
Abstract
In the literature, it is widely discussed whether the pharmaceutical industry is going through an innovation crisis. Unfortunately, no comprehensive review exists to date that outlines the main empirical findings. In this paper, we provide an extensive survey on what is currently known about the crisis and its reasons. The empirical evidence shows that project success rates decreased over time, while attrition rates, development times, and the costs per new drug increased. The reasons for these developments are quite complex. The crisis seems to have largely technological causes. The enormous scientific progress in biotechnology and related disciplines has increased the opportunities to treat diseases, but the firms have difficulties transforming these advances into new and effective medicines. More basic research is necessary and the links between science and industry should be strengthened. Some measures have recently been taken to better adapt the approval process to current developments. However, the persistently high number of consolidations in the industry seems to have largely detrimental effects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the 1990s, many important technological developments took place in the pharmaceutical industry that provoked deep changes in the previously established way to conduct research and development (R&D). The decoding of the human genome released a huge progress in biotechnology (Martin et al. 2009, p. 158). Parallel to this, advances were achieved in other scientific fields such as chemistry, pharmacology, synthetic and structural biology, or bioinformatics (Gassmann et al. 2008, pp. 33). Since the demand for drugs is rising due to aging societies and a greater access to healthcare systems (IMS Institute 2015), the new technological developments offer huge opportunities for firms and the evolution of the industry as a whole. In light of these good prospects, it is surprising that no more new drugs are being approved and launched. A discussion about whether the industry has entered an innovation crisis already began in the 1990s with some analyses on the number of approvals or project success rates (Bienz-Tadmor et al. 1992; DiMasi et al. 1995). It gained momentum with the turn of the century when the costs to develop a drug escalated, many blockbusters were about to lose patent protection, and the firms had only very few promising compounds in their pipelines (Abrantes-Metz et al. 2004; Adams and Brantner 2006; Danzon et al. 2005; Kola and Landis 2004; Munos 2009). While there have been slight signs of improvement in recent years (Evaluate 2019; Pammolli et al. 2020), it is not clear whether the industry as a whole is recovering. Thus, the discussion is still very relevant and no consensus exists yet about the true scope of the crisis, its significance, and its underlying reasons.
From an economic policy perspective, the problem is quite puzzling: Economists would usually assume that there are not enough innovations because of an underinvestment in R&D. Firms may lack incentives to invest in R&D when patent protection is ineffective or when the demand for new pharmaceutical products is low. But these causes do not seem to grasp the actual problem of the industry: Statistics show that huge sums are continuously invested in the development of new drugs (EFPIA 2017, p. 5). Another factor that is often mentioned when barriers to innovation are discussed is overregulation. However, it seems that the literature does not regard the complex regulations which firms have to fulfil to get market approval for their compounds as the main problem (Munos 2009, p. 964). These regulations may even increase the innovativeness of the industry in terms of the quality and novelty of drugs. So why are the firms not able to develop more truly innovative medicines? The problem seems to be rooted in the R&D process itself. This is quite unusual and thus especially interesting since it does not correspond to the arguments usually made when an industry generates only a few innovations. Interestingly, the discussion focused rather early on the question of whether the R&D process itself has problems and whether it is getting more and more difficult for firms to develop new drugs due to technological reasons.
More recently, another debate came up about whether management problems or factors related to the structure of the industry can be made responsible for the low R&D productivity. However, in many studies only individual reasons are considered without relating them to a broader context. Moreover, many analyses that try to verify the existence of the crisis only examine single indicators, such as project success rates, development times, or costs per new drug, and only very few of them consider their development over time. Thus, a detailed and widespread survey of the literature may help to outline and clarify our state of knowledge concerning the following questions: What problems is the industry really facing? Has the situation improved or do the problems persist? What can be done from a policy perspective? Only with the help of a more detailed analysis will it be possible to derive useful recommendations for policy and management.
The paper is structured as follows: The next section examines the current condition of the industry based on economic facts. Then, empirical studies are presented that provide possible evidence for the existence of the crisis. Subsequently, a brief overview of the pharmaceutical R&D process is given to illustrate its special characteristics. This is followed by a detailed analysis of possible reasons for the crisis mentioned in the literature. Finally, the results obtained are critically discussed and conclusions are drawn for policy, management, and science.
Stylized facts about the crisis
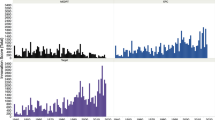
Due to the enormous scientific progress that took place in the past three decades, one would expect a significant growth in the pharmaceutical industry’s innovation rate. However, contrary to these expectations, the pharmaceutical firms were not able to substantially increase their innovation output so far. Figure 1 shows that the number of drugs approved by the US regulatory authority, the Food and Drug Administration (FDA), was largely constant between 1980 and 2010. In the literature, the peak in the approval rate in 1996 is mostly attributed to the Prescription Drug User Fee Act (PDUFA) (Berndt et al. 2005). This law was passed in 1992 and allowed the FDA to charge fees from manufacturers to fund and accelerate the review process. As a result, the FDA was able to reduce a backlog of applications, which led to a higher number of approvals in the following years (Kaitin and DiMasi 2011; Light and Lexchin 2012). Afterwards, the number of drug approvals per year declined again to its previous level. It was not until after 2010 that the rate seemed to recover slightly, with particularly high approval figures after 2016.

Figure 2 presents the five-year average of all New Chemical or Biological EntitiesFootnote 1 (NCEs or NBEs) approved on the world market and differentiated according to the nationality of the mother company. It shows decreasing approvals between 1994 and 2008 for American, European, as well as Japanese firms. However, between the periods 2009–2013 and 2014–2018, the average approval rate of US firms increased sharply. In contrast, the rates of European and Japanese firms only slightly recovered but were not able to significantly exceed their relatively low levels from the period 1999–2003.

The decline in approval counts between 1994 and 2008 is insofar remarkable since R&D expenditures increased significantly during the same time period in the US, Europe, and Japan (EFPIA 2015, p. 5). In the US, expenditures escalated from $ 11.9 to $ 40.7 billion between 1995 and 2010 (see Fig. 3). In Europe, the respective parameter rose from € 11.5 to € 27.9 billion, while in Japan, R&D investments almost doubled. This mismatch between R&D input and output has been called the ‘productivity paradox’ in the literature (Gassmann et al. 2008, p. 1).

A high activity on the input side is also documented by a strong increase in employment figures. The number of people employed in the pharmaceutical industry worldwide rose from 3.6 million in 2006 to almost 5.1 million in 2014 (IFPMA 2017, p. 44). In Europe, employment in the pharmaceutical sector grew from 500,879 to 670,088 people between 1990 and 2010, and the number of persons working in R&D increased from 76,126 to 117,035 (EFPIA 2019, p. 13). Moreover, the number of firms with an active R&D pipeline almost doubled between 2001 and 2010, from 1198 to 2207 (Informa 2019, p. 12). Thus, the figures related to the input side of the R&D process do not indicate that the industry actually is in an innovation crisis.
In general, the R&D intensity of the pharmaceutical sector is much higher than in other sectors. In 2018, the industry spent 15 percent of net salesFootnote 2 for R&D, while software and computer services only spent 8.4 percent and electronic and electrical equipment only 4.9 percent (EFPIA 2019, p. 10). High incentives to invest in R&D are triggered by the expectation that the innovation—if successfully developed—will generate a financial revenue. This expectation is raised by a high demand for pharmaceuticals on the one hand and the ability to appropriate the returns from the R&D investment on the other. Due to aging societies in industrialized countries, a higher standard of living in developing countries, and a greater access to pharmaceuticals worldwide, global drug demand has grown in recent years and is predicted to rise continuously in the following years (IMS Institute 2015). Worldwide prescription drug sales increased from $ 649 to $ 768 billion between 2008 and 2016 and are expected to grow further at 6.5 percent per year to reach $ 1060 billion in 2022 (Evaluate 2017). Reimbursement issues may restrict the demand for certain drugs, but these regulations only apply to single countries and can be compensated with the entry into other markets with better reimbursement conditions.
Incentives to invest in R&D are also created by the patent system, which can be regarded as being very effective in the pharmaceutical sector (Mansfield 1986; Scherer 2000, p. 1318). Patents are the most important form of intellectual property protection in this industry due to the public nature of the development process and the low costs of imitation (Scott Morton and Kyle 2012). In general, it is not possible to circumvent a pharmaceutical patent since the respective compound is usually precisely defined and even slight modifications constitute patent infringements as long as function and mode of action remain largely the same (Lakdawalla 2018, p. 410). But the time of effective patent protection is often quite short because patents are filed at early discovery stages and development takes several years. This was considered by the Hatch–Waxman-Act of 1984. With this act, the patent term for pharmaceutical products was extended by a maximum of five years (ibid., p. 403) and a market exclusivity provision was introduced. The latter enabled the FDA to grant periods of exclusive marketing rights after the approval of a drug to protect it from the early entry of generics (Scherer 2000, p. 1322). This provision ensures that sufficient incentives to invest in R&D remain, independent of the length of effective patent protection.
To sum up, an effective protection of intellectual property, as well as a rising worldwide drug demand, provide sufficient incentives to invest in R&D, and R&D expenditures, as well as employment figures, have actually increased during the last decades. So why is the development on the output side not in line with the development on the input side? Why is the approval rate lagging behind the large gain in R&D spending? So far, we only regarded statistics on R&D inputs and outputs at the industry level. To get a more comprehensive picture of the supposed crisis, we will review empirical studies analysing innovation productivity at the level of pharmaceutical R&D projects in the next section.
Productivity crisis in the pharmaceutical industry: Empirical evidence
In this section, we start by presenting studies analysing the success rate of pharmaceutical R&D projects, that is the share of successful projects on all projects conducted within a given time period. It can also be interpreted as the probability that a compound will be successfully developed and launched. Nevertheless, it does not tell anything about the quality of the new drugs and their market success in terms of revenues generated. To present a comprehensive overview of possible indicators, we also included studies that examine the attrition rate, the development time, and the R&D costs per new drug in our review. To identify all relevant analyses, we applied the PRISMA methodology (Moher et al. 2009). We searched for certain combinations of terms in the abstracts of the articles included in the EBSCO Academic Search Ultimate database.Footnote 3 We only considered studies in which first-hand data analyses were conducted and in which the data sets used covered R&D projects from the whole range of disease fields. The search terms and numbers of excluded and included studies are given in Table 1.
Only the studies by Backfisch (2017), DiMasi (2001), and Wong et al. (2019) analyse the development of the success rate over time. In addition to these three analyses, we, therefore, included studies in our review in which success rates for different time periods were determined. However, these studies often differ in regard to the degree of novelty or innovativeness of the compounds examined in the samples. For example, NCEs have the highest degree of novelty because they are based on a previously unknown active ingredient. Therefore, the development of these types of drugs is usually more difficult, takes longer, and is associated with higher costs than the development of already known substances.
There are only two studies examining NCEs, DiMasi et al. (1995) and DiMasi (2001). DiMasi et al. (1995) estimate different success rates for firms of different sizes and consider NCEs that entered clinical trials between 1970 and 1982. They find a success rate of 0.197 for small, 0.209 for medium-sized, and 0.279 for large firms. DiMasi (2001) distinguishes between self-originated and licensed or purchased compounds and considers NCEs that entered clinical trials between 1981 and 1992. He finds that the success rate of self-originated NCEs fell from 0.198 to 0.123 during the examined time period, whereas the rate of purchased or licensed ones rose from 0.308 to 0.373. Based on the results of both studies, it is therefore not possible to make a clear statement about how the success rate of NCEs that started clinical trials between 1970 and 1992 has developed.
Nevertheless, there are also investigations with more recent data that do not restrict their analyses to NCEs, but also include R&D projects for line-extensions, “follow-on” therapies, or “me-too” drugs. The development of these pharmaceuticals is mostly based on already known substances that have partly been examined or tested in other contexts previously. However, these studies also show major differences: Some only examine success rates for individual indications, while others determine success rates for the entire active ingredient – that means for all indications for which the compound is in development. Using the latter approach, success rates tend to be higher in general because a project is already considered to be a success when the compound has reached the next development stage or gained market approval for the first indication (Hay et al. 2014, p. 41). Possible subsequent failures in the further development of the compound for the other indications are not taken into account.Footnote 4
Unfortunately, three studies do not specify whether they analyse compounds only for single or for all indications for which they are in clinical studies. Nevertheless, we briefly describe their results to give an overview of the development of the success rate between 1983 and 2002: DiMasi et al. (2003) find a success rate of 0.215 for projects that entered clinical trials between 1983 and 1994. Abrantes-Metz et al. (2004) and Adams and Brantner (2006) both examine pipeline products that started human testing between 1989 and 2002 and estimate the success rate to be 0.264 and 0.24, respectively.
