
Abstract
Single particle tomography (SPT), also known as subtomogram averaging, is a powerful technique uniquely poised to address questions in structural biology that are not amenable to more traditional approaches like X-ray crystallography, nuclear magnetic resonance, and conventional cryoEM single particle analysis. Owing to its potential for in situ structural biology at subnanometer resolution, SPT has been gaining enormous momentum in the last five years and is becoming a prominent, widely used technique. This method can be applied to unambiguously determine the structures of macromolecular complexes that exhibit compositional and conformational heterogeneity, both in vitro and in situ. Here we review the development of SPT, highlighting its applications and identifying areas of ongoing development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION: THE NEED FOR SINGLE PARTICLE TOMOGRAPHY
Over the last five years, thanks to the development of direct detection devices (DDDs) (Milazzo et al. 2005), cryo-electron microscopy (cryoEM) single particle analysis (SPA) has transitioned from being an established, but limited, technique to being at the forefront of structural biology (Eisenstein 2016; Nogales 2016). SPA can now achieve resolutions comparable to those of typical X-ray crystal structures while maintaining the specimen in a solution-like environment, thereby avoiding dehydration and crystallization artifacts. While a very powerful technique, SPA still suffers from two primary limitations: first, it is sometimes unable to unambiguously resolve reliable structures of macromolecules exhibiting continuous conformational flexibility; second, it cannot be directly used to study macromolecules within cells or other unique structures in situ.
Single particle tomography (SPT), also known as subtomogram averaging (STA), offers a solution to both of these problems. Indeed, with per particle 3D data, it is easier to unambiguously discriminate between changes in particle orientation versus changes in particle conformation, addressing the first issue above. Furthermore, the most impactful application of SPT lies in the cellular milieu. Since tomograms are a 3D representation of the imaged specimen, with SPT it is possible to isolate individual macromolecules from a cellular tomogram. These individual “subtomograms” can then be subjected to SPA-like 3D alignment and averaging, making true in situ structural biology at nanometer resolution feasible.
The difficulty faced by SPA when studying particles undergoing large-scale continuous conformational change is the ambiguity produced by making projections of 3D objects. As conceptually outlined in Fig. 1, extremely different 3D structures can theoretically yield one or more indistinguishable projections, particularly given the high noise levels present in typical CryoEM images, both in vitro and in situ. With continuous conformational variability, it can thus be mathematically impossible to unambiguously distinguish changes in particle orientation from changes in particle conformation with only a single 2D image for each particle. These limiting factors, namely high levels of noise and conformational variability, are exacerbated for macromolecules in situ.

2D versus 3D imaging for particle classification. A simple conceptual demonstration that conformationally different particles (A and B) can yield multiple projections that are extremely similar (even if orthogonal), as shown by projections 1 and 2, particularly given the high levels of noise typical of cryoEM micrographs of either in vitro or in situ specimens. To push structures to high resolution, it is critical to distinguish between projections of structurally different molecules that might otherwise be erroneously classified together and aligned based on overall low-resolution feature similarity. Collecting more than two images, as done in tomography, possibly from a different axis (as shown by projection 3 in the figure), can help to distinguish conformational differences among particles
For macromolecules in vitro, the “tilt validation” method (Henderson et al. 2011, 2012) partially addresses the issue of confounding orientation and conformation by imaging isolated particles from two different directions to assess the reliability of orientation assignment in SPA reconstructions. However, this is a validation method and not a tool for initial analysis. The random conical tilt (RCT) (Radermacher et al. 1986) and orthogonal tilt reconstruction (OTR) (Leschziner and Nogales 2006) methods make use of this concept to reconstruct challenging structures, but have a number of other limitations. Furthermore, all these methods image the specimen from only two angles about the same axis and, again, are only applicable to isolated complexes.
In addition to allowing for the computational isolation of macromolecular complexes from cells, single particle tomography (SPT; also known as subtomogram averaging, or STA), can be viewed as an extension of the tilting concept of RTC and OTR by collecting multiple tilted views of single particles. This is literally “tomography of single particles,” as the inherent goal is to produce a tomographic 3D view for each individual particle in a system, be it in vitro or in situ. The particles are then processed through a pipeline akin to that of SPA cryoEM. That is, the tomographic single particles are (in simplified terms) aligned, classified by composition and/or conformation, and averaged as part of a standard pipeline that is applicable to both particles in vitro and in situ.
