFNβ:
-
Interferon beta
- IFN-γ:
-
Interferon-gamma
- PAMPs:
-
Pathogen-associated molecular patterns
- DAMPs:
-
Damage-associated molecular patterns
- Th1:
-
T-helper type 1
- LPS:
-
Lipopolysaccharides
- CCL5:
-
(Chemokine [C-C motif] ligand5
- CXCL10:
-
C-X-C motif chemokine 10
- TGF-β:
-
Transforming growth factor beta
- IP-10:
-
Interferon gamma-induced protein 10
- MCP-1:
-
Monocyte chemotactic protein-1
- NF-κB:
-
Nuclear factor-κB
- HSV-1:
-
Herpes simplex virus 1
- KSHV:
-
Kaposi'ssarcoma-associated herpes virus
- MV:
-
Measles virus
- CPV:
-
Cytoplasmic polyhedrosisvirus.
References
World Health Organization: Dengue and severe dengue.http://www.who.int/mediacentre/factsheets/fs117/en/,
Mackenzie JS, Gubler DJ, Petersen LR: Emerging flaviviruses: the spread and resurgence of Japanese encephalitis,West Nile and dengue viruses. Nat Med. 2004, 10: S98-S109. 10.1038/nm1144.
Chen ST, Lin YL, Huang MT, Wu MF, Cheng SC, Lei HY, Lee CK, Chiou TW, Wong CH, Hsieh SL: CLEC5A is critical for dengue-virus-induced lethal disease. Nature. 2008, 453: 672-676. 10.1038/nature07013.
Hamilton JA: Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008, 8: 533-544. 10.1038/nri2356.
Wu MF, Chen ST, Yang AH, Lin WW, Lin YL, Chen NJ, Tsai IS, Li L, Hsieh SL: CLEC5A is critical for dengue virus-induced inflammasome activation in humanmacrophages. Blood. 2013, 121: 95-106. 10.1182/blood-2012-05-430090.
Rathinam VA, Vanaja SK, Fitzgerald KA: Regulation of inflammasome signaling. Nat Immunol. 2012, 13: 333-10.1038/ni.2237.
Cheung CY, Poon LL, Lau AS, Luk W, Lau YL, Shortridge KF, Gordon S, Guan Y, Peiris JS: Induction of proinflammatory cytokines in human macrophages by influenza A(H5N1) viruses: a mechanism for the unusual severity of human disease?. Lancet. 2002, 360: 1831-1837. 10.1016/S0140-6736(02)11772-7.
Friesenhagen J, Boergeling Y, Hrincius E, Ludwig S, Roth J, Viemann D: Highly pathogenic avian influenza viruses inhibit effective immune responsesof human blood-derived macrophages. J Leukoc Biol. 2012, 92: 11-20. 10.1189/jlb.0911479.
Sieff CA: Hematopoietic growth factors. J Clin Invest. 1987, 79: 1549-1557. 10.1172/JCI112988.
Chinetti-Gbaguidi G, Staels B: Macrophage polarization in metabolic disorders: functions and regulation. Curr Opin Lipidol. 2011, 22: 365-372. 10.1097/MOL.0b013e32834a77b4.
Liu G, Yang H: Modulation of macrophage activation and programming in immunity. J Cell Physiol. 2013, 228: 502-512. 10.1002/jcp.24157.
Gordon S, Martinez FO: Alternative activation of macrophages: mechanism and functions. Immunity. 2010, 32: 593-604. 10.1016/j.immuni.2010.05.007.
Laskin DL: Macrophages and inflammatory mediators in chemical toxicity: a battle offorces. Chem Res Toxicol. 2009, 22: 1376-1385. 10.1021/tx900086v.
Wiktor-Jedrzejczak W, Gordon S: Cytokine regulation of the macrophage (M phi) system studied using the colonystimulating factor-1-deficient op/op mouse. Physiol Rev. 1996, 76: 927-947.
Naito M, Hayashi S, Yoshida H, Nishikawa S, Shultz LD, Takahashi K: Abnormal differentiation of tissue macrophage populations in 'osteopetrosis'(op) mice defective in the production of macrophage colony-stimulatingfactor. Am J Pathol. 1991, 139: 657-667.
Wiktor-Jedrzejczak W, Ratajczak MZ, Ptasznik A, Sell KW, Ahmed-Ansari A, Ostertag W: CSF-1 deficiency in the op/op mouse has differential effects on macrophagepopulations and differentiation stages. Exp Hematol. 1992, 20: 1004-1010.
Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, Sylvestre V, Stanley ER: Targeted disruption of the mouse colony-stimulating factor 1 receptor generesults in osteopetrosis, mononuclear phagocyte deficiency, increasedprimitive progenitor cell frequencies, and reproductive defects. Blood. 2002, 99: 111-120. 10.1182/blood.V99.1.111.
Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA, Maher DW, Cebon J, Sinickas V, Dunn AR: Granulocyte/macrophage colony-stimulating factor-deficient mice show no majorperturbation of hematopoiesis but develop a characteristic pulmonarypathology. Proc Natl Acad Sci U S A. 1994, 91: 5592-5596. 10.1073/pnas.91.12.5592.
Sever-Chroneos Z, Murthy A, Davis J, Florence JM, Kurdowska A, Krupa A, Tichelaar JW, White MR, Hartshorn KL, Kobzik L: GM-CSF modulates pulmonary resistance to influenza A infection. Antiviral Res. 2011, 92: 319-328. 10.1016/j.antiviral.2011.08.022.
Fixe P, Lorgeot V, Le Meur Y, Coupey L, Heymann D, Godard A, Praloran V: Development of enzymo-immunoassays (EIA) for macrophage colony-stimulatingfactor (M-CSF) and leukaemia inhibitory factor (LIF) by using the samecapture and signal generating polyclonal antibody. Cytokine. 1996, 8: 586-591. 10.1006/cyto.1996.0078.
Krakowski M, Abdelmalik R, Mocnik L, Krahl T, Sarvetnick N: Granulocyte macrophage-colony stimulating factor (GM-CSF) recruits immunecells to the pancreas and delays STZ-induced diabetes. J Pathol. 2002, 196: 103-112. 10.1002/path.1013.
Valledor AF, Comalada M, Santamaria-Babi LF, Lloberas J, Celada A: Macrophage proinflammatory activation and deactivation: a question ofbalance. Adv Immunol. 2010, 108: 1-20.
Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD: Granulocyte-macrophage colony-stimulating factor (CSF) and macrophageCSF-dependent macrophage phenotypes display differences in cytokine profilesand transcription factor activities: implications for CSF blockade ininflammation. J Immunol. 2007, 178: 5245-5252.
Fleetwood AJ, Dinh H, Cook AD, Hertzog PJ, Hamilton JA: GM-CSF- and M-CSF-dependent macrophage phenotypes display differentialdependence on type I interferon signaling. J Leukoc Biol. 2009, 86: 411-421. 10.1189/jlb.1108702.
Lacey DC, Achuthan A, Fleetwood AJ, Dinh H, Roiniotis J, Scholz GM, Chang MW, Beckman SK, Cook AD, Hamilton JA: Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by invitro models. J Immunol. 2012, 188: 5752-5765. 10.4049/jimmunol.1103426.
Franchi L, Munoz-Planillo R, Nunez G: Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012, 13: 325-332. 10.1038/ni.2231.
Schmitz N, Kurrer M, Bachmann MF, Kopf M: Interleukin-1 is responsible for acute lung immunopathology but increasessurvival of respiratory influenza virus infection. J Virol. 2005, 79: 6441-6448. 10.1128/JVI.79.10.6441-6448.2005.
Sergerie Y, Rivest S, Boivin G: Tumor necrosis factor-alpha and interleukin-1 beta play a critical role inthe resistance against lethal herpes simplex virus encephalitis. J Infect Dis. 2007, 196: 853-860. 10.1086/520094.
Liu B, Mori I, Hossain MJ, Dong L, Takeda K, Kimura Y: Interleukin-18 improves the early defence system against influenza virusinfection by augmenting natural killer cell-mediated cytotoxicity. J Gen Virol. 2004, 85: 423-428. 10.1099/vir.0.19596-0.
Fujioka N, Akazawa R, Ohashi K, Fujii M, Ikeda M, Kurimoto M: Interleukin-18 protects mice against acute herpes simplex virus type 1infection. J Virol. 1999, 73: 2401-2409.
Gram AM, Frenkel J, Ressing ME: Inflammasomes and viruses: cellular defence versus viral offence. J Gen Virol. 2012, 93: 2063-2075. 10.1099/vir.0.042978-0.
Chen YC, Wang SY: Activation of terminally differentiated human monocytes/macrophages by denguevirus: productive infection, hierarchical production of innate cytokines andchemokines, and the synergistic effect of lipopolysaccharide. J Virol. 2002, 76: 9877-9887. 10.1128/JVI.76.19.9877-9887.2002.
Jessie K, Fong MY, Devi S, Lam SK, Wong KT: Localization of dengue virus in naturally infected human tissues, byimmunohistochemistry and in situ hybridization. J Infect Dis. 2004, 189: 1411-1418. 10.1086/383043.