Four studies calculate success rates for individual indications for which the compounds are being tested: Arora et al. (2009) estimate a success rate of 0.34 for projects that began clinical trials between 1980 and 1994. Hay et al. (2014) find a success rate of 0.104 for projects that started tests in humans between 2003 and 2011. According to Thomas et al. (2016), the success rate of projects that entered clinical development between 2006 and 2015 is 0.096. Thus, taken together, the results of these three studies indicate a sharp decrease in the success rate concerning clinical trials for individual indications between 1980 and 2015. However, the findings by Wong et al. (2019) are less clear cut. According to the latter analysis, the success rate falls from 0.112 to 0.052 between 2005 and 2013 but then rises again to 0.067 in 2014 and 0.138 in 2015.
Hay et al. (2014) and Backfisch (2017) calculate success rates of entire compounds, which means they do not differentiate between individual indications. Hay et al. (2014) estimate a success rate of 0.153 for compounds that entered clinical trials between 2003 and 2011. Backfisch (2017) also includes projects in preclinical development in the analysis and finds that the success rate almost halved from 0.069 to 0.036 between 1995 and 2010. However, since preclinical projects are also included in the sample, the success rate tends to be lower than in the analysis of Hay et al. (2014). This is due to the fact that a further selection takes place before entering clinical trials and many projects are already discontinued at the end of the preclinical phase.
Unfortunately, other factors further limit the comparability of the results from the studies presented above. Firstly, some studies focus only on projects developed in the US, whereas others look at global R&D activities. Abrantes-Metz et al. (2004), Arora et al. (2009), Hay et al. (2014), and Thomas et al. (2016) only consider projects that were in clinical trials in the US, while Adams and Brantner (2006), Backfisch (2017), DiMasi (2001), DiMasi et al. (2003), and Wong et al. (2019) examine drugs that entered clinical testing anywhere in the world.Footnote 5 Secondly, the data sets of the studies differ substantially in terms of the number and size of the included firms that were responsible for the development of the projects: Abrantes-Metz et al. (2004), Arora et al. (2009), Backfisch (2017), and Wong et al. (2019) contain broad samples with a variety of firms of different sizes, whereas DiMasi et al. (1995), DiMasi (2001), and DiMasi et al. (2003) include only a small number of firms in their investigations.
Having the limitations of comparability in mind, we can cautiously infer the development of the success rate from 1980 to 2015. Overall, the studies indicate that the success rate decreased between 1980 and 2013 (Arora et al. 2009; Backfisch 2017; Hay et al. 2014; Wong et al. 2019). However, some evidence points to a slight increase during the subperiod 1983–2002 (Abrantes-Metz et al. 2004; Adams and Brantner 2006; DiMasi et al. 2003). The analysis of Wong et al. (2019) suggests a recovering success rate for the years 2014 and 2015.
To complement the evidence on the development of the success rate, we now look at the attrition rate of pharmaceutical R&D projects. Footnote 6This indicator is measured as the number of projects that were terminated in a certain development phase during a specific time period, related to the number of all projects that were in this development phase during that period. A higher attrition rate shows that fewer projects reach the next development phase. DiMasi (2001) compares the attrition rate of projects that entered clinical trials anywhere in the world between 1981 and 1986 with that of projects that were first in clinical studies between 1987 and 1992. He shows that the attrition rate of projects in phase I clinical trials increased between the two periods, while it remained unchanged for projects in phase II, and decreased for those in phase III. Pammolli et al. (2011) present evidence for rising attrition rates within all stages of development for projects started between 1990 and 2004 in the United States, Europe, and Japan. In the updated version of their paper, they compare attrition rates of projects entering any development phase during the three different periods 1990–1999, 2000–2009, and 2010–2013. They find that attrition rates increased between the first two observation periods for all development phases, while they significantly decreased between the second and the third period for all phases except phase III, for which only a small number of observations was available (Pammolli et al. 2020). However, the authors also show that even if attrition rates have decreased after 2010, they have remained above the levels of the period 1990–1999.
Another important indicator of changes in R&D productivity is the development of average project duration over time. Longer development times indicate that there are problems in the innovation process and that new drugs cannot be brought to market as quickly as desired. Furthermore, R&D costs increase when the development of drugs takes longer. DiMasi (2001) and Pammolli et al. (2020) present evidence that the firms in the industry managed to identify and terminate potentially unsuccessful projects earlier during the R&D process. But for successfully approved drugs, the time required for clinical development has grown. The duration from phase I to the submission of a registration application increased from 68.6 to 72.1 months for projects that entered clinical studies between 1983 and 1994. Thus, the decrease of total development time from phase I to approval from 98.9 to 90.3 months was solely based on a faster drug review by the FDA (DiMasi et al. 1991, 2003). Kaitin and DiMasi (2011) confirm these results with drugs approved in the US between 1980 and 2009. While the time required for clinical development increased from 5.7 to 6.4 years, regulatory approval times decreased from 2.8 to 1.2 years. The latest study by Pammolli et al. (2020) shows a further increase in development times after 2010, especially in phase III clinical trials. Accordingly, the time it takes to develop a drug remains a matter of concern.
Finally, another important indicator is the estimated cost per approved drug, which is calculated based on actual costs of successfully approved drugs and estimated costs of discontinued projects.Footnote 7 While out-of-pocket costs are obtained by simply adding up the estimated expenditures over the whole development time, capitalized costs also include the costs of capital at a given interest rate per year. DiMasi et al. (1991) estimate out-of-pocket costs per approved drug for NCEs that entered clinical studies between 1970 and 1982 to be $ 114 million and capitalized costs to be $ 231 million (in 1987 dollars). In a study conducted by the US congress, the capitalized costs determined in DiMasi et al. (1991) are recalculated to be $ 359 million (in 1990 dollars) by using an interest rate that varies over the drug development lifecycle (OTA 1993). DiMasi et al. (2003) calculate capitalized costs per approved drug for compounds that entered clinical testing between 1983 and 1994 to be $ 802 million (in 2000 dollars). Adams and Brantner (2006) use data on compounds that started clinical development between 1989 and 2002 and find average out-of-pocket costs to be $ 282 million (in 2000 dollars), while capitalized costs are estimated to be $ 868 million. Adams and Brantner (2010) use the phase durations and success rates from their previous study and recalculate capitalized cost to be $ 1214 million (in 1999 dollars). Munos (2009) takes the $ 802 million estimate by DiMasi et al. (2003) and complements it with assumptions concerning the development of the success and the inflation rate in the period 2000–2009. He determines capitalized costs per approved drug to be $ 1754 million (in 2000 dollars). DiMasi et al. (2016) estimate capitalized costs of compounds that started with clinical development between 1995 and 2007 to be $ 2558 million (in 2013 dollars). Finally, Wouters et al. (2020) find that capitalized costs per approved drug ranged from $ 1801 million to $ 2215 million in the period 2009–2018 (in 2018 dollars). Taken together, there is strong evidence that capitalized and out-of-pocket costs per approved drug increased sharply since the end of the 1980s.
However, some studies indicate that this increase in total development costs is largely based on a surge in the share of costs dedicated to clinical trials. DiMasi et al. (2016) show that for projects started in the 1980s, the share of preclinical development costs was 67 percent of total capitalized costs per approved drug. This share decreased to 43 percent for projects started between 2000 and 2015. A recent study by Wouters et al. (2020) confirms these findings and shows that for drugs approved between 2009 and 2018, preclinical development costs accounted for only 39 percent of total development costs. Therefore, there seems to be evidence that the costs of drug development have increased particularly during the clinical phase.
To sum up, many empirical studies analyse different indicators that may, in total, reflect the pharmaceutical industry’s development. However, the comparability of the empirical findings over time is limited since the samples underlying the analyses differ in regard to regional focus, number and size of included firms, and novelty of examined drugs. This applies to all indicators presented above, but in particular to the development of the success rate. Nevertheless, aside from these limitations, the studies show that the success rate decreased between 1980 and 2013, even if there was a slight increase in the subperiod from 1989 to 2002. In the years 2014 and 2015, the success rate seems to have recovered somewhat. However, whether this development indeed took place as described by the studies presented above and whether it is sustainable should be investigated and confirmed by further long-term research. It seems that the attrition rate of pharmaceutical R&D projects has also improved recently, whereas the time required to successfully develop a drug still continues to grow. The latter rather indicates that problems in the innovation process persist, which retard the development of new medicines. Moreover, the costs per approved drug have escalated dramatically since the end of the 1980s. There is also evidence that the share of costs dedicated to clinical compared to preclinical development has strongly increased. Taken together with the number of new drug approvals that is only recovering recently and that is still far lower than it could be, given the extraordinarily good framework conditions, there is strong reason to assume that the industry is indeed in a productivity crisis. The rising costs and the increasing time required for the successful development of drugs are mostly attributed to the comprehensive requirements that are associated with the approval of medicines. Because of these high requirements, which exist in most industrialized countries, the R&D process in the pharmaceutical industry shows considerable peculiarities compared to other sectors. In the next section, this process is therefore described in more detail and the individual development steps are explained. This should serve as a basis for being able to better understand and discuss the possible reasons for the crisis.
The drug approval process
In general, the R&D process in the pharmaceutical industry can be divided into two superordinate phases: drug discovery and drug development.
In the past, drug discovery was largely a random process. In the 1990s, it became more systematic with the shift from the so-called physiology-based to the target-based approach (Sams-Dodd 2013, p. 211). Since then, the search for drug candidates has essentially been based on basic research that aims at the understanding of cell mechanisms and their relation to diseases (Seyhan 2019, p. 3). Usually, the process starts with the discovery or identification of a target molecule – a protein, DNA or RNA – that is directly or indirectly involved in a certain disease (Lakdawalla 2018, p. 399). To validate the identified target, it has to be demonstrated that its modulation has a therapeutic effect (Drews 2000, p. 551). Then, a so-called assay is developed with which many active substances can be screened. The aim is to find at least one agent that binds to the target and changes its function in the desired way. Often, a small group of potential candidates is determined, the so-called lead compounds. These compounds undergo preclinical testing in vitro or in vivo to examine their pharmacological and toxicological characteristics. After that, one is usually selected that can proceed to clinical tests in humans (Posey Norris et al. 2014, p. 10). A patent application is often filed once the compound has been chosen. When preclinical tests with the drug candidate are completed successfully, the inventor can submit an Investigational New Drug Application (IND) to the FDA.Footnote 8 If the application is granted, the inventor is allowed to begin testing the compound in humans (Lakdawalla 2018, pp. 399).
Drug development is structured in three main phases. It starts with phase Ia in which a single dose is applied to 20–80 healthy volunteers (Seyhan 2019, p. 3). This is followed by phase Ib in which many increasing doses are administered to determine safety, pharmacokinetics, and pharmacodynamics. These trials are also increasingly used to conduct proof of concept studies in which, ideally, mechanism of action and concept are confirmed (Posey Norris et al. 2014, p. 11). In phase II, the transition from healthy individuals to patients takes place (Seyhan 2019, p. 3). The compound is tested on 100–300 patients to obtain important data on its efficacy and to reveal possible side effects. Moreover, the optimal dose is determined and a risk–benefit-profile is established (Lakdawalla 2018, p. 400). In phase III, the effectiveness of the compound is examined in comparison to a placebo or an already approved and used drug (ibid., p. 401). In general, the tests are carried out on 1000–5000 patients. Ideally, so-called randomized controlled trials (RCTs) are applied in which patients are randomly assigned to either the treatment or control group (Seyhan 2019, p. 3). Once phase III trials have been successfully completed, the inventor can submit a New Drug Application (NDA) to the FDA. The NDA is reviewed by the regulatory authority and if it is granted, the drug can be launched on the market. Usually, a compound is initially approved for one indication, but approval can be extended to other indications.Footnote 9 Moreover, drugs are often subject to further testing after they have been launched (Lakdawalla 2018, p. 402). These phase IV trials are conducted to demonstrate that the medicines also work effectively and safely under real-world conditions, outside the artificial setting of the clinical trials. In addition, comparative studies are carried out to determine the benefits and costs compared to other forms of therapy (Seyhan 2019, p. 3).
Only when a potential drug candidate successfully passes preclinical research and all three phases of clinical trials can it ultimately receive marketing authorization. In each individual phase, many factors determine whether development can be successfully continued or not. The empirical studies presented in Sect. 3 indicate that it may have become more difficult for firms to survive the complex approval process and to bring innovative new drugs to market. The possible reasons for this development discussed in the literature will be explained in more detail in the next section.
