
Abstract
The structural, nonlinear optical, electronic and thermodynamic properties of andirobin molecule were carried out by density functional theory at the B3LYP, WB97XD level and at the Restricted Hartree–Fock level by employing 6–311G(d,p) basis set. The obtained values of bond lengths, bond angles, 1H NMR and 13C NMR are in good agreement with experimental values. The dipole moment and first static hyperpolarizability show that andirobin can be applied in nonlinear optical devices. HOMO–LUMO energy gap values were found to be greater than 4 eV and lead us to the conclusion that this molecule can be used as insulator in many electronic devices. The thermal energy (E), molar heat capacity at constant volume \((C_{v}\)) and entropy (S) were also calculated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Currently, studies on the calculation of the electronic and physico-chemical properties of organic systems are increasingly developed [1]. The development of increasingly high-performance atomistic simulation tools has enabled researchers such as chemists and physicists to model molecular structures that can be synthesized in laboratories and that can meet the demands of the materials market. Thus, materials can be designed and produced in 2D or 3D representations with electrical properties such as electrical conductivity and HOMO–LUMO energy gap to describe their insulating, semiconducting or conducting character. Molecules with a disordered spatial distribution can be simulated starting from a configuration corresponding to a global extremum whose energy is in the range of energies containing the local minimum or point of stability sought. More and more sophisticated modeling methods based on the solution of the Schrödinger equation such as ab-initio methods, perturbation methods and density functional theory allow to explore and obtain information from the microscopic states of materials.
Theoretical studies carried out by modeling or numerical simulations serve as a guide for experimenters by providing information that is difficult to access experimentally. In general, it is difficult to access the atomic scale experimentally. Modeling therefore makes it possible to obtain invaluable predictions of the properties of a molecular structure more quickly and at less cost than test methods. One of the major challenges of simulations based on quantum methods is to determine the properties of materials, including organic systems derived from medicinal plants, at the atomic scale in order to refine physical understanding and push back the limits of emerging technologies.
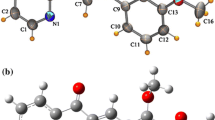
In Meliaceae, there are several substances whose active principle is found more in limonoids. Among them, one can findandirobin (systematic name: methyl-2{(1R,2R)-2-[(1aS, 4S, 4aS, 8aS)-4-(fluran-3-yl)-4a-methyl-8-methylene-2-oxooctahydrooxireno[2,3-d]isochromen-7-yl]-2,6,6-trimethyl-5-oxocyclohex-3-en-1-yl}acetate) (Fig. 1) of molecular formula C27H32O7 and a molar mass of 468,54 g/mol. It is a molecule with anti-inflammatory and analgesic properties, as well as anti-plasmodial activity [2, 3].The crystal structure of andirobin [4] with the labelling of the atoms is shown in Fig. 1.This compound crystallizes in the space group P212121 and the lattice dimensions of its orthorhombic structure are as follow: a = 88,125 Å b = 12,590 Å, c = 21,939 Å. The Nuclear magnetic resonance (NMR) of this compound has also been reported [4, 5]. Vibrational spectroscopy and nuclear magnetic resonance (NMR) studies of active substances extracted from medicinal plants are essential to provide detailed information on their structural and vibrational properties [6,7,8,9]. However, to our knowledge, studies on this compound using Hartree Fock and DFT methods have not yet been reported in literature.

The crystal structure of andirobin
In this study, have we reported the structural, electronic, thermodynamic and nonlinear optical properties of andirobin to highlight other fields of application of this organic molecule.The paper is organized as follows. In Sect. 2, we describe the computational details and the theoretical framework related to this study. The results and discussions are presented in Sect. 3. The last section is the conclusion.
2 Computational details and theoretical framework
2.1 Computational details
Quantum chemical calculations were performed on andirobin using HF and DFT methods at the B3LYP and wB97XD level with the 6–311G (d,p) basis set as implemented in Gaussian 09 W software [10]. This was done in orderto predict the energies, vibrational frequencies and quantum chemical properties of the studied molecular system. No geometric restriction was applied during the optimization procedure.