Balsitis SJ, Coloma J, Castro G, Alava A, Flores D, McKerrow JH, Beatty PR, Harris E: Tropism of dengue virus in mice and humans defined by viral nonstructuralprotein 3-specific immunostaining. Am J Trop Med Hyg. 2009, 80: 416-424.
Ho LJ, Wang JJ, Shaio MF, Kao CL, Chang DM, Han SW, Lai JH: Infection of human dendritic cells by dengue virus causes cell maturation andcytokine production. J Immunol. 2001, 166: 1499-1506.
Verreck FA, de Boer T, Langenberg DM, Hoeve MA, Kramer M, Vaisberg E, Kastelein R, Kolk A, de Waal-Malefyt R, Ottenhoff TH: Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2macrophages subvert immunity to (myco)bacteria. Pro Natl Acad Sci U S A. 2004, 101: 4560-4565. 10.1073/pnas.0400983101.
Dungan LS, Mills KH: Caspase-1-processed IL-1 family cytokines play a vital role in driving innateIL-17. Cytokine. 2011, 56: 126-132. 10.1016/j.cyto.2011.07.007.
Mills KH, Dungan LS, Jones SA, Harris J: The role of inflammasome-derived IL-1 in driving IL-17 responses. J Leukoc Biol. 2013, 93: 489-497. 10.1189/jlb.1012543.
Peral de Castro C, Jones SA, Ni Cheallaigh C, Hearnden CA, Williams L, Winter J, Lavelle EC, Mills KH, Harris J: Autophagy regulates IL-23 secretion and innate T cell responses througheffects on IL-1 secretion. J Immunol. 2012, 189: 4144-4153. 10.4049/jimmunol.1201946.
Netea MG, Kullberg BJ, Van der Meer JW: Circulating cytokines as mediators of fever. Clin Infect Dis. 2000, 31 (Suppl 5): S178-S184.
Dinarello CA: Infection, fever, and exogenous and endogenous pyrogens: some concepts havechanged. J Endotoxin Res. 2004, 10: 201-222.
Mills KH: Induction, function and regulation of IL-17-producing T cells. Eur J Immunol. 2008, 38: 2636-2649. 10.1002/eji.200838535.
Martina BE, Koraka P, Osterhaus AD: Dengue virus pathogenesis: an integrated view. Clin Microbiol Rev. 2009, 22: 564-581. 10.1128/CMR.00035-09.
Mustafa AS, Elbishbishi EA, Agarwal R, Chaturvedi UC: Elevated levels of interleukin-13 and IL-18 in patients with denguehemorrhagic fever. FEMS Immunol Med Microbiol. 2001, 30: 229-233. 10.1111/j.1574-695X.2001.tb01575.x.
Bozza FA, Cruz OG, Zagne SM, Azeredo EL, Nogueira RM, Assis EF, Bozza PT, Kubelka CF: Multiplex cytokine profile from dengue patients: MIP-1beta and IFN-gamma aspredictive factors for severity. BMC Infect Dis. 2008, 8: 86-10.1186/1471-2334-8-86.
Ichinohe T, Pang IK, Iwasaki A: Influenza virus activates inflammasomes via its intracellular M2 ionchannel. Nat Immunol. 2010, 11: 404-410.
Lamkanfi M: Emerging inflammasome effector mechanisms. Nat Rev Immunol. 2011, 11: 213-220. 10.1038/nri2936.
Acknowledgements
The authors thank I-Shuen Tsai and Wan-Chen Wu for their technical support. Thiswork was supported by the National Science Council (NSC 101-2325-B-010-006 andNSC 101-2321-B-010-003), Academia Sinica, Thematic Research Project(AS-101-TP-B06-2), the Infection and Immunity Center, National Yang-MingUniversity, Taiwan (a grant from the Ministry of Education, Aim for the TopUniversity Plan), Taipei Veterans General Hospital (V102E4-003), Cheng HsinGeneral Hospital (102F218C13), Veterans General Hospitals, and the UniversitySystem of Taiwan Joint Research Program (VGHUST102-G6-2-1).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MFW and STC were involved in literature review, discussion, and drafting of themanuscript. SLH initiated the concept and wrote the draft of this manuscript. All ofthe authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wu, MF., Chen, ST. & Hsieh, SL. Distinct regulation of dengue virus-induced inflammasome activation in humanmacrophage subsets. J Biomed Sci 20, 36 (2013). https://doi.org/10.1186/1423-0127-20-36
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1423-0127-20-36