Possible reasons for the crisis
For this part of our literature review, we also used the PRISMA methodology to identify studies that discuss possible causes of the crisis. We searched for specific combinations of terms in the EBSCO Academic Search Ultimate database. We only included studies in the qualitative synthesis that focused on the last decades and that discussed possible reasons in relation to the industry as a whole. Therefore, we excluded analyses that only proposed measures to increase innovation output or that looked at single countries or individual firms. The search terms and the number of excluded and included studies are given in Table 2.
The potential causes of the crisis discussed in the literature can be largely grouped into four main classes: scientific or technological reasons, regulatory causes, management problems, and factors related to the structure of the industry and its organization. We will explore these types of reasons in more detail in the following subsections.
Problems related to science and technology
A possible reason that has already been discussed for some time is the so-called “low hanging fruit” or “mining out” problem (Cockburn 2006, p. 14; Danzon and Keuffel 2014, p. 424). It says that the comparably easy scientific problems have already been solved in the past and only the more complex diseases are left which are not entirely understood yet, more difficult to investigate in regard to biochemistry and disease pathology, and more challenging to cure (Cockburn 2006, p. 14). On the one hand, this may be partially true for some therapeutic areas: for example, some neurological disorders may be more complex to treat than certain cardiovascular diseases. On the other hand, technological opportunities are not finite and have increased sharply in recent years due to the exceptional scientific progress in biotechnology and related disciplines (ibid., p. 17). Thus, the knowledge stock today is greater than ever before. However, the firms seem to have difficulties exploiting these advances and transforming them into new and effective medicines. So far, it is not really clear why. Has the potential that the new scientific findings can contribute significantly to the cure of human diseases been overestimated? Or is the knowledge gained so far simply not enough? Or is it sufficient but major obstacles exist concerning its transfer to clinical applications?
Biotechnology can mainly influence traditional drug development in three ways: First, drugs can be developed that stem from living organisms – so-called large-molecule compounds such as monoclonal antibodies (Drews 2000, p. 547). Second, the technology can provide additional tools and improved techniques for the development of medicines, also for chemical ones. For example, improved biotech-based assays resulted in the creation of high-throughput screening platforms (Hopkins et al. 2007, p. 5). And finally, it can contribute to the understanding of illnesses. Thereby, it allows the identification of many more target molecules on which certain compounds can exert their effects (ibid., p. 7).
After the decoding of the human genome in the 1990s, a transformation of the industry through a “genomic revolution” was widely anticipated (Martin et al. 2009, p. 158). It was assumed that the previously used technological paradigm, chemistry-based drug development, had already entered the maturity phase of its lifecycle. Therefore, only decreasing marginal returns to R&D could be achieved with its use. In contrast, biotechnology was only at the beginning of its development but was said to have the potential to replace chemistry as dominant design (Cockburn 2006, p. 16). However, it was supposed that the incumbent firms would have to undertake considerable efforts during the transition phase to be able to adopt the new technology (Fagerberg 2005, p. 14). This would initially lead to rising R&D costs. But with the increasing dissemination of the new technology, the costs would decrease (or at least stabilize) and a large number of innovations would be brought to market (Cockburn 2006, pp. 16).
Some authors believe that these expectations were largely exaggerated. Munos (2016, pp. 588) argues that diseases do not all have clear genetic causes. Instead, many seem to depend on external influences. For example, evidence exists that the microbiome plays an important role in many disorders. Hopkins et al. (2007) show that biotech rather has an incremental impact on technological change because the technology builds on previous research methods instead of disrupting them. Drews (2000, p. 551) believes that expectations were realistic in principle, but that the estimate of the required time span in which the new technology should develop its potential was unrealistic. Other authors point out that biotech already had a strong influence on drug development so far and that it can be seen as the major growth engine in the pharmaceutical industry today (Evens 2016; Waldman and Terzic 2016). For example, Evens (2016, p. 283) highlights that the share of approvals of NBEs by the FDA increased steadily between 1990 and 2014 and even accounted for 37% of all approvals between 2010 and 2014. Overall, there seems to be a general consensus that biotechnology can make a significant contribution to drug development. However, it is still unclear how large this contribution will actually be.
Agreement also seems to exist that the knowledge necessary for the development of drugs is still very incomplete, despite the scientific advances of the last decades. Many processes in the human organism are largely unexplored. For example, it is not clear what role genes play in normal physiology (Cockburn 2006, p. 18). Human biology seems to be much more complicated than previously thought (Munos 2016, p. 589). Some authors emphasize that new disciplines such as proteomics, metabolomics, transcriptomics, microbiomics, or connectomics are becoming increasingly important for drug discovery, but that research in most of these fields is still largely in its infancy (Gassmann et al. 2008, pp. 33). Moreover, there are also significant gaps in our knowledge of human diseases (Sams-Dodd 2013, p. 212). The causes and mechanisms of many complex illnesses, in particular, are still unknown (Munos 2016, p. 589). A frequently cited example is Alzheimer’s: Although 350 compounds have already been tested against the neurological disorder, its etiology is still unclear (Munos 2016, p. 589; Posey Norris et al. 2014, p. 5). Thus, various authors highlight that much more scientific research is necessary and knowledge from different disciplines should be combined in a more structured way (Munos 2010, p. 534; Posey Norris et al. 2014, p. 13; Seyhan 2019, pp. 5).
Other authors point out that there are major obstacles in transferring existing knowledge to clinical applications (Butler 2008; Mankoff et al. 2004). There seems to be a gap between basic and clinical research that has been referred to in the literature as the “Valley of Death” (Bowen and Casadevall 2015; Roberts et al. 2012). Translational medicine aims at the transfer of results from basic science to the treatment of human disorders. This includes all steps of the development process described in Sect. 4, from target identification to testing of potential drug candidates in humans (Seyhan 2019, p. 2). Drug development is a very complicated, time-consuming, and costly process in which different stakeholders from academia, industry, or government are involved (Cowlrick et al. 2011). Whether the transfer is possible depends on various factors. Important determinants are primarily the quality of the findings from basic and preclinical research and the methods available for their processing and transmission (Posey Norris et al. 2014, p. 16; Seyhan 2019, p. 5). However, some empirical studies show that many published results from biomedical science are misleading, not as robust as stated, or cannot be replicated (Ioannidis 2005, 2016). A growing awareness that there are qualitative problems with basic research has led some authors to speak of a “reproducibility crisis” (Begley and Ioannidis 2015; Scannell and Bosley 2016). Other authors point out that many new targets which could be identified in the past were only poorly validated (Garnier 2008; Morgan et al. 2012). Empirical evidence shows that the main reasons why compounds fail in clinical trials are a lack of efficacy or a poor safety profile (Hay et al. 2014; Scannell et al. 2012). This indicates that weaknesses in the validation of targets and the selection of compounds for further development exist (Bunnage 2011, p. 335). Some authors highlight that especially animal models may be responsible for these deficiencies (Akhtar 2015; Garnier 2008). Many of the results from animal studies cannot be directly applied to human trials. This is probably because animal models can only partially mimic complex human diseases (Posey Norris et al. 2014, p. 15). Furthermore, the efficacy end points used are often quite different from those measured in humans. Thus, dosages and risk–benefit ratios can differ substantially (Chu 2006). The prediction of the effectiveness of a compound based on these models may therefore be risky (Paul et al. 2010, p. 211). However, other authors question whether the main part of the problem is actually based on animal models or whether other factors, such as a lack of understanding of the compound's pharmacokinetic and pharmacodynamic properties, are more decisive (Morgan et al. 2012, p. 419).
Regulatory reasons
As described in Sect. 4, the R&D process in the pharmaceutical industry consists of individual, precisely defined steps that must be carried out in a relatively strict sequence. The high requirements associated with drug approval represent significant entry barriers for new firms (Scherer 2010, p. 554). Moreover, there is some evidence that the complexity of these requirements has even increased in recent decades: A study of the association of the Pharmaceutical Research and Manufacturers of America shows that, for example, the number of admission criteria, the workload per trial, and the number of pages of the approval protocols rose after 2000 (PhRMA 2016, p. 37).Footnote 10
Two factors are mainly made responsible for the increase in regulatory requests: Firstly, it is of lesser value to develop a drug for the treatment of a certain disease when a safe and effective therapy already exists. Then, it is more difficult to demonstrate that the new drug has advantages over the existing one. Over time, the steady improvement of medicines may result in a continuous increase of approval hurdles. In the literature, this phenomenon is called the “better than the Beatles” problem (Scannell et al. 2012, p. 193).Footnote 11
Secondly, after each safety scandal, the regulatory authorities gradually lowered their risk tolerance (Ruffolo 2006, p. 101). For example, after the problems that emerged with Vioxx,Footnote 12 the FDA issued the Amendments Act in 2007 which enabled the authority to demand the submission of risk evaluations before approval and to require additional clinical studies of already approved medicines when safety problems emerge (Kaitin and DiMasi 2011, p. 184).
On the one hand, challenging standards ensure a high quality of drugs and are thus good for consumers. In general, access to pharmaceuticals needs to be regulated since effectiveness and safety are critical to patients’ health but not immediately apparent (Danzon and Keuffel 2014, p. 407). In a free market, manufacturers would probably not carry out enough tests, there would be insufficient evidence on the quality of drugs, and it would be far too costly and time-consuming for individual patients or physicians to collect this information (ibid., p. 429). Strict regulatory standards may even raise incentives to invest in R&D since the companies are going to face less competition when they manage to launch their medicines and can therefore expect higher revenues. Furthermore, they force firms to be more critical in the selection of compounds and to develop really innovative drugs. For example, Thomas (1996) examines the share of sales that companies from nine leading drug-developing nations achieved outside their home markets in 1985. He finds that the higher the regulatory standards in one country, the larger are the domestic companies’ sales abroad. Therefore, he concludes that higher standards encourage firms to focus their R&D activities on drugs of superior effectiveness that—if approved—are particularly competitive internationally. In this regard, higher standards seem to be good for innovation.
On the other hand, there seems to be a certain level over which standards should not be further increased. Some authors emphasize that the risk–benefit ratio should not shift to a nearly unachievable level (Ruffolo 2006, p. 102; Scannell et al. 2012, p. 194). All medicines have a certain level of risk. It is clear that this level should be kept small, but it is not possible to develop drugs that are absolutely safe (Scherer 2000, p. 1315). Therefore, regulators often find themselves caught in the dilemma of reducing uncertainty about possible side effects of a compound and providing patients with timely access to it (Woodcock 2012, p. 378). Moreover, some authors emphasize that the decisions of the regulatory authorities are based on highly imperfect information since the quality of the data gained in clinical trials is often poor (Manski 2009; Seyhan 2019). Trials are not carried out long enough to investigate rare but serious side effects such as heart attacks or strokes. Instead, only surrogate end points are used such as, for example, progression-free survival for cancer therapies. In addition, samples are not randomly assembled in reality since patients have to participate voluntarily in the trials (Lakdawalla 2018, p. 418). Therefore, the probability that errors occur seems to be generally high. Type I errors (approving a drug that is not safe or effective) can theoretically be remedied, for example, the drug can be withdrawn from the market or its use restricted.Footnote 13 In contrast, type II errors (rejecting a drug when it is actually safe and effective) may be more permanent (Manski 2009).
An indication that standards have rather been too low (at least in some therapeutic fields) is given by the discussion on “me-too” drugs: Many authors claim that a lot of approved drugs are not or only marginally better than existing therapy (Light and Lexchin 2012; Light and Warburton 2011; Munos and Chin 2011). A study conducted by Prescrire International (2003) shows that only 3 percent of the 2693 new drugs assessed between 1981 and 2002 provided significant therapeutic gain over already launched medicine. However, me-too drugs can also emerge because several companies – triggered by advances in basic research – are simultaneously pursuing R&D projects in a certain therapeutic area (Aronson and Green 2020, p. 2117). These projects can all be equally innovative, but one of them will be the first to gain approval (Lakdawalla 2018, p. 421). But the latecomers may also be beneficial for consumers since they compete with already existing therapies and provide alternatives for patients who do not respond very well to the previously registered medicine (Aronson and Green 2020, p. 2114). Thus, total benefits to society increase (Gagne and Choudhry 2011, p. 711). However, an indication that firms partially invest more in R&D than would be optimal for society is that many me-too drugs can be found in therapeutic classes with high sales, such as antihypertensive drugs, antibiotics, or antidepressants (CBO 2006, p. 12).