2.2 Theoretical framework
The calculated Raman activities (\(S_{i}\)) were converted into relative Raman intensities \((I_{i}\)) using the relation given as follow [11]:
where f is a normalization factor for all peak intensities, \(\nu_{0}\) represents the wavenumber of the exciting laser, \(\nu_{i}\) is the wave number of the i vibrational mode, c stands for the speed of light, h and \(k_{B}\) are the Planck and Boltzmann constants, and T represents the temperature.
The HOMO and LUMO energies were used to determine the chemical potential, electronegativity, electrophilicity index, maximum charge transfer and chemical hardness, which are known as very important global reactivity descriptors and are calculated as follows [12]:
Chemical potential
Chemical hardness
Electronegativity
Electrophilicity index
Maximum charge transfer
The optimized form of our molecule obtained using the above parameter was then used to determine NLOs properties and also by applying the formulas below which can also be found in the literature [13,14,15].
3 Results and discussions
3.1 Optimized geometric structure
The optimized structure of our molecule with the adopted atomic labelling scheme is shown in Fig. 2. The total electronic energies obtained respectively using RHF, B3LYP and wB97XD with 6–311 G(d, p) are \({\text{E}}_{{{\text{RHF}}}}\) = − 1565.38830231 hartree,\({\text{ E}}_{{{\text{B}}3{\text{LYP}}}}\) = − 1575.019289 hartree and \({\text{E}}_{{{\text{WB}}97{\text{XD}}}}\) = − 1574.56299017 hartree. Furthermore, no imaginary frequency was observed after optimization of our system. This led us to the conclusion that the optimized molecular systems are stable at all the levels and basis set used. From the above result we can conclude that the andirobin molecule is more stable with the functional B3LYP.

The optimized structure of the andirobin molecule
3.2 Structural properties
The optimized structural parameters such as bond lengths and bond angles of title molecule obtained using RHF, B3LYP and wB97XD level with the 6–311 G (d, p) basis set are given in the Table 1. The general analysis of all bond lengths and angles in comparison with the available experimental data is shown in Figs. 3 and 4 respectively. From Table 1, it can be observed that the calculated bond lengths and angles obtained at the level and at the basis set mentioned above are very close to the experimental values [4].We can also notice that these values are slightly higher than their corresponding experimental values. Furthermore, we observed that the values of bond lengths obtained at B3LYP are slightly greater than those obtained at the RHF and wB97XD level. These differences are found between 0.0044 Å and 0.0086 Å for C–O, 0.0009 Å and 0.0169 Å for C–C and 0.1154 Å and 0.1466 Å for C–H. The bond angles of the andirobin molecule are slightly different when we move from one method to another. It can be seen that the C–C–C bond angle varies from 105.1554° to 128.265°, from 105.2159° to 128.6359° and from 105.475° to 128.0052° respectively at the B3LYP, RHF and WB97XD level of theory. It can be seen in Fig. 3 that the curves for the C–O and C–C bonds coincide with each other, whereas a shift is observed for the curves of the C–H bond. This is due to the fact that the bond lengths obtained by simulation are slightly greater than those obtained experimentally. Indeed, the tuning factor \(\left( {a_{cc} = \frac{{Val_{exp} - Val_{th} }}{{Val_{exp} }} \times 100} \right)\) is between [− 15.7; 0.98]. In Fig. 4, there is a strong coincidence of the curves for the angles C–C–H, C–C–C, C–O–C and O–C–C. On the other hand, the curves obtained for the angles H–C–H, O–C–H and O–C–O are offset from each other. This can be explained by the fact that most of the experimental values are slightly higher than those obtained by simulations. The tuning factor is included in the ranges [0.25, 3.04] for H–C–H, [0.31, 7.28] for O–C–H and [− 1.98, 0.85] for O–C–O.

Variation of the bond lengths calculated by B3LYP, wB97XD and RHF. The experimental data [4] are also illustrated for comparison

Variation of the angles calculated by B3LYP, wB97XD and RHF. The experimental data [4] are also illustrated for comparison
Experimentally, andirobin was found to crystallize in an orthorhombic structure. After optimization of our molecular structure, we found that the bond length and bond angles are closed to the experimental results. We can therefore conclude that the optimized geometrical structure of andirobin is close to the experimental results. In this case, the lattice parameters vary slightly with optimization, and therefore, the optimized molecular structure will crystallize in the orthorhombic structure.