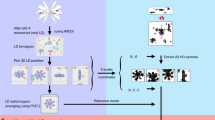
Although SPT has opened the window to increasing the resolution of structural biology in situ by averaging repeating features in cellular tomograms, this method also suffers from significant limitations. SPT builds on cryo-electron tomography (cryoET), which historically has been considered a low-resolution technique. In cryoET, a set of images (i.e., a “tiltseries”) is collected for each specimen area by tilting the specimen stage through a range of angles, usually about a single axis (e.g., ±60° in increments of 1°–5° or more). Very high cumulative electron dose (~50–120 e/A2) has been the norm to obtain sufficient contrast in each image of a tiltseries to permit accurate alignment, with the side effect of destroying high-resolution information progressively through the series. Each tiltseries can then be computationally reconstructed into a 3D tomogram representing the 3D structure of the imaged area (Fig. 2). The resolution of raw tomograms is highly anisotropic and remains somewhat ill-defined (Cardone et al. 2005), but is generally estimated to range from roughly 50–150 Å (depending on the specimen and on data collection parameters). While this is sufficient to resolve cellular organelles and identify large macromolecular complexes, it is not sufficient to resolve macromolecular structure in detail. Nonetheless, each individual subtomogram does contain some high-resolution information, which, upon averaging with other ostensibly identical particles, can be recovered to yield structures at much higher resolution, depending on imaging conditions.

CryoET schematic. A In cryoET, the ice-embedded specimen, typically shaped as a slab, is tilted through a wide range of angles in the electron microscope and an image is recorded at each angle. B This collection of images around a common axis constitutes a “tiltseries.” C The images in a tiltseries can be computationally aligned to their common axis and reconstructed into a 3D tomogram by weighted back-projection or other methods. D Subtomograms representing a 3D view of individual macromolecules can be extracted from the reconstructed tomogram, then aligned and averaged (Fig. 7). (Partially inspired by Grünewald et al. 2002)
The recent development of direct electron detectors, phase plate technology, and improved contrast transfer function (CTF) correction methodologies for cryoET have made it possible to achieve images with higher contrast and resolution, respectively, using much lower dose. Additionally, when averaging subtomograms, fewer images may be collected for each tiltseries, permitting the use of higher dose per image without impacting total dose. Indeed, these improvements have led to several recent cryoSPT studies approaching (Cassidy et al. 2015; Galaz-Montoya et al. 2016a; Khoshouei et al. 2016; Kudryashev et al. 2016; Li et al. 2016) or achieving (Bharat et al. 2015; Pfeffer et al. 2015a, b; Schur et al. 2013, 2015a, b; Mattei et al. 2016) subnanometer resolution, with the highest-resolution structure being solved to ~4 Å (Schur et al. 2016). Simplistic CTF correction (not accounting for defocus gradients) of cellular data has also demonstrated measurable improvements through SPT of microtubules in situ (Grange et al. 2017). Some of the most impressive results in cryoSPT during the last few years are shown in Fig. 3. Perhaps the greatest remaining limiting factor for in situ experiments is specimen thickness, which limits electron penetration, making it impractical to study specimens thicker than roughly 0.5–1.0 μm. The study of thicker eukaryotic cells requires significant physical manipulation, such as slicing the specimen into thin sections (Al-Amoudi et al. 2004).

Sampling of notorious cryoSPT studies. This figure is a sampling of cryoSPT structures published at different resolutions, prepared directly from the corresponding deposited (emdatabank.org) maps and models. A The structure of M-MPV CANC Gag (EMD-2488) was the first one solved to subnanometer resolution by cryoSPT. B Asano et al. undertook the study of proteasomes inside intact neurons, resolving multiple states for the 26S proteasome (EMD-2830 is shown), making use of a Volta phase plate (VPP). C Nuclear pore complexes (NPCs) are some of the most challenging specimens studied by cryoSPT due to their large size, extreme conformational flexibility, and the need for a lipid environment. Eibauer et al. solved the structure of the X. laevis NPC at unprecedented resolution for this specimen (EMD-3005). A comparable resolution was recently achieved for another nuclear pore complex (not shown; Kosinski et al. 2016). D A recent proof-of-concept study demonstrated that the VPP could be used in cryoSPT experiments to solve the structure of particles without any symmetry, such as the ribosome, to subnanometer resolution, using a relatively small number of particles (N = 1400; EMD-3418). E The highest-resolution structure by cryoSPT to date is that of the immature HIV-1 CA-SP1 lattice (EMD-4015), which allowed building an atomic model (PDB-5L93)
Continued development of computer technology has also played a critical role in improving the resolution achievable by cryoSPT. A single cellular tomogram (including those with repeating features that can be averaged) can exceed 64 GB in size at full resolution; therefore, the reconstruction of hundreds of tomograms is extremely computationally intensive, particularly if iterative reconstruction methods are used without downsampling. Per-particle computational costs (i.e., preprocessing, alignment, classification, and averaging of subtomograms) are several orders of magnitude greater for SPT than for SPA. However, Moore’s Law (Schaller 1997) has finally caught up with this field, and it is now practical to compute hundreds of tomographic reconstructions and average tens of thousands of subtomograms making use of algorithms that previously would have been untenable (Agulleiro et al. 2012).