A related but slightly different discourse focusses on incrementally modified or so-called “follow-on” drugs. These treatments constitute about two-thirds of all drugs approved by the FDA (Frank 2003, p. 327). Generally, they cause fewer R&D costs, require less time, and may provide significant benefits to consumers, for example due to better dosing requirements. Nevertheless, they are criticized because firms are often able to demand high prices for these drugs although they are only marginally different from older and cheaper ones. Thus, the price difference may not correspond to the additional value provided by the new product (Lakdawalla 2018, p. 422). Doctors and consumers have generally weak incentives to consider the prices of drugs since medicines are usually reimbursed by health insurances. However, attempts have been undertaken in recent times to reinforce these incentives, for example through the introduction of multitier copayment structures (CBO 2006, p. 48).Footnote 14
A certain qualitative and innovative level can better be ensured when clinical tests are conducted against existing therapy, like it is done in Europe, and not against placebos, like in the US (Danzon and Keuffel 2014, p. 420). However, the former is more difficult and costly because quality differences are likely to be smaller, which makes larger trials necessary to provide statistically significant results (CBO 2006, p. 24). In recent decades, policy in the US, as well as in Europe, has taken a number of measures to increase the incentives of firms to develop truly innovative or especially needed drugs (Baird et al. 2014). In the US, several expedited approval programs have been created for drug candidates that treat a serious condition, are expected to provide significant improvements over existing therapy, or address an unmet medical need. A priority review procedure was already introduced with the PDUFA in 1992 and helped to reduce review times from ten to six months (Darrow et al. 2014, p. 1253). In the same year, the FDA also enacted the “accelerated approval” process that allows applications to be conditionally approved based on surrogate end points. A “fast-track” pathway was created in 1997 that provided an expedited development time due to a staggered submission of applications and a more intensive support by the FDA (Baird et al. 2014, p. 560). And finally, the “breakthrough therapy” program was established in 2012 that enabled the approval of drugs already after phase II, when clinical evidence shows substantial treatment advantages over existing therapy (Darrow et al. 2014, p. 1254). It covers all elements of the fast-track pathway and a more intensive support from the FDA, in which even agency executives are involved. Similar programs have been introduced by the European Medicines Agency (EMA) as well, such as the “accelerated assessment” pathway which facilitates shorter review times for medicines of major interest to the public. In addition, the “approval under exceptional circumstances” was created for situations in which comprehensive effectiveness and safety data cannot be provided, for example due to the rareness of the disease. And finally, the “conditional marketing authorization” was introduced which permits approval of drugs for the treatment of life-threatening diseases already after phase II, provided that a favorable risk–benefit profile can be demonstrated.Footnote 15
These measures to facilitate early access to medication were supported by several reimbursement provisions. In the US, Medicare created the so-called “coverage with evidence development” path which ensures that the new drugs approved by the FDA through one of the accelerated processes are reimbursed (Mohr and Tunis 2010). A condition for its application is that real-world data is collected to reduce uncertainties about benefits and possible harms (Baird et al. 2014, p. 566). In contrast, incentives to develop medicines for already overcrowded therapy classes are diminished by the fact that significantly fewer drugs of these classes have to be included in the reimbursement lists of the public healthcare programs (ibid., p. 567). Furthermore, to be able to negotiate a higher price with insurers or pharmacy benefit managers, firms in the US must increasingly provide evidence that their new product is of better quality than already existing drugs (Jommi et al. 2020, p. 21). In contrast to the US, drug prices are more strictly regulated by law in most European countries. In Germany, Italy, and France, forms of value-based pricing are deployed on the basis of the clinical quality of the products (ibid., p. 19). More far-reaching measures have been undertaken, for example, in the United Kingdom. Here, comparisons based on cost effectiveness are possible and even relevant for securing reimbursement by the National Health Service. When the National Institute for Health and Clinical Excellence (NICE) determines that a medicine is not cost effective at the current price, the National Health Service can deny access to the drug (Miller 2012, p. 218).
However, in the more recent discussion the focus is set on the question of whether even more far-reaching measures are necessary to make the development process more efficient and to provide patients faster access to medication. In particular, it is discussed whether forms of staggered approval or “adaptive licensing” in conjunction with the use of real-world data should be applied more broadly (Corrigan-Curay et al. 2018; Eichler et al. 2015; Sherman et al. 2016; Woodcock 2012).Footnote 16 In contrast to the different accelerated access pathways described above, these concepts aim at the flexibilization of the development process and the generation of data through the entire life span of a drug. Therefore, they also include reimbursement questions and a greater monitoring of the use of the medicines in practice (Eichler et al. 2015, p. 235). Both the FDA and the EMA conducted pilot projects to explore ways to implement these measures (Baird et al. 2014, pp. 561). In Europe, this resulted in the introduction of the adaptive pathway approach in 2016 (EMA 2016a, 2016b). The FDA developed a framework to evaluate the use of real-world evidence in post-approval studies or in the registration of further indications of already approved drugs in 2018 (FDA 2018c). Additionally, the US agency issued new guidance on the use of adaptive designs in November 2019 (FDA 2019b). In September 2020, the EMA also published a draft guideline on registry-based studies, on which the public could comment over the following three months (EMA 2020). When finalized, these guidelines are intended to support the use of registry-based studies as a source of real-world evidence.
Recent empirical work shows that both the staggered approval options and the use of real-world evidence are increasingly applied in the US as well as in Europe, particularly in the field of oncology (Bolisis et al. 2020; Bothwell et al. 2018). However, it remains to be seen to what extent these measures will have a positive impact on the overall number of registrations.
Problems related to the management of the R&D process
Some authors criticize that the pharmaceutical industry concentrates too much on the development of drug candidates that have the potential to generate a lot of sales (Cockburn 2006, p. 19; Eichler et al. 2015, p. 241; Seyhan 2019, p. 5). The focus on blockbuster drugs and thus the concentration on the most lucrative markets became possible with target-based drug development. In comparison to the previously used random selection of compounds, it made research more purposive, controllable, and easier to explain to managers and investors (Martin et al. 2009, p. 151). However, the blockbuster strategy may have become too risky today. Usually, in therapy classes with high patient populations many drugs are already available and competition is fierce. Therefore, new compounds often require larger clinical trials to demonstrate advantages over existing therapies. This causes higher development costs. However, when these costs arise, it is still uncertain whether the drug will actually be approved and successfully marketed and whether it will meet sales forecasts (Cockburn 2006, p. 19). Moreover, in the case of chronic diseases, patient populations are often very heterogenous (Posey Norris et al. 2014, p. 1). That means that a high risk exists that the compound will show poor effectiveness in some patient subgroups or that safety problems will arise (Eichler et al. 2015, p. 241).
Other authors claim that the focus on target-based drug discovery has strongly restricted creativity concerning other discovery methods (Bowen and Casadevall 2015; Sams-Dodd 2013). Moreover, it caused some sort of “reductionism”: To make screening more effective, many firms want their researchers to search for compounds that act only on a single target molecule (Bowen and Casadevall 2015, p. 11,335). Experience from the past however shows that this is not always a good strategy. For example, major advances in the treatment of HIV were only achieved when different compounds were combined (Richard and Wurtman 1997). The objective to act on just one target may not suffice because the human organism functions over many pathways (Sams-Dodd 2013, p. 213). Moreover, some diseases stem from a faulty network of receptors, genes, and proteins, all of which contribute to the pathology (Munos 2016, pp. 588). Hence, the link between a single target molecule and a disease state may be weaker than previously thought (Scannell et al. 2012, pp. 194). This could play a particularly important role in regard to complex diseases such as nervous system disorders. In recent decades, even fewer new drugs were approved in this therapeutic class than in others (Posey Norris et al. 2014, p. 1). Therefore, some scholars propose that drug development should consider from the outset that a broader approach might be necessary to achieve success (ibid., p. 7).
Sometimes the R&D process may be simply not managed very well, which may be reflected in a slow recruitment of patients for clinical studies, a low quality of the recorded data, or a poor communication with regulators (Buonansegna et al. 2014, p. 193; Cockburn 2006, p. 18). Some authors emphasize that, in addition to project management and negotiation skills, data management competences are becoming increasingly important for drug development (Posey Norris et al. 2014; Seyhan 2019). They suggest that data, protocols, and other R&D processes should be made publicly available. Moreover, negative results and reasons for failure should be published as well (Posey Norris et al. 2014, p. 8). This would be helpful not just for review purposes, but could also aid in the development of new creative ideas and could prevent failures from being repeated (Seyhan 2019, p. 9).
Furthermore, there is evidence that managers are at a high risk of making imperfect decisions. Especially in preclinical development, decisions are usually made intuitively since the high level of uncertainty during this development phase makes it difficult to apply portfolio management approaches (Betz 2011, p. 609). But studies show that experts assess the benefits and risks of further development of a compound quite differently (Cowlrick et al. 2011, p. 321). This indicates that individual decisions are largely based on incomplete information, personal bias, and varying levels of expertise in the respective disease area (Seyhan 2019, p. 11). Thus, there is a considerable risk that the development of an actually marketable compound is wrongly discontinued or that the development of an unmarketable substance is further pursued. Both scenarios have negative consequences for the firm as well as for its clients. The problems are even reinforced by the long period of time between discovery and market launch. Feedback on the outcome of a selected strategy can only be obtained with a considerable time lag (Schmid and Smith 2004, p. 25). However, involving teams in such a decision seems to enhance results and reduce failures (Disis and Slattery 2010, p. 1).
Some authors emphasize that interaction and collaboration between scientists and clinicians are particularly important for translational medicine to work well (Disis and Slattery 2010; Posey Norris et al. 2014; Seyhan 2019, p. 2). Most of the basic research is conducted outside firms, in universities or public research institutions (Coombs and Metcalfe 2002, p. 262). Thus, it is crucial for firms to cultivate strong links with these institutions to stay informed about new scientific findings and to get access to the knowledge generated there (Belderbos et al. 2016). However, it seems that cooperation between academia and industry has not yet taken place to a sufficient extent (Seyhan 2019, p. 2). Additionally, to be able to use the knowledge generated externally, a firm has to care for its absorptive capacity (Cohen and Levinthal 1990). Existing routines that are based on cumulative and embedded firm-specific knowledge may constrain the ability to absorb new knowledge coming from outside (Fagerberg 2005, p. 11). This is especially the case when the new findings significantly question the existing know-how of the firm.
Reasons related to the structure of the industry and its organization
Traditionally, the pharmaceutical industry is composed of a core of large firms and a significant fringe of smaller ones. The level of concentration is generally low (Danzon and Keuffel 2014, p. 441). A firm may just obtain a dominant position in a submarket since the knowledge needed in one of these markets is usually quite specific and cannot be easily used in other submarkets. And this dominance may be difficult to defend since the current success does not guarantee a promising pipeline in the future (Malerba and Orsenigo 2015, pp. 670).
With the biotech boom at the end of the 1970s, many small spin-out and start-up companies entered the industry (Kinch 2014, p. 1689). This seriously challenged the traditional pharmaceutical firms that had mainly relied on synthetic chemistry so far. To stay competitive and to gain access to the new technologies, they increasingly entered into mergers and acquisitions. Kinch and Moore (2016) analyse the development of the number of firms that contributed to an FDA-approved drug over time. Their analysis shows that company formations strongly increased after 1970, but declined again after the turn of the millennium. Simultaneously, the number of consolidations rose sharply and remained at a high level until 2015. As a result, the total number of successful companies fell significantly between 2001 and 2015, to a level similar to that in 1945 (ibid., pp. 644). And more recent data show that this trend is even accelerating (Kinch et al. 2021).
Some authors suggest that the surge in consolidations in the 1980s was the response of the established pharmaceutical firms to the shock triggered by the scientific advances in biotechnology (Grabowski and Kyle 2012, p. 557; Kinch and Moore 2016, p. 644). The resulting pressure was compounded by extremely high R&D costs, expiring patents, impending generic competition, and the need to have promising candidates in the pipeline (Henderson 2000, p. 11). However, the fact that there are fewer innovating firms does not necessarily mean that R&D competences are lost and the innovation capacity of the industry as a whole is deteriorated (Kinch 2014, p. 1688). A broader portfolio of R&D projects and the bundling of diverse resources and competences can spur innovation since more opportunities for mutual learning and knowledge spillovers exist (Cockburn and Henderson 2001). Moreover, large firms have more financial resources and are better equipped to build the necessary infrastructure to conduct a costly and complex clinical trial. There is also evidence that the established pharmaceutical firms are benefiting from their previous experience with the high regulatory requirements (Danzon et al. 2005). This experience has even been characterized as the “key complementary assets” needed to develop an innovation (Coombs and Metcalfe 2002; Pisano 2006). Additionally, it is particularly expensive and challenging to obtain market approval in different countries because approval standards lack international harmonization (Garnier 2008, p. 70). However, large international companies, in turn, can better organize their R&D activities according to local comparative advantages (Pammolli et al. 2011, p. 436). And finally, they are better able to carry out extensive marketing activities, which increases post-approval profits and can have a positive impact on R&D investments (Lakdawalla 2018, p. 442).