3.3 Vibrational frequencies and assignments
Vibrational frequency analysis is a useful and important method used to estimate different movements of atoms and groups of atoms present in a molecular structure. The vibrational frequencies were calculated in this work from the optimized geometry of the title compound using RHF, B3LYP and WB97XD level with 6–311G (d,p) basis set. The experimental and computed vibrational frequencies, IR and Raman Intensities and their probable assignments with PED are presented in Table 2. The experimental and simulated vibrational spectra of andirobin are shown in Fig. 5.

Experimental data [9] and calculated infrared spectra of andirobin with B3LYP, wB97XD and RHF methods
The FT–IR absorption spectra (experimental Fig. 5) of andirobin [9] samples showed about eight intense absorbance bands ranging from 4000 to 500 cm−1. The result showed the stretching vibration at 1738.7 cm−1 and 1667.8 cm−1 attributed to C=O and C=C, respectively. On the other hand, the bending vibration at 1244.5 cm−1is a characteristic of the C–O bonds present in the andirobin molecule. Furthermore, for the stretching vibration bands, we observed values of 2978.2 cm−1, 3127.5 and 3450.0 cm−1characteristic of chromophores Csp3-H, Csp2-H and O–H. While the zone corresponding to bending vibrations showed bands of mean intensity at 1030.5 cm−1, 821.2 cm−1 and 611.4 cm−1attributable to the chromophores CO (primary alcohol), C=C (vinylidene) and C=C (disustituated alkene).
We also observed that the calculated harmonic vibrational frequencies are bigger than those obtained experimentally. Similar results have also been found byothers co-workers [16, 17]. However, we observe that the IR curves of the RHF, B3LYP and wB97XDmethods with the 6–311 G(d,p) basis set are slightly offset from each other. This difference is mainly due to the fact that the RHF does not take into account the electronic correlation unlike the B3LYP and wB97XD.
3.4 Vibrational study of functional groups
3.4.1 C–H Vibration
In literature, it is known that C–H stretching vibrations in alkanes and aromatic structures are located in the regions of 2850–3000 cm−1 and 3000–3100 cm−1, respectively [18]. From Fig. 5, the FT–IR bands for C–H stretching vibrations are observed at 2978 cm−1. This value is in agreement with experimental data in the literature [19, 20]. Using the Hartree–Fock and DFT methods, we observed CH stretching vibrations at 3050.51–2912.7 cm−1,while the in-plane and out-of -plane bending vibrations involving CH bonding were observed between 1049 and 988 cm−1 and 775–593 cm−1.
3.4.2 C=O Vibration
The C=O bond in the ester gives rise to characteristic bands in the IR spectrum covering the spectral range from 1730 to 1750 cm−1 [20]. In the andirobin molecule, we observe a C=O stretching vibration in the frequency range 1838–2021 cm−1 in good agreement with the experimental data. In-plane and out-of-plane bending vibrations are observed in the frequency ranges 1545–533 cm−1and 1463–859 cm−1 for infrared spectra.
3.5 NMR Study
The GIAO (Gauge Invariant Atomic Orbitals) method [21,22,23] was used to predict the 1H and 13C chemical shifts at the RHF, B3LYP and wB97XD levels with the 6–311 G (d, p) basis set. The TMS (Tetramethylsilane) was used to convert chemical shielding to chemical shift. The following equation was used for this purpose:
where i is the atom type. The experimental and calculated values of 13C and 1H chemical shift are presented in Tables 3 and 4. The positions of the atoms were numbered as in Fig. 1. From Table 3, it can be observed that RHF provides results of 13Cchemical shift, which are very close to the experimental values than those obtained using B3LYP and wB97XD with the exception of the carbon atoms directly linked to the oxygen atoms. The same observation is made for the 1H chemical shift as shown in Table 4. For more clarity, we show the experimental 13C NMR and 1H NMR spectra of andirobin obtained by Sidjui et al. [9] and represented respectively in Figs. 6 and 7. The experiment and calculations were carried out in a chloroform solvent. The comparison between the experimental and theoretical calculations is shown in Figs. 1 and 2 for the 13C and 1H NMR respectively.