HISTORY AND APPLICATIONS OF SINGLE PARTICLE CRYO-ELECTRON TOMOGRAPHY
The mathematical foundations underlying 3D image reconstruction date to 1917 when Johann Radon demonstrated that a function could be precisely reconstructed from an infinite set of its projections (Hawkes 2007). Since then, many mathematical techniques have been devised to reconstruct a 3D model from a set of 2D projection images, for medical imaging and a wide range of other applications, including transmission electron microscopy (TEM) tomography.
The theory underlying electron tomography (ET) and its application to study biological specimens (De Rosier and Klug 1968a, b; Hart 1968; Hoppe et al. 1968), including individual metal-stained macromolecules (Hoppe et al. 1974) and averages of a few subvolumes (Knauer et al. 1983; Oettl et al. 1983), were first demonstrated decades ago. However, there were many experimental and computational barriers to widespread adoption of ET at that time.
The development of cryo-electron microscopy (cryoEM) was a major breakthrough that demonstrated that biological specimens, including cells and “single particles,” could be better preserved in vitrified water solutions, free from crystallization and staining artifacts (Dubochet et al. 1981, 1988), in a close-to-native state. CryoEM was first applied to 2D protein crystals (Taylor and Glaeser 1974) and was demonstrated for isolated particles (viruses) a decade later (Adrian et al. 1984). It took many more years before Walz et al. completed the first cryoSPT experiments in 1997, in their study of thermosomes in vitro. They showed that the structure of this chaperonin could be determined without missing wedge artifacts by computing tomograms of the specimen in solution, extracting volumes (i.e., subtomograms) containing individual thermosomes, and averaging them after correct alignment. Since then, the publication rate of studies using SPT has been accelerating. Figure 4 shows the number of yearly structures solved by SPT and deposited in the Electron Microscopy Data Bank (EMDB) from 2004 to 2016, as well as the best resolution achieved in each of those years. A few structures that were not deposited to the EMDB were solved in 1997, 1998, and 2003, as noted in previous reviews (Schmid 2011; Kudryashev et al. 2012). The resolution averages presented in Fig. 4 exclude structures for which no resolution was reported (pink line). Of note, averages of at least two particles should always report an estimate of the resolution, however modest it might be. This will facilitate the interpretation of features in the structures being published and in the figures being displayed.

Increase in yearly structures deposited in the EMDB by SPT from 2004 to 2016 and resolution improvement trend. While the highest resolution achieved each year has continued to improve, the average resolution is improving only very gradually
Early SPT relied largely on undocumented ad hoc scripts (Winkler and Taylor 1999; Pascual-Montano et al. 2002; Förster et al. 2005; Nicastro et al. 2006; Schmid et al. 2006; Winkler 2007; Schmid and Booth 2008). However, the methodology has gradually become more accessible with the surge of open-access software for SPT, such as AV3 (Förster and Hegerl 2007), PEET (Nicastro et al. 2006) (available through IMOD (Kremer et al. 1996)), Jsubtomo (Huiskonen et al. 2010), PyTom (Hrabe et al. 2012), Dynamo (Castaño-Díez et al. 2012), EMAN2 (Galaz et al. 2012; Murray et al. 2014; Galaz-Montoya et al. 2015), and RELION (Bharat et al. 2015).
The evolution of cryoET can be followed through reviews in this discipline over the last couple of decades, (e.g., Koster et al. 1997; Baumeister 2002; Förster et al. 2005; Crowther 2010; Fernández 2012). CryoET can now be routinely applied to study macromolecules in solution (Medalia et al. 2002a) and in their native cellular context (Medalia et al. 2002b; Ortiz et al. 2006, 2010; Brandt et al. 2010; Schwartz et al. 2012). Challenging specimens whose structures have been solved by cryoSPT include complexes that exhibit extensive structural heterogeneity, such as carboxysomes (Schmid et al. 2006), dynein interacting with microtubules along axonemes (Nicastro et al. 2006), and pleomorphic viruses (Harris et al. 2006, 2013; Huiskonen et al. 2010; Schmid et al. 2012). CryoSPT has also been used to study viruses infecting their host cells (Hu et al. 2013; Peralta et al. 2013; Sun et al. 2014; Riedel et al. 2017; Murata et al. 2017), even in transient conformations along their assembly pathway inside cells (Dai et al. 2013). Much smaller complexes, such as the proteasome in different conformational states, have also been visualized in situ, inside neurons (Asano et al. 2015) (Fig. 3). Other complex systems whose structures have been best characterized using SPT are flagellae (Koyfman et al. 2011; Carbajal-González et al. 2013; Zhao et al. 2013), polysomes in situ (Brandt et al. 2010), membrane-bound ribosomes (Pfeffer et al. 2012, 2015a, b), nuclear pore complexes (Stoffler et al. 2003; Maimon et al. 2012; Eibauer et al. 2015; Kosinski et al. 2016), and other membrane-bound complexes (Davies et al. 2011; Eibauer et al. 2012; Dalm et al. 2015; Nans et al. 2015; Briegel et al. 2016; Sharp et al. 2016), as well as amyloid protein aggregates interacting with chaperones (Shahmoradian et al. 2013; Darrow et al. 2015), among others. A recent review (Asano et al. 2016) and a book chapter (Wan and Briggs 2016) describe in detail the technical aspects of carrying out SPT analyses.