But many empirical studies rather indicate that mergers and acquisitions have negative effects on the innovative ability of the firms involved (Danzon et al. 2007; De Man and Duysters 2005; LaMattina 2011; Ornaghi 2009). They require extensive financial resources and reduce the funds available for R&D (Hall 1999; Hitt et al. 1991). Entire research sites are being eliminated afterwards (LaMattina 2011, p. 560). R&D projects are terminated on a larger scale than usual, especially those of the acquired or smaller firm (Laermann-Nguyen 2015). This reduces the extent of parallel research conducted at the firm and the industry level (Comanor and Scherer 2013). There is also evidence that firms use mergers to escape innovation competition by buying others with potentially competing projects in the pipeline and terminating their development (Cunningham et al. 2021). Furthermore, the restructuring following a consolidation may damage important intangible resources through the exit of key personnel or the inadequate integration of both firms’ R&D departments (Ernst and Vitt 2000; Granstrand and Sjölander 1990). R&D may be disrupted for approximately three years while scientists try to get used to the new organization and deal with the accompanying uncertainties (Ruffolo 2006, p. 100). Using the example of the merger between Pfizer and Wyeth, LaMattina (2011, p. 560) demonstrates that the progress of drug candidates in the development process can be significantly slowed down after a merger.
In contrast, strategic alliances and licensing deals seem to have much more positive effects on innovation (Grabowski and Kyle 2012, p. 552). With both strategic instruments, firms can revive their pipelines if patents of existing drugs threaten to expire (ibid., p. 567). Especially with alliances, they can explore new ways of working and get familiar with new technologies (Mittra 2007, p. 289). Substantial beneficial effects on R&D productivity are possible through the sharing of technological knowledge and the specialization of activities.
Deeds and Hill (1996, p. 42) show that the innovation rate of a firm and the number of strategic alliances it has entered are linked by an inverted U-shaped relation. This means that strategic alliances have a positive impact on innovation, but the effect begins to decrease as the number of alliances increases. Danzon et al. (2005) find that compounds developed in an alliance have a higher probability to reach the next development stage, at least for those in phase II and III clinical trials. More recent studies demonstrate that strategic alliances are particularly important for the development of breakthrough innovations (Dong et al. 2017; Dong and McCarthy 2019). A whole network of alliances can be even more advantageous, whereby its composition seems to play an important role and not too many parties should be involved. Moreover, it seems to be particularly beneficial when a university, which has special competences in relevant research fields, participates in the network (Dong and McCarthy 2019, p. 676).
With licensing deals, companies are able to “cherry pick” promising compounds (Mittra 2007, p. 293). These licensed-in compounds seem to have a higher probability of success than self-originated ones (Kola and Landis 2004, p. 713). But inefficiencies may also emerge with the use of both strategies due to market imperfections, such as information asymmetries or transaction costs. Nevertheless, in comparison to mergers and acquisitions, they represent lower cost and risk alternatives.
In light of escalating R&D costs and the lack of new drug approvals, some authors even question whether the fully integrated pharmaceutical company is still an adequate business model (Kaitin and DiMasi 2011, p. 184). They, therefore, suggest that a paradigm shift should take place towards more open forms of R&D (Munos 2010; Shaw 2017). These may be better suited to combine all the relevant know-how and integrate the different technology strands necessary to develop truly innovative drugs (Munos 2009, p. 966). Maybe the knowledge gained so far is just too complex for any company to bundle and employ it on its own (Gassmann and Reepmeyer 2005, p. 241; Seyhan 2019, p. 14).
Empirical evidence shows that open innovation models are enjoying ever wider acceptance in the industry and are increasingly used in different stages of drug development (Munos 2010, p. 536; Shaw 2017, p. 147). Examples include the sharing of genome sequences, crowdsourcing platforms such as InnoCentive or Scientist.com or the “Drugs for Neglected Diseases initiative” (DNDi). This initiative is an independent network, founded by various university institutes and non-governmental organizations, with the aim to develop drugs for the treatment of understudied, mostly tropical diseases. It mainly operates virtually and outsources all its R&D activities through public–private partnerships (Munos 2016, p. 590; Seyhan 2019, p. 15).Footnote 17 However, these models also bear some risks. For example, information may be accidentally revealed or firms may lose their competitive advantage through the disclosure of their intellectual property (Da Silva 2019).
Discussion
Our literature analysis has shown that the success rate of pharmaceutical R&D projects declined between 1980 and 2013, while there was a subperiod from 1989 to 2002 with a modest increase. More recent data demonstrate that the rate is slightly recovering again. However, the results of the different studies are only comparable to a limited extent. Therefore, further analyses with longitudinal data are necessary to obtain more reliable evidence on the development of the success rate over time. The attrition rate also rose sharply between 1980 and 2010, but has somewhat decreased after 2010, even though it is still above the level of the 1990s. Furthermore, the duration of the clinical development phase has increased considerably since the 1980s and seems to be rising even further. Finally, overall drug development costs escalated dramatically in recent decades, and this trend is also continuing. Taken together, these developments strongly indicate that the industry is indeed in an innovation crisis. Another sign of the existence of the crisis is that the approval rate is only recovering recently but is far lower than one would expect given the extraordinarily high R&D investments and the enormous scientific progress of the last decades. However, other reasons for the development of the indicators may also come into consideration. A falling success and a rising attrition rate can be caused by certain developments that do not indicate a decline in innovation performance. For example, the innovative activity of the industry has generally intensified, as shown in the higher total number of R&D projects conducted by firms (Informa 2019, p. 12). Thus, the lower success rate may be simply based on the fact that more backup compounds are started in early development stages and these are later discontinued when the lead compound turns out to be safe and effective. Findings by Arora et al. (2009) demonstrate that firms conduct large research programs with many different projects aimed at treating the same disease to raise the likelihood that one of them will eventually be successful. The authors call this phenomenon “portfolio effect” (ibid., p. 1648). Since it is only important for a firm to get one drug launched on the market, the development of the other compounds is terminated. As a result, the success rate is decreasing, although this does not reflect the existence of a productivity crisis. However, the importance of parallel research and backup compounds is still largely unexplored. Thus, with our current level of knowledge, it is impossible to assess to what extent these strategies have contributed to rising costs, declining success rates, and growing attrition rates. Further research on this topic is therefore necessary.
Nevertheless, the increase in the time required for clinical development cannot be directly explained by a higher amount of parallel research. Companies have an incentive to bring their drugs to the market as quickly as possible, as otherwise R&D costs rise and the remaining time of effective patent protection is reduced. But the longer development time may also be caused by an increased focus of the firms on more difficult disease fields. For example, Kaitin and DiMasi (2011) show that longer clinical development times are partially due to a higher number of compounds developed in therapy classes with relatively long average development times. This result is confirmed by Pammolli et al. (2011). They note that firms are increasingly directing their R&D activities to disease fields with unmet therapeutic need, less validated targets, and new mechanisms of action. And the concentration on therapy classes with a generally lower success probability and a higher risk of failure seems to have even increased after 2010 (Pammolli et al. 2020). However, if the development of the indicators is based on strategic firm decisions, it does not point to the existence of an innovation crisis. Nevertheless, this demonstrates that much more research is necessary to uncover the true reasons for the development of the indicators. According to the current state of knowledge, it is difficult to provide clear evidence on the existence of the crisis. While several signs point in this direction, they should be generally supported and validated by further analyses. In this context, studies on the quality and novelty of the approved drugs would also be informative. For example, if the approval rate is constant at a low level, but the approved drugs are more innovative and of higher quality, the crisis would be less severe. Future research should therefore differentiate between the diverse types of drugs and use appropriate indicators to measure their degree of novelty and quality.
There is also some evidence that the higher total number of R&D projects is based on the increased entry of small firms in the industry. Backfisch (2018) finds that the share of projects conducted by small firms has grown, while these firms generally have a lower success rate than their larger rivals. Thus, the productivity crisis may also be driven by the overall lower ability of small firms to successfully develop new drugs. This could be a cause of concern, but again, more research is needed to confirm these results. Analyses based on more recent data that examine the number of small firms in the industry and their success rate over time would be particularly instructive.
Our review further shows that many possible reasons for the crisis are discussed in the literature. Some of them may indeed matter, others seem to be less relevant. In our opinion, one important cause is based on the technological change induced by the biotech boom. Many scientific discoveries and technological advances have been generated in recent decades, but both the traditional pharmaceutical firms and the biotech entrants have enormous difficulties transforming this progress into new and effective medicines. Considerable knowledge gaps still exist, for example, in regard to certain processes of the organism, specific cell mechanisms, the importance of genes, the pathology of diseases, and the biological function of target molecules. Much more basic research is necessary and ways must be found to better combine the knowledge from the different scientific disciplines. Moreover, more translational research and a stronger collaboration between basic researchers and clinical scientists are particularly needed. A higher public funding of these research areas could help to alleviate the problems. In addition, economic policy should take measures to strengthen cooperation between science and industry. Some studies indicate that there are problems with validating target molecules and selecting appropriate compounds for further development. These problems can have various causes. For example, the methods used for validation may be unsuitable for certain diseases or insufficient knowledge of the underlying disease mechanism may lead to incorrect decisions. Further research is also needed in this regard and more differentiated tools should be developed, based on a deep understanding of the disease in question and its complex mode of action in the human body. The development of better biomarkers could be particularly helpful for stratifying patient groups. Better phenotyping of the patient population could make a significant contribution to improving outcomes, accelerating development, and reducing costs.
Another important reason for the low R&D productivity seems to be the very high requirements for drug approval. There is general agreement in the literature that high standards are good for innovation and that they ensure quality in terms of safety and efficacy. However, they are becoming problematic when certain groups of firms have growing difficulties to satisfy them. This might be the case for small firms or entrants. Furthermore, the requirements might not be suitable for all technologies, disease fields, or therapy classes. For example, existing standards which are based on trials with large samples may not correspond to the development of more personalized therapies. Perhaps a more elaborated system with varying requirements for different therapy classes is necessary. Additionally, more flexibility seems to be important to facilitate a rapid adjustment to new scientific and technological developments. Some measures have already been undertaken by the authorities in this regard. A broader application of adaptive licensing in conjunction with the stronger phenotyping of patients and the use of real-world data to confirm safety and effectiveness are very promising paths. However, more research is necessary on the question of how the use of these concepts can be further advanced and what impact their application will have.
Another detrimental factor seems to be the high number of consolidations in the industry. In contrast to alliances and licensing deals, the empirical literature finds predominantly negative effects of mergers on innovation. Empirical evidence indicates that the large pharmaceutical firms buy their smaller rivals to fill gaps in the own pipeline or to prevent their own drug candidates from the threat of future competition. Maybe the traditional players in the industry even impeded a faster diffusion and implementation of the biotechnological advances with this strategy. Overall, the number of successful innovators in the industry has strongly decreased due to the high merger activity, and this trend even seems to be accelerating in recent times. There are also indications that firms are increasingly licensing or acquiring promising compounds rather than pursuing own R&D activities (Kinch et al. 2014, p. 1038) and that the total number of companies involved in R&D (successful or not) is falling (Kinch et al. 2021, p. 240). This may indeed threaten the innovative capability of the industry in the long run. Therefore, the innovation effects of mergers and acquisitions should attract stronger notice from competition authorities. Moreover, more research is needed on the role of established firms in the dissemination and implementation of new technologies.
To keep up with new technological developments and to strengthen one’s own R&D capabilities, it seems to be much better to participate in collaborations or alliance networks. The extremely high R&D costs and problems in developing new drugs have even raised the question of whether the traditional R&D model is still efficient or whether more open forms of R&D are necessary. This seems to be a promising path, and open innovation models are increasingly gaining acceptance in the industry. These models offer great potential simply because many unused approaches can be revisited and can be applied in other settings. Additionally, assumptions can be tested on a larger scope than in a single firm. However, using these models can also pose some risks and challenges. These usually arise from the tension between the ownership of intellectual property and the sharing of returns from the jointly developed product or generated knowledge. Governments and authorities could, for example, develop guidelines on how to ensure access to the results from the open processes and how to share the costs and risks of development. In doing so, they could make a significant contribution to the diffusion of open innovation models.
Conclusion
In this article, we carried out a comprehensive literature review regarding the possible existence of an innovation crisis in the pharmaceutical industry. The aim of this work was to outline the current state of knowledge about whether a crisis indeed exists and, if so, what reasons could be made responsible for it. Therefore, we examined empirical studies on various indicators and discussed numerous possible reasons from an economic point of view. Such a comprehensive analysis has not been carried out previously.