Experimental data 13C NMR spectrum of andirobin (CDCl3) [9]

Experimental data1H NMR spectrum of andirobin (CDCl3) [9]
The following figures show the comparison between the experimental [9] and calculated values of 13C and 1H chemical shift. The regression line was plotted for each method using the equation \( \delta_{cal} = a\delta_{exp} + b\), where a and b are given in Figs. 8 and 9.The values of r2 show that our results are close to experimental results. The linear correlation coefficient calculated as R-square found in Figs. 8 and 9 also confirms this.

Comparison of experimental [9] and theoretical 13C chemical shifts of andirobin calculated at the RHF, B3LYP and wB97XD levels with 6–311 G(d,p) basis set

Comparison of experimental [9] and theoretical 1H chemical shifts of andirobin calculated at the RHF, B3LYP and wB97XD levels with 6–311 G(d,p) basis set
3.6 Electronic properties
3.6.1 Frontier molecular orbitals
The molecular mechanism of the reactivity of the molecule can be described by the frontier molecular orbitals; these are two particular types of molecular orbitals: HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) [24]. The HOMO–LUMO energy gap and the global reactivity descriptors of the molecule are given in Table 5. The values of these parameters were determined using B3LYP, wB97XD and RHF methods with the 6–311 G(d,p) basis set. Figure 10 shows the atomic compositions of the frontier molecular orbitals. In this Fig. 10, the red color stands for surfaces of high electron density and the green color stands for surfaces of low electron density. From the data obtained using the three methods, we find that the HOMO binds to the furan group while the LUMO is located on a ring comprising the C=O and C=C bonds which means that the electron transfer would take place from the furan group to the C=O and C=C groups. The energy gap of the andirobin (5.06 eV) obtained by B3LYP is lower than that obtained using RHF (11.89 eV) and wB97XD (9.11 eV) methods with 6–311 G(d, p) basis set. In addition, from Table 5, we observe that η decreases from the RHF method to the wB97XD and B3LYP method which means that the molecule is more reactive with the B3LYP. This can also be confirmed by a decrease in the energy gap and an increase in softness as well as by the value of the electrophilicity index which increases. The gap being greater than 4 eV, it can be concluded that andirobin can be used as an insulator for the design of electronic devices.

HOMO and LUMO orbitals of andirobin for B3LYP, wB97XD and RHF methods
3.6.2 Atomic charge analysis of andirobin
The atomic charge analysis plays an important role in the study of molecular systems because atomic charges have an effect on dipole moment, molecular polarizability, electronic structure and many other molecular properties of the system [24]. It also makes it possible to predict the types of bonds that may [25] be formed.
The natural atomic charge values calculated from the Mulliken population analysis of andirobin using the DFT and HF methods are illustrated in Fig. 11.

Analysis of the atomic charge of andirobin
We notice that all the hydrogen and oxygen atoms have a positive charge and a negative charge, respectively. On the other hand, the carbon atoms do not all have the same sign charge; this is due to the presence of C–H and C–O bonds. The presence of these different signs of the charge makes it possible the formation of intramolecular and intermolecular interactions through the C–H, C=C, C=O and O–H bonds which are the different functional groups of the molecule.
3.7 Thermodynamic properties
Thermodynamics parameters of andirobin were calculated using B3LYP, wB97XD and RHF methods with 6–311 G(d,p) basis set and are listed in Table 6. The total energy of a molecule is the sum of the translational, rotational, vibrational and electronic energies:
The difference between the values calculated by two methods is marginal. Relationships between partition functions and various thermodynamic functions have been used to evaluate the partition function due to the degrees of freedom of translation, rotation and vibration of the movements of the molecule [25]. The thermodynamic parameters such as energy (E), Molar heat capacity at constant volume \( (C_{v}\)) and entropy (S) were calculated for andirobin at room temperature using RHF, B3LYP and wB97XD methods. The calculated values can be found in Table 6. We observed from this table that the total energy (E) of the molecule decreases slightly when we move from the RHF to wB97XD and B3LYP.We also found that the heat capacity at constant volume \((C_{v}\)) and the entropy (S) are higher at the B3LYP/6-311G (d,p) level. This can be explained by the fact that the vibrational part of these parameters are higher with B3LYP.To our knowledge, no thermodynamic property had yet been studied on the andirobin molecule to compare our results.