CHALLENGES IN SINGLE PARTICLE TOMOGRAPHY
While cryoSPT is now being successfully applied to many biological problems that could not be addressed in near-native conditions a few years ago, it is also important to understand its current limitations and their underlying sources.
Radiation damage
A fundamental limitation in any cryoEM/ET study is the unavoidable fact that the specimen is being destroyed as it is being imaged. Thus, the permissible dose is limited to preserve detailed features (Glaeser 1971; Grubb 1974). This problem is exacerbated in tomography, since many tilt images of the same specimen area must be collected, and yet each tilt image must contain sufficient information for accurate tiltseries alignment to yield a high-fidelity reconstruction. How to optimally allocate the total cumulative dose among all the images of a tiltseries depending on the particular goals of a study (i.e., “dose fractionation”) has been a longstanding problem in cryoET (McEwen et al. 1995) and is still being actively researched (Hagen et al. 2017). A recent clever technique to turn the radiation sensitivity of biological specimens into an advantage is the concept of “bubblegrams” (Cheng et al. 2012; Wu et al. 2012) and “tomo-bubblegrams” (Fontana et al. 2015), in which the varying radiation sensitivity of different molecular species can be used to localize and identify substructures, while still preserving high-resolution detail in the early portion of the exposure. Aside from such unorthodox tricks, however, radiation damage remains the primary limiting factor in cryoEM and cryoET (Cosslett 1978; Glaeser and Taylor 1978; Baker and Rubinstein 2010). Indeed, radiation damage is a complex phenomenon that remains under active investigation, since it depends not only on the chemical composition of the specimen but also on multiple data collection parameters such as cumulative dose, imaging temperature (Comolli and Downing 2005; Iancu et al. 2006; Bammes et al. 2010), and dose rate (Chen et al. 2008; Karuppasamy et al. 2011), among others. The recent development of DDDs has reduced the impact of radiation damage by permitting the recovery of a larger fraction of information at lower cumulative doses owing to the detector’s higher detective quantum efficiency (DQE) and improved modulation transfer function (MTF) (Milazzo et al. 2010).
Dose fractionation, ice thickness, and beam-induced specimen motion
The SNR and contrast of an electron micrograph also depend on the thickness of the ice in which the specimen is embedded. In cryoET, the effective ice thickness scales with the secant of the tilt angle (Fig. 5). Images from tilt angles any higher than ±65° are often unusable and can decrease the quality of the tomographic reconstruction if included. The inability to collect a complete tomographic tiltseries (tilting through ±90°) causes the so-called “missing wedge” artifact. This term refers to the wedge-shaped region of Fourier space that is empty due to missing tilt images. This artifact produces anisotropic resolution in tomograms (Radermacher 1988) and poses one of the most critical problems for alignment and classification of subtomograms, as discussed further below.

The effective ice thickness of slab-shaped specimens increases with tilt angle. Even for relatively thin specimens (A), in cryoET the path of the electron beam through the slab-shaped specimen (green arrows, labeled d, d’, and d’’) increases dramatically with tilt angle (B, C). This limits the thickness t of cellular material that can be studied by cryoET and the maximum tilt angle at which usable data can be collected. Indeed, greater ice thickness degrades image quality and contrast as it increases charging, blurring, and multi-scattering events that cause images to be noisier
When the specimen is too thick for beam penetration, it can be sectioned with a diamond knife (Al-Amoudi et al. 2004) or milled thin with a focused ion beam (FIB) (Marko et al. 2007) prior to imaging. While this enables the study of thicker specimens by cryoET, cryo-sectioning is, unfortunately, an extremely challenging technique that yields sections laden with artifacts, from compression and curving to crevasses and cracks (Al-Amoudi et al. 2003, 2005). On the other hand, while FIB milling produces less severe artifacts, it destroys the bulk of the specimen, and it can be very difficult to focus the milling process on the region of interest. An alternative method to study thick frozen-hydrated specimens is X-ray tomography (Wang et al. 2000; Le Gros et al. 2005), albeit typically at resolutions an order of magnitude lower than cryoET.