Our evaluation of the empirical studies shows that the framework conditions for innovation in the industry are generally good: technological advances have led to new opportunities for the treatment of diseases, the patent system provides sufficient incentives to invest in R&D, and global drug demand and healthcare expenditures are growing. Nonetheless, the success rate of pharmaceutical R&D projects decreased during the last decades, while the attrition rate, the average development time, and the cost per new drug increased. While there is evidence that the success rate and the attrition rate have recovered slightly in recent years, growing development times and escalating costs remain a cause of concern. Thus, the empirical studies indicate that the pharmaceutical industry is indeed in an innovation crisis. However, further research on the long-term development of the indicators would be desirable to confirm these findings.
The actual causes of the crisis seem to be very complicated. On the one hand, the knowledge needed to develop truly innovative drugs seems to be more complex than previously assumed. Perhaps it is simply too extensive for a single firm to bundle and apply it on its own. Additionally, already existing insights are highly fragmented and a considerable lack of knowledge still exists in many relevant areas. On the other hand, there seems to be a gap between basic and clinical research, which may even have widened with the advent of biotechnology and the other scientific disciplines. The strong regulation of drug approval also seems to have played its part in creating and deepening this gap. There are indications that particularly small firms and entrants, which often make new scientific and technological discoveries, have difficulties in meeting the high approval requirements. Therefore, it has become increasingly important for them to enter into alliances with their larger rivals which have the necessary experience and financial resources to successfully bring a compound through clinical trials. It appears that the current regulation of the development process has given the traditional pharmaceutical firms an advantage over their smaller competitors. Additionally, there have been many mergers and acquisitions in the industry in recent decades. As a result, both the number of successful innovators and the number of all firms that carry out own R&D activities have declined. These developments are worrisome and can endanger the innovative capability of the industry in the long run. Apart from their potentially negative impact on innovation, mergers can lead to greater bargaining power, more market power, and thus to a greater influence on prices. This in turn can cause significant problems for regulators, insurers, policy makers, and patients.
To improve the industry’s ability to innovate, various measures are therefore necessary. First, more basic and translational research and a better integration of findings from different scientific disciplines are needed. Policymakers should adopt measures and provide the necessary funding to bridge the gap between basic and clinical research. Thereby, better opportunities should be created for industry and science to cooperate in the development of drugs. Second, a more precise adaptation of regulatory standards to the different conditions in the individual therapy classes and a more flexible design of the development process are necessary. Measures such as adaptive licensing, a stronger phenotyping of patients, and the use of real-world evidence are promising concepts. However, more research concerning their optimal application and their impact on the total number of approvals would be desirable. Thereby, possibilities should be examined as to how especially small firms and entrants can be supported during development. Finally, competition policy should pay more attention to the possible innovation and long-term effects of mergers. In this context, more research on the impact of the high merger activity on the industry as a whole and the role of the established firms in implementing and disseminating new scientific and technological advances would also be useful.
From a purely technological point of view, it is still unclear whether biotechnology has the potential to completely replace traditional chemistry. The full extent of its influence is not yet foreseeable, and it will certainly take much more time to be able to overlook the entire potential. Perhaps biotechnology will become the prevalent technology in certain therapeutic areas. This also depends on whether the demand for chemical drugs in these fields will stay at constant levels or will decline over the long run. At the same time, enormous progress has also taken place in other relevant scientific areas in recent decades and very promising technologies have emerged. Personalized medicine, new cancer immunotherapies, RNA interference technologies, biosensing, mobile health, big data, remote monitoring and artificial intelligence are technological advances that provide a fruitful background for innovative medicines. Some of these technologies may bring radical changes to the entire model of drug delivery. Others may replace the classical “one drug for all” scheme. Nevertheless, many of the new technological developments seem to be rather complementary to the use of classical chemical drugs. For example, for personalized medicine it is still necessary to develop a compound that has a desired effect on a specific target receptor. Genetic engineering may lead to fewer and less severe diseases, which will probably reduce the necessity to be on medication. However, it is unclear in which time horizon and to what extent this development can or will take place. It seems to be rather a long-term issue and it is uncertain whether it will work equally well for all disease groups. The prediction of both growing prescription drug sales as well as an increasing future demand shows that the classical medicines are still in a strong position and are likely to maintain this status in the next years. Perhaps chemical drugs, advanced biologics, nucleic acid therapeutics, cell therapies, and implantables will all be requested and offered side by side in the future.
Availability of data and material
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Code availability
Not applicable.
Notes
A New Chemical Entity is a chemical drug that contains no active moiety that has previously been approved by the FDA in another application (FDA 2018a). In contrast, a New Biological Entity is a new biological product used to prevent or treat a disease, such as a protein, peptide, antibody, virus, or a vaccine (Branch and Agranat 2014, p. 8751).
Net sales are calculated as gross sales minus customer discounts, sales returns, and allowances.
Included sources were Business Source Premier, EBSCO eBook Collection, ERIC, GreenFILE, MEDLINE, EBSCO Open Dissertations, and Library, Information Science & Technology Abstracts.
Hay et al. (2014) apply both project definitions and actually find a lower success rate for the single indications of drug candidates than for all indications (0.104 compared to 0.153).
DiMasi et al. (1995) do not indicate where the clinical trials in their data set take place. However, their sample only contains US firms.
The studies presented here were found during the extensive systematic search for studies on the success rate
The latter are determined by the help of actual R&D spending per successfully approved drug, project duration, and success rate per development phase (DiMasi et al. 1991, p. 115).
.
Since most of the literature refers to regulatory standards in the US, we will focus our description on the approval process determined by the FDA. However, quite similar standards exist in Europe and Japan (Scherer 2000, p. 1309).
The indication is defined “… by a disease area, a line of therapy, and a patient population” (Lakdawalla 2018, p. 402).
Certainly, this largely represents the point of view of the affected firms. From an economic standpoint, higher regulatory demands can be reasonable what will be discussed in more detail below.
It differs from the “low hanging fruit” problem as follows: The latter means that the relatively simple problems have already been solved and only the more challenging tasks remain. In contrast, the former argues that the solutions developed so far reduce the value of possible future solutions (Scannell et al. 2012, p. 193).
In September 2004, Merck & Co. withdrew its successful drug, a treatment of osteoarthritis and acute pain, from the market because of concerns about increased risks of suffering a heart attack or stroke when used during a long time. Merck withheld information about these risks from doctors and patients in over 88,000 cases (Topol 2004).
Of course, the consequences in practice cannot simply be remedied, especially in the case of safety deficiencies.
In general, copayment amounts are higher for substances with relatively little therapeutic value (Gilman and Kautter 2008, pp. 478).
An elaborated overview of the different accelerated access pathways established in the US and the EU is provided by Baird et al. (2014).
Adaptive licensing means that a drug is partially approved based on limited evidence about effectiveness and safety and that approval is later expanded or restricted, when more evidence is available, generated either through randomized clinical trials or based on real-world data (Woodcock 2012, p. 378).
Outsourcing is also an alternative model of drug development which is increasingly used in the industry, not only by small start-ups but also by established pharmaceutical firms (Kinch and Moore 2016, p. 651).
References
Abrantes-Metz R, Adams C, Metz A (2004) Pharmaceutical development phases: A duration analysis. Bureau of Economics, Federal Trade Commission, Working Paper No. 274. https://doi.org/10.2139/ssrn.607941. Accessed 15 Jan 2020
Adams C, Brantner V (2006) Estimating the cost of new drug development: Is it really $802 million? Health Aff 25(2):420–428. https://doi.org/10.1377/hlthaff.25.2.420
Adams C, Brantner V (2010) Spending on new drug development. Health Econ 19(2):130–141. https://doi.org/10.1002/hec.1454
Akhtar A (2015) The flaws and human harms of animal experimentation. Camb Q Healthc Ethics 24(4):407–419. https://doi.org/10.1017/S0963180115000079
Aronson J, Green A (2020) Me-too pharmaceutical products: History, definitions, examples, and relevance to drug shortages and essential medicines lists. Br J Clin Pharmacol 86(11):2114–2122. https://doi.org/10.1111/bcp.14327
Arora A, Gambardella A, Magazzini L, Pammolli F (2009) A breath of fresh air? Firm type, scale, scope, and selection effects in drug development. Manage Sci 55(10):1638–1653. https://doi.org/10.1287/mnsc.1090.1055
Backfisch M (2017) Have pharmaceutical R&D project success rates decreased? A critical review and new empirical results. MAGKS Discussion Paper No. 46–2017, Philipps-University Marburg, School of Business and Economics. https://www.uni-marburg.de/fb02/makro/forschung/magkspapers/paper_2017/46-2017_backfisch.pdf. Accessed 19 Feb 2020
Backfisch M (2018) The development of firm size and innovativeness in the pharmaceutical industry between 1989 and 2010. MAGKS Discussion Paper No. 13–2018. Philipps-University Marburg, School of Business and Economics. https://www.uni-marburg.de/fb02/makro/forschung/magkspapers/paper_2018/13-2018_backfisch.pdf. Accessed 19 Febr 2020
Baird L, Banken R, Eichler H et al (2014) Accelerated access to innovative medicines for patients in need. Clin Pharmacol Ther 96(5):559–571. https://doi.org/10.1038/clpt.2014.145
Begley C, Ioannidis J (2015) Reproducibility in science - improving the standard for basic and preclinical research. Circ Res 116(1):116–126. https://doi.org/10.1161/CIRCRESAHA.114.303819
Belderbos R, Gilsing V, Suzuki S (2016) Direct and mediated ties to universities: “Scientific” absorptive capacity and innovation performance of pharmaceutical firms. Strateg Organ 14(1):32–52. https://doi.org/10.1177/1476127015604734
Berndt E, Gottschalk A, Philipson T, Strobeck M (2005) Industry funding of the FDA: Effects of PDUFA on approval times and withdrawal rates. Nat Rev Drug Discov 4:545–554. https://doi.org/10.1038/nrd1774
Betz U (2011) Portfolio management in early-stage drug discovery – a traveler’s guide through uncharted territory. Drug Discov Today 16:609–618. https://doi.org/10.1016/j.drudis.2011.04.012