3.8 Non-linear optical properties
Materials with appropriate nonlinear responses to incident light can be exploited to modify propagation characteristics such as the frequency, amplitude or phase of transmitted electromagnetic radiation. These materials make it possible to adapt the nature of light to particular applications [26, 27]. Currently, many studies are being done on the determination of the non-linear optical properties of organic molecules. Indeed, organ systems generally have excellent electrical response properties such as hyperpolarizability and polarizability at the microscopic level. The large values of the non-linear responses may be due to the very rapid response or relaxation time of the material, as well as to the movement of π electrons in electronic clouds between the donor and acceptor groups. In this context, some parameters of nonlinear optical properties (NLO) such as dipole moment (µ), mean polarizability (\(\alpha_{0}\)), anisotropy (∆α), first order hyperpolarizability (\( \beta_{0}\)) were calculated in this work with the RHF, B3LYP and wB97XD level using 6–311 G(d,p)basis set by applying the relations (7–10) mentioned above. Table 7 shows these values.We observe that \( \alpha_{0}\), ∆α and \( \beta_{0} \) increase when we move from RHF to wB97XD and from wB97XD to B3LYP. It is also noted that these values increase as the energy gap decreases. We can therefore conclude that the increase of these values (\(\alpha_{0}\), ∆α and \( \beta_{0}\)) improves the reactivity of our system. This can be explained by the strong dynamics of charge carriers within these structures. The large \(\alpha_{0}\) values of andirobin with B3LYP suggest that the interaction of light or other electromagnetic fields polarizes the charge distribution and modifies the propagated field. The calculated values of polarizability (\(\alpha_{0} )\) and first static hyperpolarizability (\( \beta_{0} )\) obtained from Gaussian output are in atomic units. These values were then converted into electrostatic unit (esu) for comparison (for \(\alpha_{0}\): 1 a.u = \(0.1482 \times 10^{ - 24}\) esu, for \( \beta_{0}\): 1 a.u = 8.6393 \(\times 10^{ - 33} esu\)) [17]. For a given molecule, when these values (µ and β) are greater than those of urea (\(\mu\) = 3.8851Dand \( \beta_{0}\) = 372.8 × 10−33esu) [17, 28, 29], the molecule has good active NLO properties. Similarly when we compare the values of µ and \( \beta_{0}\) of andirobin with the experimental values of urea, our values are much higher than those of urea. We can conclude that, in addition to its insulating character, the andirobin molecule can also have applications in the field of nonlinear optics, especially in the development of many technologies such as optical information processing, telecommunications, integrated optics, optical computers and laser technology [14, 30,31,32].
4 Conclusion
This study has allowed us to characterize the structural, nonlinear optical, electronic and thermodynamic properties of andirobin. We have found that with regard to the structural properties, the parameters of the optimized geometrical structure (bond lengths and bond angles) obtained by numerical computations correspond correctly to those found experimentally. A good agreement with the experiment was also found for 1H NMR and 13C NMR. Concerning the electronic properties obtained, andirobin is an insulator because its energy gap is greater than 4 eV. Moreover, the nonlinear optical properties reveal that andirobin is a polar molecule (μ ≠ 0). Similarly, the value of the first order hyperpolarizability is higher than that of urea, which allowed us to say that andirobin is a molecule that can be used for nonlinear optical applications. Therefore, our results obtained on the andirobin molecule indicate that this compound is a potential candidate for the emergence of the technology in many devices.