The conductivity of holey and/or continuous amorphous carbon support films commonly used in cryoET is decreased at cryogenic temperatures, resulting in the accumulation of charge during imaging. Charging and beam-induced motion are exacerbated by specimen curvature (i.e., frozen menisci)(Chen et al. 2008) and ice thickness (Brink et al. 1998), and are therefore more prominent and more likely to occur in cryoET the higher the tilt angle (Galaz-Montoya et al. 2016a). Although DDDs permit collecting images as short movies, which can be corrected for beam-induced motion (Brilot et al. 2012), different particles in the imaged specimen may move in different directions (Campbell et al. 2012). Strategies have emerged to correct for this in SPA cryoEM, where individual particles can be motion corrected (Scheres 2014). However, extensive dose fractionation in cryoET with DDDs typically yields movie frames at each tilt angle with considerably lower contrast and higher noise than movie frames in SPA cryoEM. Aligning whole movie frames can produce micrographs that are only locally unblurred, sometimes blurring areas of the image that were not originally blurry. This can happen both in thick, cellular tomograms (Fig. 6), as well as in tomograms of isolated macromolecules (not shown). Single particles are not always readily detectable in the individual frames of movies in cryoET images. Furthermore, cryoET images often cannot be completely broken down into discrete particles because the specimen is continuous (e.g., cells). Therefore, local unblurring strategies that can correct extremely low-dose images anisotropically and without discontinuities are needed to maximize productive data usage in cryoET, as recently proposed for SPA cryoEM (Zheng et al. 2016). Given the extensive dose fractionation and greater radiation damage due to total cumulative dose, accomplishing this for cryoET is foreseeably more challenging than for SPA cryoEM. To a first approximation, gold fiducials might serve the purpose of guiding local motion correction for extremely low-dose cryoET movie frames, in addition to serving their normal role in tiltseries alignment.

Incomplete, local unblurring of cryoET images by whole-frame motion correction. Image of a mouse platelet at 57° tilt without (A) and with (B) motion correction applied to 21 frames collected with a K2 DDD on a JEM3200FSC microscope. Blurring is anisotropic both before (Ai and Aii) and after (Bi and Bii) whole-frame motion correction. While motion correction by iterative frame alignment (Galaz-Montoya et al. 2016b) improves the overall image (B), the extent of improvement varies in different parts of the image (Bii). Unexpectedly, a region that was not originally blurry (Ai) becomes blurry after motion correction (Bi) while a different region is effectively unblurred (Aii versus Bii). This suggests that different parts of the specimen are subject to divergent apparent motions, possibly due to charging effects and/or motion perpendicular to the imaging plane, and therefore local motion correction methods are needed for cryoET images, similar to those applied per particle in SPA cryoEM
Tiltseries alignment
Gold fiducials are commonly used as markers in cryoET so that the precise 3D orientation of each image in a tiltseries can be determined. Unfortunately, such gold markers have been shown to undergo independent beam-induced motions and therefore their positions are not strictly consistent and predictable across a tiltseries (Comolli and Downing 2005; Noble and Stagg 2015). This effect varies considerably among specimens and can be a limiting factor in reconstruction quality. Furthermore, gold fiducials cause extremely strong artifacts (“streaks”) that can obscure features in tomograms and interfere with subtomograms alignment. Strategies to regularize gold fiducial artifacts have been proposed (Song et al. 2012; Maiorca et al. 2014; Han et al. 2015), some of which are available in IMOD, but do not seem to be routinely applied. Alternatively, many fiducial-less alignment methods have been developed (Liu et al. 1995; Brandt et al. 2001; Renken and McEwen 2003; Castaño-Díez et al. 2007) but usually require that the specimen itself exhibits high-contrast features. These procedures are still prone to error since specimens can also undergo deformations with cumulative dose, and in most cases gold fiducials yield better tiltseries alignment. While subnanometer resolution can be achieved in cryoSPT with standard fiducial-based alignment (Schur et al. 2013), circumventing the known errors inherent in this methodology can improve resolution (Bartesaghi et al. 2012). Indeed, if made easily applicable, overcoming the limitations of fiducial-based alignment (Iwasaki et al. 2005; Bartesaghi et al. 2012; Zhang and Ren 2012) might facilitate achieving subtomogram averages beyond the 8–12 Å resolution range routinely for various types of specimens.