Bienz-Tadmor B, DiCerbo P, Tadmor G, Lasagna L (1992) Biopharmaceuticals and conventional drugs: Clinical success rates. Nat Biotechnol 10:521–525. https://doi.org/10.1038/nbt0592-521
Bolisis W, Fay M, Kühler T (2020) Use of real-world data for new drug applications and line extensions. Clin Ther 42(5):926–938. https://doi.org/10.1016/j.clinthera.2020.03.006
Bothwell L, Avorn J, Khan N, Kesselheim A (2018) Adaptive design clinical trials: a review of the literature and ClinicalTrials.gov. BMJ Open 8(2):e018320. https://doi.org/10.1136/bmjopen-2017-018320
Bowen A, Casadevall A (2015) Increasing disparities between resource inputs and outcomes, as measured by certain health deliverables, in biomedical research. Proceed Natl Acad Sci USA 112(36):11335–11340. https://doi.org/10.1073/pnas.1504955112
Branch S, Agranat I (2014) “New drug” designations for new therapeutic entities: new active substance, new chemical entity, new biological entity, new molecular entity. J Med Chem 57(21):8729–8765. https://doi.org/10.1021/jm402001w
Bunnage M (2011) Getting pharmaceutical R&D back on target. Nat Chem Biol 7:335–339. https://doi.org/10.1038/nchembio.581
Buonansegna E, Salomo S, Maier A, Li-Ying J (2014) Pharmaceutical new product development: why do clinical trials fail? R&D Manag 44(2):189–202. https://doi.org/10.1111/radm.12053
Butler D (2008) Translational research: Crossing the valley of death. Nature 453:840–842. https://doi.org/10.1038/453840a
CBO (2006) Research and Development in the Pharmaceutical Industry. Congress of the United States, Congressional Budget Office. https://www.cbo.gov/sites/default/files/cbofiles/ftpdocs/76xx/doc7615/10-02-drugr-d.pdf. Accessed 23 Aug 2018
Chu W (2006) Has new technology hampered R&D productivity? William Reed Business Media Ltd. Crawley, England. https://www.outsourcing-pharma.com/Article/2006/03/30/Has-new-technology-hampered-R-D-productivity?utm_source=copyright&utm_medium=OnSite&utm_campaign=copyright. Accessed 12 Aug 2018
Cockburn I (2006) Is the pharmaceutical industry in a productivity crisis? Innov Policy Econ 7:1–32. https://doi.org/10.1086/ipe.7.25056188
Cockburn I, Henderson R (2001) Scale and scope in drug development: unpacking the advantages of size in pharmaceutical research. J Health Econ 20(6):1033–1057. https://doi.org/10.1016/S0167-6296(01)00108-4
Cohen W, Levinthal D (1990) Absorptive capacity: a new perspective on learning and innovation. Adm Sci Q 35(1):128–152. https://doi.org/10.2307/2393553
Comanor W, Scherer F (2013) Mergers and innovation in the pharmaceutical industry. J Health Econ 32(1):106–113. https://doi.org/10.1016/j.jhealeco.2012.09.006
Coombs R, Metcalfe S (2002) Innovation in pharmaceuticals: perspectives on the co-ordination, combination and creation of capabilities. Technol Anal Strateg Managt 14(3):261–271. https://doi.org/10.1080/095373202200002104
Corrigan-Curay J, Sacks L, Woodcock J (2018) Real-world evidence and real-world data for evaluating drug safety and effectiveness. JAMA 320(9):867–868. https://doi.org/10.1001/jama.2018.10136
Cowlrick I, Hedner T, Wolf R, Olausson M, Klofsten M (2011) Decision-making in the pharmaceutical industry: analysis of entrepreneurial risk and attitude using uncertain information. R&D Management 41(4):321–336. https://doi.org/10.1111/j.1467-9310.2011.00649.x
Cunningham C, Ederer F, Ma S (2021) Killer Acquisitions. J Polit Econ 129(3):649–702. https://doi.org/10.1086/712506
Danzon P, Nicholson S, Pereira N (2005) Productivity in pharmaceutical-biotechnology R&D: the role of experience and alliances. J Health Econ 24(2):317–339. https://doi.org/10.1016/j.jhealeco.2004.09.006
Danzon P, Epstein A, Nicholson S (2007) Mergers and acquisitions in the pharmaceutical and biotech industries. Manag Decis Econ 28:307–328. https://doi.org/10.1002/mde.1343
Danzon P, Keuffel E (2014) Regulation of the Pharmaceutical-Biotechnology Industry. In: Rose N (ed) Economic Regulation and Its Reform: What Have We Learned? University of Chicago Press, Chicago, pp 407–484
Darrow J, Avorn J, Kesselheim A (2014) New FDA breakthrough-drug category – implications for patients. N Engl J Med 370:1252–1258. https://doi.org/10.1056/NEJMhle1311493
Da Silva M (2019) Open innovation and IPRs: Mutually incompatible or complementary institutions? J Innov Knowl 4(4):248–252. https://doi.org/10.1016/j.jik.2018.03.010
Deeds D, Hill C (1996) Strategic alliances and the rate of new product development: An empirical study of entrepreneurial biotechnology firms. J Bus Ventur 11(1):41–55. https://doi.org/10.1016/0883-9026(95)00087-9
De Man AP, Duysters G (2005) Collaboration and innovation: A review of the effects of mergers, acquisitions and alliances on innovation. Technovation 25(12):1377–1387. https://doi.org/10.1016/j.technovation.2004.07.021
DiMasi J (2001) Risks in new drug development: Approval success rates for investigational drugs. Clin Pharmacol Ther 69(5):297–307. https://doi.org/10.1067/mcp.2001.115446
DiMasi J, Hansen R, Grabowski H, Lasagna L (1991) Cost of innovation in the pharmaceutical industry. J Health Econ 10(2):107–142. https://doi.org/10.1016/0167-6296(91)90001-4
DiMasi J, Grabowski H, Vernon J (1995) R&D costs, innovative output and firm size in the pharmaceutical industry. Int J Econ Bus 2(2):201–219. https://doi.org/10.1080/758519309
DiMasi J, Hansen R, Grabowski H (2003) The price of innovation: new estimates of drug development costs. J Health Econ 22(2):151–185. https://doi.org/10.1016/S0167-6296(02)00126-1
DiMasi J, Grabowski H, Hansen R (2016) Innovation in the pharmaceutical industry: New estimates of R&D costs. J Health Econ 47:20–33. https://doi.org/10.1016/j.jhealeco.2016.01.012
Disis M, Slattery J (2010) The road we must take: Multidisciplinary team science. Sci Transl Med. 2(22):22cm9. https://doi.org/10.1126/scitranslmed.3000421
Dong J, McCarthy K (2019) When more isn’t merrier: Pharmaceutical alliance networks and breakthrough innovation. Drug Discovery Today 24(3):673–677. https://doi.org/10.1016/j.drudis.2019.01.002
Dong J, McCarthy K, Schoenmakers W (2017) How central is too central? Organizing interorganizational collaboration networks for breakthrough innovation. J Prod Innov Manag 34(4):526–542. https://doi.org/10.1111/jpim.12384
Drews J (2000) Quo vadis, biotech? (Part 1). Drug Discov Today 5(12):547–553. https://doi.org/10.1016/S1359-6446(00)01580-4
EFPIA (2010) The Pharmaceutical Industry in Figures – 2010 Edition. Report of the European Federation of Pharmaceutical Industries and Associations. https://www.sfee.gr/wp-content/uploads/2015/05/Figures_2010_Final-20100611-001-EN-v1_0.pdf. Accessed 10 March 2020
EFPIA (2014) The Pharmaceutical Industry in Figures – Key Data 2014. Report of the European Federation of Pharmaceutical Industries and Associations. https://www.phar-in.eu/wp-content/uploads/2014/05/Figures_2014_Final.pdf. Accessed 9 March 2020
EFPIA (2015) The Pharmaceutical Industry in Figures – Key Data 2015. Report of the European Federation of Pharmaceutical Industries and Associations. https://www.efpia.eu/media/25822/2015-the-pharmaceutical-industry-in-figures.pdf. Accessed 10 March 2020
EFPIA (2017) The Pharmaceutical Industry in Figures – Key Data 2017. Report of the European Federation of Pharmaceutical Industries and Associations. https://www.efpia.eu/media/219735/efpia-pharmafigures2017_statisticbroch_v04-final.pdf. Accessed 21 Jan 2020
EFPIA (2018) The Pharmaceutical Industry in Figures – Key Data 2018. Report of the European Federation of Pharmaceutical Industries and Associations. https://efpia.eu/media/361960/efpia-pharmafigures2018_v07-hq.pdf. Accessed 10 March 2020
EFPIA (2019) The Pharmaceutical Industry in Figures – Key Data 2019. Report of the European Federation of Pharmaceutical Industries and Associations. https://www.efpia.eu/media/413006/the-pharmaceutical-industry-in-figures.pdf. Accessed 10 March 2020
Eichler H, Baird L, Barker R et al (2015) From adaptive licensing to adaptive pathways: delivering a flexible life-span approach to bring new drugs to patients. Clin Pharmacol Ther 97(3):234–246. https://doi.org/10.1002/cpt.59
EMA (2016a) Final report on the adaptive pathways pilot. European Medicines Agency. https://www.ema.europa.eu/en/documents/report/final-report-adaptive-pathways-pilot_en.pdf. Accessed 10 March 2020
EMA (2016b) Guidance for companies considering the adaptive pathways approach. European Medicines Agency. https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/guidance-companies-considering-adaptive-pathways-approach_en.pdf. Accessed 10 March 2020
EMA (2020) Guideline on registry-based studies – Draft. European Medicines Agency. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-registry-based-studies_en.pdf. Accessed 20 Febr 2021
Ernst H, Vitt J (2000) The influence of corporate acquisitions on the behavior of key inventors. R&D Manag 30(2):105–119. https://doi.org/10.1111/1467-9310.00162
Evaluate (2017) Evaluate Pharma, World Preview 2017, Outlook to 2022. Evaluate Ltd. http://www.evaluate.com/PharmaWorldPreview2017. Accessed 12 Aug 2018
Evaluate (2019) Evaluate Vantage 2020 Preview. Evaluate Ltd. https://www.evaluate.com/thought-leadership/vantage/evaluate-vantage-2020-preview#download. Accessed 27 Aug 2020
Evens R (2016) Pharma success in product development – does biotechnology change the paradigm in product development and attrition. AAPS J 18(1):281–285. https://doi.org/10.1208/s12248-015-9833-6
Fagerberg J (2005) Innovation: A guide to the literature. In: Fagerberg J, Mowery D (eds) The Oxford Handbook of Innovation. University Press, Oxford, pp 1–26
FDA (2018a) New Drugs at FDA: CDER’s New Molecular Entities and New Therapeutic Biological Products. US Food and Drug Administration. https://www.fda.gov/drugs/development-approval-process-drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products. Accessed 10 March 2020
FDA (2018b) Summary of NDA Approvals & Receipts, 1938 to the Present. US Food and Drug Administration. https://www.fda.gov/about-fda/histories-product-regulation/summary-nda-approvals-receipts-1938-present. Accessed 9 March 2020
FDA (2018c) Framework for FDA’s Real-World Evidence Program. US Food and Drug Administration. https://www.fda.gov/media/120060/download. Accessed 20 Febr 2021
FDA (2019a) Advancing Health through Innovation, New Drug Therapy Approvals 2019. Center for Drug Evaluation and Research. US Food and Drug Administration. https://www.fda.gov/media/134493/download. Accessed 9 March 2020
FDA (2019b) Adaptive Designs for Clinical Trials of Drugs and Biologics, Guidance for Industry. US Food and Drug Administration. https://www.fda.gov/media/78495/download. Accessed 20 Febr 2021
Frank R (2003) New estimates of drug development costs. J Health Econ 22(2):325–330. https://doi.org/10.1016/S0167-6296(03)00002-X
Gagne J, Choudhry N (2011) How many “me-too” drugs is too many? J Am Med Assoc 305(7):711–712. https://doi.org/10.1001/jama.2011.152
Garnier JP (2008) Rebuilding the R&D engine in big pharma. Harv Bus Rev 86(5):68–76
Gassmann O, Reepmeyer G (2005) Organizing pharmaceutical innovation: From science-based knowledge creators to drug-oriented knowledge brokers. Creat Innov Manag 14(3):233–245. https://doi.org/10.1111/j.1467-8691.2005.00344.x
Gassmann O, Reepmeyer G, von Zedtwitz M (2008) Leading Pharmaceutical Innovation: Trends and Drivers for Growth in the Pharmaceutical Industry, 2nd edn. Springer, Berlin
Gilman B, Kautter J (2008) Impact of multitiered copayments on the use and cost of prescription drugs among medicare beneficiaries. Health Serv Res 43(2):478–495. https://doi.org/10.1111/j.1475-6773.2007.00774.x
Grabowski H, Kyle M (2012) Mergers, acquisitions, and alliances. In: Danzon P, Nicholson S (eds) The Oxford Handbook of the Economics of the Biopharmaceutical Industry. University Press, Oxford, pp 552–579
Granstrand O, Sjölander S (1990) The acquisition of technology and small firms by large firms. J Econ Behav Organ 13(3):367–386. https://doi.org/10.1016/0167-2681(90)90006-Y
Hall B (1999) Mergers and R&D revisited. Paper prepared for the Quasi-Experimental Methods Symposium. Econometrics Laboratory, UC Berkeley. https://www.researchgate.net/profile/Bronwyn-Hall-3/publication/2353839_Mergers_and_RD_Revisited/links/0c96051f981c9ec90e000000/Mergers-and-R-D-Revisited.pdf. Accessed 10 March 2020
Hay M, Thomas D, Craighead J, Economides C, Rosenthal J (2014) Clinical development success rates for investigational drugs. Nat Biotechnol 32:40–51. https://doi.org/10.1038/nbt.2786