References
Pope M, Swenberg CE (1984) Electronic processes in organic solids. Ann Rev Phys Chem 35:613–655
Miyake T, Ishimoto S, Ishimatsu N, Higuchi K, Minoura K, Kikuchi T, Tanaka R (2015) Carapanolides T-X from Carapa guianensis (Andiroba) seeds. Molecules 20:20955–20966. https://doi.org/10.3390/molecules201119737
Inoue T, Ohmori S, Kikuchi T, Yamada T, Tanaka R (2018) Carapanosins D-F from the seeds of andiroba (Carapa guianensis, Meliaceae) and their effects on LPS-activated NO production. Molecules 23(7):1778. https://doi.org/10.3390/molecules23071778
Jittaniyom C, Sommit D, Muangsin N, Pudhom K (2012) Andirobin from X moluccensis. Acta Cryst E68:o2550–o2551. https://doi.org/10.1107/S1600536812027705
Ollis WD, Ward AD, De Oliveira MH, Zelnik R (1970) Andirobin. Tetrahedron 26(7):1637–1645. https://doi.org/10.1016/S0040-4020(01)93014-5
De Toledo TA, da Silva LE, Botelho TC, Ramos RJ, de Souza PT, Teixeira AMR, Freire PTC, Bento RRF (2012) Characterization of flavonoid 3-Methoxyquercetin performed by FT-IR and FT-Raman spectroscopies and DFT calculations. J Mol Struct. https://doi.org/10.1016/j.molstruc.2012.06.058
Melo IRS, Teixeira AMR, Sena DM, Santos HS, Albuquerque M, Bandeira PN, Rodrigues AS, BrazFilho R, Gusmao GOM, Silva JH, Faria JLB, Bento RRF (2014) FT-Raman and FTIR-ATR spectroscopies and DFT calculations of triterpene acetyl aleuritolic acid. J Mol Struct 1058:221–227. https://doi.org/10.1016/j.molstruc.2013.11.023
Cheng Y-B, Chien Y-T, Lee J-C, Tseng C-K, Wang H-C, Lo I-W, Wu Y-H, Wang S-Y, Wu Y-C, Chang F-R (2014) Limonoids from the Seeds of Swietenia macrophylla with inhibitory activity against dengue virus 2. J Nat Prod 77:112367–112374. https://doi.org/10.1021/np5002829
Sidjui LS, Nganso YOD, Toghueo RMK, Brussine NK, Wakeu BNK, Joel T, Dameue JT, Mkounga P, Adhikari A, Lateef M, Folefoc GN, Muhammad SA (2018) Kostchyienones A and B, new antiplasmodial and cytotoxicity of limonoids from the roots of Pseudocedrela kotschyi (Schweinf.) Harms Z. Natur forsch. 73(3–4):153–160. https://doi.org/10.1515/znc-2017-0102
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA Nakatsuji H, Carito M, Li X, Hratchian HP, Izmaylov AF, Bloino J, zheng G (2009) Gaussian 09, Revision A. 02. Gaussian, Inc., Wallingford CT
Fonkem C, Ejuh GW, Tchangnwa NF, Yossa KR, Ndjaka JMB (2020) Theoretical study of optoelectronic properties of the molecule 2-cyano-3-[4-(diphenylamino) phenyl] acrylic acid. J Iran Chem Soc 17:533–543. https://doi.org/10.1007/s13738-019-01790-4
Hossian MI, Kumer A, Begum SH (2018) Synthesis and characterization of ammonium benzoate and its derivative based ionic liquids and their antimicrobial StudiesAsian. J Phys Chem Sci 3:1–9. https://doi.org/10.9734/AJOPACS/2018/39148
Tchangnwa NF, Ejuh GW, Ndjaka JMB (2017) Theoretical study of optoelectronic and thermodynamic properties of molecule 4-[2-(2-N, N-dihydroxy amino thiophene)vinyl]benzanamine: Influence of hydroxyl position. Mater Lett 202:89–95. https://doi.org/10.1016/j.matlet.2017.05.064
Mveme CDD, Tchangnwa NF, Ejuh GW, Yossa Kamsi RA, Ndjaka JMB (2020) Frequency down-conversion of multiline CO laser into the THz range with ZnGeP2 crystal. Opt Quantum Electron 52:1
Fankam JBF, Ejuh GW, Tchangnwa Nya F, Ndjaka JMB (2020) Study of electronic structure, optoelectronics, linear and nonlinear optical properties and chemical descriptors of dibromodinitrofluorescein isomers in gas phase and solvent media using Ab Initio and DFT Methods. Chin J Phy 66:461–473. https://doi.org/10.1016/j.cjph.2020.05.015
Yossa Kamsi RA, Ejuh GW, Mkounga P, Ndjaka JMB (2020) Study of the molecular structure, electronic and chemical properties of Rubescin D molecule. Chin J Phys 63:104–121. https://doi.org/10.1016/j.cjph.2019.10.016