Tomographic reconstruction methodologies
A century ago, Johann Radon postulated that a function could be precisely reconstructed from an infinite set of its projections (Radon 1917). This concept was a stepping-stone in the development of computed tomography (CT), which has found applications in medical imaging, astronomy, and TEM (Van Heel 1987). Indeed, a variety of methods have been developed to reconstruct the 3D structure of biological specimens from a set of 2D TEM projection images in known orientations. Mathematically, all of these methods are based on the central section theorem (DeRosier and Klug 1968a, b; Crowther et al. 1970; Crowther 1971), according to which the Fourier transform (FT) of a 2D projection from a 3D specimen corresponds to a section through the center of the specimen’s representation in Fourier space. Thus, one can construct the 3D FT of the specimen by combining the FTs of all the 2D projection images inserted into a 3D FT volume, and computing the inverse Fourier transform of the volume (i.e., “direct Fourier inversion”) (Ludtke et al. 1999; Belnap 2015). Although this approach is conceptually simple, interpolation in Fourier space is non-trivial, and different strategies produce different artifacts. While Fourier inversion has become the standard approach in SPA (Penczek et al. 2004; Penczek 2010), it is not widely used in cryoET (Heymann and Belnap 2007).
The most popular reconstruction method for cryoET due to its speed and relatively easy implementation for large volumes is weighted back-projection (WBP), a real-space equivalent of the central section theorem (Gilbert 1972; Radermacher 2007). It consists of literally “projecting back” the densities of 2D projections as rays into a 3D reconstruction volume. Appropriate weighting of the back-projected densities is needed to avoid the implicit low-pass filtering effect of WBP. However, WBP yields reconstructions with very poor contrast and strong “streaking” artifacts compared to iterative algebraic reconstruction methods such as the simultaneous iterative reconstruction technique (SIRT) (Gilbert 1972) or algebraic reconstruction techniques (ART) (Marabini et al. 1998). On the other hand, iterative algebraic methods are typically much slower, can diverge for some datasets or sometimes destroy high-resolution information, and determining a priori the optimal number of iterations to use and other parameters is not generally possible.
The problem of tomographic reconstruction is still being actively researched, with many novel methods described over the last decade (Díez et al. 2007; Batenburg and Sijbers 2009; Wan et al. 2011; Kunz and Frangakis 2014; Turoňová et al. 2015; Zhou et al. 2015; Chen et al. 2016). A few methods have been proposed to recover some of the missing information in limited-angle tomography, for example, by using convex projections (Carazo and Carrascosa 1987). Recently, the reconstruction method proposed by Chen and Förster (2014) was shown to restore some of the missing information by iterative extrapolation. Total variation minimization (TMV, or “regularization”) based on compressed sensing (CS) (Donoho 2006) has been applied successfully to ET in material sciences, yielding structures with minimal missing wedge artifacts and improved contrast (Saghi et al. 2011; Goris et al. 2012; Leary et al. 2013). Variations of this method have also been demonstrated for cryoET specimens (Aganj et al. 2007; Song et al. 2012). Recently, the iterative compressed-sensing optimized non-uniform (ICON) reconstruction (Deng et al. 2016) method demonstrated that CS can restore missing information in noisy cryoET data of both cells and isolated macromolecules, thereby minimizing missing wedge artifacts and yielding measurably better reconstructions than WBP.
At present, no single optimal algorithm has emerged, and the vast majority of users adopt whichever algorithm is most conveniently available or recommended by the software they have selected for tiltseries alignment. Reconstruction methods need to be carefully chosen depending on the goals of the study in question and the data collection parameters. For example, the success of TMV has been reported to depend on the tilt scheme used during data collection, while SIRT is less sensitive to variations in total dose or tilt scheme (Chen et al. 2014). It is important to note that certain algorithms are optimized for direct interpretation of tomograms but can destroy high-resolution information and are therefore suboptimal for averaging subtomograms if achieving higher resolution is the end goal. For example, SIRT delivers tomograms with much better contrast than WBP at the expense of introducing artifacts at high resolution. Furthermore, some algorithms rely on the individual images in a tiltseries having high contrast, such as the filtered iterative reconstruction technique (FIRT) (Chen et al. 2016), which does not seem to provide any advantages over WBP when applied to cryoET data. Interestingly, it has also been proposed that several reconstruction techniques can be applied sequentially to guide the choice of optimal parameters. For example, an initial SIRT reconstruction can guide the selection of the regularization parameter for TMV reconstruction, which can in turn help to choose adequate gray values to run a final reconstruction with the discrete algebraic reconstruction technique (DART) (Goris et al. 2013).