Henderson R (2000) One point of view: Drug industry mergers won’t necessarily benefit R&D. Res Technol Manag 43(4):10–11
Hitt M, Hoskisson R, Ireland D, Harrison J (1991) Effects of acquisitions on R&D inputs and outputs. Acad Manag J 34(3):693–711. https://doi.org/10.5465/256412
Hopkins M, Martin P, Nightingale P, Kraft A, Mahdi S (2007) The myth of the biotech revolution: An assessment of technological, clinical and organizational change. Res Policy 36(4):566–589. https://doi.org/10.1016/j.respol.2007.02.013
IFPMA (2017) The Pharmaceutical Industry and Global Health. Facts and Figures 2017. International Federation of Pharmaceutical Manufacturers & Associations. https://www.ifpma.org/wp-content/uploads/2017/02/IFPMA-Facts-And-Figures-2017.pdf. Accessed 19 Jan 2020
IMS Institute (2015) Global Medicines Use in 2020 – Outlook and Implications. IMS Institute for Healthcare Informatics. https://www.iqvia.com/-/media/iqvia/pdfs/institute-reports/global-medicines-use-in-2020. Accessed 15 Jan 2020
Informa (2019) Pharma R&D Annual Review 2019. Pharmaprojects, Pharma Intelligence, Informa Ltd. https://pharmaintelligence.informa.com/~/media/informa-shop-window/pharma/2019/files/whitepapers/pharma-rd-review-2019-whitepaper.pdf. Accessed 19 Jan 2020
Ioannidis J (2005) Why most published research findings are false. PLoS Med 2(8):e124. https://doi.org/10.1371/journal.pmed.0020124
Ioannidis J (2016) Why most clinical research is not useful. PLoS Med 13(6):e1002049. https://doi.org/10.1371/journal.pmed.1002049
Jommi C, Armeni P, Costa F, Bertolani A, Otto M (2020) Implementation of Value-based Pricing for Medicines. Clin Ther 42(1):15–24. https://doi.org/10.1016/j.clinthera.2019.11.006
Kaitin K, DiMasi J (2011) Pharmaceutical innovation in the 21st century: new drug approvals in the first decade, 2000–2009. Clin Pharmacol Ther 89(2):183–188. https://doi.org/10.1038/clpt.2010.286
Kinch M (2014) The rise (and decline?) of biotechnology. Drug Discover Today 19(11):1686–1690. https://doi.org/10.1016/j.drudis.2014.04.006
Kinch M, Haynesworth A, Kinch S, Hoyer D (2014) An overview of FDA-approved new molecular entities: 1827–2013. Drug Discov Today 19(8):1033–1039. https://doi.org/10.1016/j.drudis.2014.03.018
Kinch M, Moore R (2016) Innovator organizations in new drug development: assessing the sustainability of the biopharmaceutical industry. Cell Chem Biol 23(6):644–653. https://doi.org/10.1016/j.chembiol.2016.05.013
Kinch M, Kraft Z, Schwartz T (2021) Sources of innovation for new medicines: questions of sustainability. Drug Discov Today 26(1):240–247. https://doi.org/10.1016/j.drudis.2020.10.026
Kola I, Landis J (2004) Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 3:711–716. https://doi.org/10.1038/nrd1470
Laermann-Nguyen U (2015) Pharmaceutical mergers and their effect on parallel R&D paths. Paper presented at the DRUID Academy Conference in Rebild/Aalborg, Denmark. January 21–23, 2015. https://conference.druid.dk/acc_papers/rtrg5rn16q8kq7rcma21v87em7hk.pdf. Accessed 19 Jan 2020
Lakdawalla D (2018) Economics of the pharmaceutical industry. J Econ Lit 56(2):397–449. https://doi.org/10.1257/jel.20161327
LaMattina J (2011) The impact of mergers on pharmaceutical R&D. Nat Rev Drug Discov 10:559–560. https://doi.org/10.1038/nrd3514
Light D, Lexchin J (2012) Pharmaceutical research and development: what do we get for all that money? BMJ 345:e4348. https://doi.org/10.1136/bmj.e4348
Light D, Warburton R (2011) Demythologizing the high cost of pharmaceutical research. BioSocieties 6:34–50. https://doi.org/10.1057/biosoc.2010.40
Malerba F, Orsenigo L (2015) The evolution of the pharmaceutical industry. Bus Hist 57(5):664–687. https://doi.org/10.1080/00076791.2014.975119
Mankoff S, Brander C, Ferrone S, Marincola F (2004) Lost in translation: obstacles to translational medicine. J Transl Med 2(14):1–5. https://doi.org/10.1186/1479-5876-2-14
Mansfield E (1986) Patents and innovation: an empirical study. Manag Sci 32(2):139–256. https://doi.org/10.1287/mnsc.32.2.173
Manski C (2009) Adaptive partial drug approval: a health policy proposal. Econom’ Voice. https://doi.org/10.2202/1553-3832.1449
Martin P, Hopkins M, Nightingale P, Kraft A (2009) On a Critical Path – Genomics, the Crisis of Pharmaceutical Productivity, and the Search for Sustainability. In: Atkinson P, Glasner P, Lock M (eds) The Handbook of Genetics & Society: Mapping the New Genomic Era. Routledge, New York, pp 145–162
Miller D (2012) Value-based pricing: examples of healthcare system reforms from the UK and US and implications for industry. Pharm Med 26:217–222. https://doi.org/10.1007/BF03262478
Mittra J (2007) Life science innovation and the restructuring of the pharmaceutical industry: merger, acquisition and strategic alliance behaviour of large firms. Technol Anal Strateg Manag 19(3):279–301. https://doi.org/10.1080/09537320701281540
Moher D, Liberati A, Tetzlaff J, Altmann D (2009) Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med 6(7):e1000097. https://doi.org/10.1371/journal.pmed.1000097
Mohr P, Tunis S (2010) Access with evidence development: The US experience. Pharmacoeconomics 28(2):153–162. https://doi.org/10.2165/11531050-000000000-00000
Morgan P, Van Der Graaf P, Arrowsmith J et al (2012) Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug Discov Today 17(9/10):419–424. https://doi.org/10.1016/j.drudis.2011.12.020
Munos B (2009) Lessons from 60 years of pharmaceutical innovation. Nat Rev Drug Discov 8:959–968. https://doi.org/10.1038/nrd2961
Munos B (2010) Can open-source drug R&D repower pharmaceutical innovation? Clin Pharmacol Ther 87(5):534–536. https://doi.org/10.1038/clpt.2010.26
Munos B (2016) Biomedical innovation: lessons from the past and perspectives for the future. Clin Pharmacol Ther 100(6):588–590. https://doi.org/10.1002/cpt.456
Munos B, Chin W (2011) How to revive breakthrough innovation in the pharmaceutical industry. Sci Transl Med 3(89):89cm16. https://doi.org/10.1126/scitranslmed.3002273
Ornaghi C (2009) Mergers and innovation in big pharma. Int J Ind Organ 27(1):70–79. https://doi.org/10.1016/j.ijindorg.2008.04.003
OTA (1993) Pharmaceutical R&D: Costs, Risks, and Rewards. US Congress, Office of Technology Assessment, OTA-H-522. US Government Printing Office, Washington, D.C. https://govinfo.library.unt.edu/ota/Ota_1/DATA/1993/9336.PDF. Accessed 15 Jan 2020
Pammolli F, Magazzini L, Riccaboni M (2011) The productivity crisis in pharmaceutical R&D. Nat Rev Drug Discov 10(6):428–438. https://doi.org/10.1038/nrd3405
Pammolli F, Righetto L, Abrignani S, Pani L, Pelicci P, Rabosio E (2020) The endless frontier? The recent increase in R&D productivity in pharmaceuticals. J Transl Med 18(162):1–14. https://doi.org/10.1186/s12967-020-02313-z
Paul S, Mytelka D, Dunwiddie C et al (2010) How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov 9:203–214. https://doi.org/10.1038/nrd3078
PhRMA (2016) Biopharmaceutical Research Industry Profile 2016. Pharmaceutical Research and Manufacturers of America. Washington, D.C. http://phrma-docs.phrma.org/sites/default/files/pdf/biopharmaceutical-industry-profile.pdf. Accessed 11 Jan 2020
Pisano G (2006) Science Business: The Promise, the Reality and the Future of Biotech. Harvard Business School University Press, Boston, MA
Posey Norris S, Pankevich D, Davis M, Altevogt B (2014) Improving and Accelerating Therapeutic Development for Nervous System Disorders. Forum on Neuroscience and Nervous System Disorders. Workshop Summary. Institute of Medicine of the National Academies of Sciences Engineering Medicine, Washington, D.C. https://doi.org/10.17226/18494
Prescrire International (2003) A review of new drugs and indications in 2002: Financial speculation or better patient care? Prescrire Int 12(64):74–77. https://english.prescrire.org/en/SummaryDetail.aspx?IssueId=64. Accessed 05 Febr 2020
Richard J, Wurtman M (1997) What went right: Why is HIV a treatable infection? Nat Med 3:714–717. https://doi.org/10.1038/nm0797-714
Roberts S, Fischhoff M, Sakowski S, Feldman E (2012) Perspective: Transforming science into medicine: How clinician-scientists can build bridges across research’s “valley of death.” Acad Med 87(3):266–270. https://doi.org/10.1097/ACM.0b013e3182446fa3
Ruffolo R (2006) Why has R&D productivity declined in the pharmaceutical industry? Expert Opin Drug Discov 1(2):99–102. https://doi.org/10.1517/17460441.1.2.99
Sams-Dodd F (2013) Is poor research the cause of the declining productivity of the pharmaceutical industry? An industry in need of a paradigm shift. Drug Discov Today 18(5/6):211–217. https://doi.org/10.1016/j.drudis.2012.10.010
Scannell J, Blanckley A, Boldon H, Warrington B (2012) Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discovery 11:191–200. https://doi.org/10.1038/nrd3681
Scannell J, Bosley J (2016) When quality beats quantity: decision theory, drug discovery, and the reproducibility crisis. PLoS One 11(2):e0147215. https://doi.org/10.1371/journal.pone.0147215
Scherer F (2000) The pharmaceutical industry. In: Culyer AJ, Newhouse JP (eds) Handbook of Health Economics I. Elsevier, Amsterdam, pp 1298–1336
Scherer F (2010) Pharmaceutical innovation. In: Hall BH, Rosenberg N (eds) Handbook of the Economics of Innovation I. North-Holland, Amsterdam, pp 539–547
Schmid E, Smith D (2004) Is pharmaceutical R&D just a game of chance or can strategy make a difference? Drug Discov Today 9(1):18–26. https://doi.org/10.1016/S1359-6446(04)02951-4
Scott Morton F, Kyle M (2012) Markets for Pharmaceutical Products. In: Pauly M, McGuire T, Pita Barros P (eds) Handbook of Health Economics II. North-Holland, Amsterdam, pp 763–823
Seyhan A (2019) Lost in translation: the valley of death across preclinical and clinical divide – identification of problems and overcoming obstacles. Transl Med Commun. https://doi.org/10.1186/s41231-019-0050-7
Shaw D (2017) Is open science the future of drug development? Yale J Biol Med 90(1):147–151
Sherman R, Anderson S, Pan G et al (2016) Real-world evidence — what is it and what can it tell us? N Engl J Med 375(23):2293–2297. https://doi.org/10.1056/NEJMsb1609216
Thomas L (1996) Industrial Policy and International Competitiveness in the Pharmaceutical Industry. In: Helms R (ed) Competitive Strategies in the Pharmaceutical Industry. The American Enterprise Institute, Washington, D.C., pp 107–129
Thomas D, Burns J, Audette J, Carroll A, Dow-Hygelund C, Hay M (2016) Clinical Development Success Rates 2006–2015. Biotechnology Innovation Organization. https://www.bio.org/sites/default/files/legacy/bioorg/docs/Clinical%20Development%20Success%20Rates%202006-2015%20-%20BIO,%20Biomedtracker,%20Amplion%202016.pdf. Accessed 23 Febr 2021
Topol E (2004) Failing the public health – rofecoxib, merck, and the FDA. N Engl J Med 352:1707–1709. https://doi.org/10.1056/NEJMp048286
Waldman S, Terzic A (2016) Bioinnovation Enterprise: An engine driving breakthrough therapies. Clin Pharmacol Ther 99(1):8–13. https://doi.org/10.1002/cpt.272
Wong C, Siah K, Lo A (2019) Estimation of clinical trial success rates and related parameters. Biostatistics 20(2):273–286. https://doi.org/10.1093/biostatistics/kxx069
Woodcock J (2012) Evidence vs. access: can twenty-first-century drug regulation refine the tradeoffs? Clin Pharmacol Therap 91(3):378–380. https://doi.org/10.1038/clpt.2011.337
Wouters O, McKee M, Luyten J (2020) Estimated research and development investment needed to bring a new medicine to market, 2009–2018. JAMA 323(9):844–853. https://doi.org/10.1001/jama.2020.1166
Funding
Open Access funding enabled and organized by Projekt DEAL. No funding was received to assist with the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
Idea: Ute Laermann-Nguyen, Martin Backfisch; Literature search and data analysis: Ute Laermann-Nguyen, Martin Backfisch; Writing – original draft preparation: Ute Laermann-Nguyen, Martin Backfisch; Writing – review and editing: Ute Laermann-Nguyen. Visualization: Ute Laermann-Nguyen.
Corresponding author
Ethics declarations
Conflicts of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
A former version of this essay is part of the cumulative PhD thesis of Martin Backfisch: Innovation, Search, and the Success of R&D Projects in the Pharmaceutical Industry, Philipps University Marburg, 2019.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Laermann-Nguyen, U., Backfisch, M. Innovation crisis in the pharmaceutical industry? A survey. SN Bus Econ 1, 164 (2021). https://doi.org/10.1007/s43546-021-00163-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s43546-021-00163-5