Yossa Kamsi RA, Ejuh GW, Tchoffo F, Mkounga P, Ndjaka JMB (2019) Electronic structure, spectroscopic (IR, Raman, UV Vis, NMR), Optoelectronic, and NLO properties investigations of Rubescin E (C31H36O7) molecule in gas phase and chloroform solution using Ab Initio and DFT Methods. Adv Cond Matt Phys. 2019:1–22. https://doi.org/10.1155/2019/4246810
Jeyavijayan S (2015) Molecular structure, vibrational spectra, NBO analysis, first hyperpolarizability, and HOMO–LUMO studies of 2-amino-4- hydroxypyrimidine by density functional method. J Mol Struct 1085:137–146
Zahra A, Mehdi DE, Esmail V, Manzarbanou A, Saeideh Y, Ali K (2017) A structural study of fentanyl by DFT calculations, NMR and IR spectroscopy. J Mol Struct 1128:552
Rastogi VK, Palafox MA, Tanwar RP, Mittal L (2002) Spectrochim. Acta A. 58:403
Silverstein RM, Basseler GC, Morill TC (1962) Spectrometric identification of organic compounds. J Chem Edu 39:546
Bühl M, Kaupp M, Malkina OL, Malkin VG (1999J) The DFT route to NMR chemical shifts. Comput Chem 20:91
Perera SA, Nooijen M, Bartlett RJ (1996) Electron correlation effects on the theoretical calculation of nuclear magnetic resonance spin–spin coupling constants. J Chem Phys 104:3290
Karabacak M, Kurt M, Cinar M, Coruh A (2009) Experimental (UV, NMR, IR and Raman) and theoretical spectroscopic properties of 2-chloro-6-methylaniline. Mol Phys 107:253
Scrocco E, Tomasi J, Lowdin P (1978) Advances in quantum chemistry. Acad Press 11:115
Chemla DS, Zyss J (1987) Nonlinear Optical Properties of Organic Molecules and Crystals. Academic Press, 1 Chap. 2, 18. ISBN 9780121706111, 9780323148153
Tasal E, Kumalar M (2012) Ab initio Hartree–Fock and density functional theory investigations on the conformational stability, molecular structure and vibrational spectra of 5-chloro-3-(2-(4-methylpiperazin-1-yl)-2-oxoethyl)benzo[d]thiazol-2(3H)-one drug molecule. Spectrochim Acta A Mol Biomol Spectrosc 95:282. https://doi.org/10.1016/j.saa.2012.04.081
Soscún H, Castellano O, Bermúdez Y, Toro-Mendoza C, Marcano A (2002) Linear and nonlinear optical properties of pyridine N-oxide molecule. J Mol Struct 592:19
Thanthiriwatte KS, de Silva KN (2002) Non-linear optical properties of novel fluorenyl derivatives:ab initio quantum chemical calculations. J Mol Struct 617:169
Karakas A, El Kouari Y, Migalska-Zalas A, Sahraoui B (2012) Photon Lett Pol 4(1):17–19
Ortyl E, Chan SW, Nunzi JM, Kucharski S (2006) Second harmonic generation by all-optical poling and its relaxation in the polymer films containing azo sulfonamide chromophores. Opt Mater 29:268–272
Andraud C, Brotin T, Garcia PF, Goldner P, Bigot B, Collet A (1994) Theoretical and experimental investigations of the nonlinear optical properties of vanillin, polyenovanillin, and bisvanillin derivatives. J Am Chem Soc 116:2094–2102
Acknowledgements
We are thankful to the Council of Scientific and Industrial Research (CSIR), India for financial support through Emeritus Professor scheme (Grant No. 21(0582)/03/EMR-II) to Prof. A.N. Singh of the Physics Department, Bahamas Hindu University, India which enabled him to purchase the Gaussian Software. We are most grateful to Emeritus Prof. A.N. Singh for donating this software to Prof Geh Wilson Ejuh, University of Dschang, IUT-FV Bandjoun, Cameroon.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abe, M.T.O., Nzia, C.L., Sidjui, L.S. et al. Predictive calculation of structural, nonlinear optical, electronic and thermodynamic properties of andirobin molecule from ab initio and DFT methods. SN Appl. Sci. 3, 768 (2021). https://doi.org/10.1007/s42452-021-04749-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-021-04749-4