Contrast transfer function determination and correction for tilted specimens
The contrast transfer function (CTF) (Erickson and Klug 1971) of the electron microscope modifies the amplitude of the signal in cryoEM micrographs (Toyoshima and Unwin 1988) in an oscillatory, resolution-dependent manner. While it is a function of several parameters, the only one that varies significantly during an imaging session is the defocus. In SPA cryoEM, CTF correction is a well-established, largely automated, and straightforward process, with various approaches achieving comparable results. Indeed, the “CTF challenge” recently compared many of the multiple algorithms available to perform CTF correction in SPA cryoEM (Marabini et al. 2015). On the other hand, as the defocus is directly related to the specimen height in the column with respect to the focal plane in the electron microscope, ET specimens produce a CTF that varies across the imaging plane due to the tilted geometry and, to a lesser extent, due to the thickness of the specimen. Compensating for these defocus gradients in images of cryoET tiltseries requires more complicated correction strategies than those implemented for SPA cryoEM. Without CTF correction, the resolution of subtomogram averages will typically be limited to 20–100 Å, depending on the imaging parameters. Resolving the structure of macromolecules by cryoSPT to better than 20 Å resolution is not yet a routine procedure, with the yearly average resolution being above this threshold (Fig. 4).
Several approaches have been implemented to determine the CTF and/or correct for it in cryoET data (Mindell and Grigorieff 2003; Winkler and Taylor 2003; Fernandez et al. 2006; Xiong et al. 2009; Zanetti et al. 2009; Voortman et al. 2011), with the most successful approach so far being that by (Schur et al. 2013). The latter approach achieved cryoSPT in vitro at subnanometer resolution for the first time, and has continued to yield structures at even higher resolution when combined with modern instrumentation, such as DDDs, and algorithmic improvements in image processing. The paramount achievement of Schur et al. 2013 using images taken with a charge-coupled device (CCD) was initially heavily dependent on stage eucentricity and stability during data collection and accurate autofocusing, as well as on a high particle density and automated data collection. Thin ice was also essential, as the specimen was assumed to be co-planar throughout the tomograms. So, while this was an effective proof of concept, these conditions might not be straightforward to achieve in a typical lab for all specimens. A year prior, a hybrid methodology combining concepts and data processing strategies from SPA cryoEM and cryoSPT resolved GroEL at subnanometer resolution as well (Bartesaghi et al. 2012), but this hybrid approach cannot be easily applied to cellular data. Another successful method (Eibauer et al. 2012) also recently achieved subnanometer resolution (Bharat et al. 2015), making use of a pair of additional high-contrast images collected away from the imaging area to interpolate the defocus in the region of interest. While this method was successful, it significantly increased data collection and processing complexity and assumed that the cryoEM support grid was flat (though not necessarily parallel to the imaging plane). Unfortunately, grid bending and “cryo-crinkling” of the carbon support mesh are common artifacts (Booy and Pawley 1993). Therefore, there is no guarantee that interpolation of the defocus at the imaging site by measuring the defocus in adjacent sites several microns away will always be accurate. Indeed, increased exposure near the imaging area due to lengthy focusing routines can induce deformations that compromise the accuracy of tiltseries alignment (Khoshouei et al. 2016).
Recently, a per-particle CTF correction method in 3D for cryoSPT was proposed (Galaz-Montoya et al. 2015) and demonstrated in vitro at near subnanometer resolution, without making any of the aforementioned assumptions (accurate defocusing during data collection, thin ice, unbent specimen, etc.), using only a few hundred icosahedrally symmetric virus particles (Galaz-Montoya et al. 2016a). Most importantly, the defocus gradient due to tilting was fitted by directly measuring the power spectrum in strips of constant defocus, similar to the method proposed by Fernández et al. (2006), except that it was done on each individual image (i.e., different images in a tiltseries were never combined and the defocus gradient was linearly fit on a per-image basis, instead of relying on a single value from the central region of the image to compute the gradient). Indeed, DDDs now allow measuring the defocus directly from each image in a tiltseries, even at high tilt, which is essential under experimental settings for which the actual defocus might significantly differ from the target defocus and vary widely across the images of a tiltseries.
Performing CTF correction in 3D for tomographic reconstructions, including per-particle corrections (Bharat et al. 2015), has further been demonstrated to yield improvements compared to corrections considering 2D information only (Kunz and Frangakis 2016), as first theoretically proposed for virus reconstructions in SPA cryoEM nearly two decades ago when the depth of field started to become a resolution-limiting factor (Jensen and Kornberg 2000). The method proposed by Jensen and Kornberg to compensate for the depth of field was later generalized mathematically (Kazantsev et al. 2010).
The missing wedge
The effective thickness of ET specimens increases with the secant of the tilt angle, meaning that at 60° the specimen is twice as thick than at 0°, and at 70° it is nearly three times as thick. This effect degrades image quality rapidly at higher tilt angles and, in most cases, ~60° is the highest tilt worth expending dose on. This means that typical ET tiltseries span only \(\sim{2}/{3}\) of complete tomographic angular sampling. The missing angular range is termed the “missing wedge,” and leads to a variety of 3D reconstruction artifacts (Radermacher 1988), where features or particles are distorted in different ways, depending on their orientation with respect to the missing wedge.
Data collection methods alternative to canonical single-axis ET have been proposed to reduce the deleterious effects of the missing wedge in cryoET, such as dual-axis tomography (Penczek et al. 1995; Mastronarde 1997; Tong et al. 2006; Xu et al. 2007), conical tilt (Lanzavecchia et al. 2005), and multiple-axis tomography (Messaoudi et al. 2006). While these techniques reduce the missing wedge (to a missing pyramid, a missing cone, or a smaller missing region in general depending on how many tiltseries around different axes are combined), complete coverage is still not achieved. Furthermore, dose fractionation becomes increasingly problematic when two or more tiltseries are collected from the same imaging area, compromising the ability to align the images in the tiltseries accurately due to an exceedingly low SNR in individual images. In a recent proof-of-concept study, FIB milling was used to shape cellular material into a needle that could be fully rotated (i.e., from −90° to 90°) (Narayan et al. 2012) and imaged by scanning electron microscopy (SEM). This study demonstrated that atom probe tomography (APT) (Miller et al. 2012) can be applied to chemically map freeze-dried cells with a thin metal coat in 3D. However, this was a unique experiment on unique equipment; it remains to be demonstrated whether an analogous approach could become widely applicable to frozen-hydrated cells using cryoET. In this direction, a recent study (Saghi et al. 2016) combined needle-shape FIB milling of the specimen with SEM imaging and tomographic reconstruction using CS. While these are exciting advances, added complications in data collection, storage, and processing preclude multiple-axis cryoET and APT of biological specimens from being routinely applied. A more promising proof of concept visualized bacterial cells by cryoET using a novel cylindrical holder (Palmer and Löwe 2014).
Missing wedge compensation for subtomogram classification and alignment
In cryoSPT, subtomograms need to be correctly aligned to each other or to a common reference before they can be averaged coherently. Preventing “missing wedge bias” is an essential step to accomplish this. Without correction, the missing wedge is the strongest feature in individual subtomograms and tends to bias the alignment of any two given subvolumes, favoring orientations with maximum density overlap as opposed to optimizing the overlap of matching structural features (Fig. 7).

Missing wedge bias in Fourier space. A Cartoon representation of a molecule to image (left), its representation as a subtomogram in Fourier space after limited-angle tomographic reconstruction (center), and the effects that the “missing wedge” of information due to limited-angle imaging and dose fractionation due to limited tolerable electron dose have on the subtomogram in real space (i.e., individual subtomograms suffer from elongation and high levels of noise). B, C Since the comparison metric most commonly used in SPT is cross correlation, greater density overlap tends to increase the similarity score during alignment. This “missing wedge bias” towards favoring larger density overlaps can cause misalignment of subtomograms; however, it can be compensated for by cross correlation map normalization or other methods. D After averaging many correctly aligned subtomograms in different orientations, the missing wedge can be “filled in”, and the associated elongation artifacts disappear while improving the SNR and the resolution of the macromolecular complex of interest. Note : The Fourier transform of the Superman logo would appear visually as an abstract and somewhat random pattern, in which it would be difficult to discern correct feature overlaps. For this reason, we have represented the image in Fourier space using the real-space image as a proxy to facilitate interpretation of the impact of missing wedge bias
Given the current state of SPT, accurate classification of noisy subtomograms with a missing wedge remains one of the biggest challenges. The missing wedge, as well as missing information between tilts, can make accurate classification statistically impossible in specific cases. Popular techniques used in cryoEM SPA such as multivariate statistical analysis (MSA) (Van Heel and Frank 1981; Frank et al. 1982; Van Heel 1984) can be tricky to apply to SPT, because the missing wedge, which is often the strongest feature, is difficult to exclude when the particles have already been rotated for alignment. A classification technique for subtomograms based on 2D reprojections has been proposed to overcome the uncertainties in classification introduced by the missing wedge (Yu et al. 2010, 2013). While this method would still be, in principle, subject to the degeneracy problem that arises in SPA cryoEM (Fig. 1), it may allow for fast, initial 2D classification of particles from cellular tomograms without the concern of overlapping densities.
Multiple methods have been proposed to identify and compensate for the missing wedge. Those based on normalization of cross correlation maps are the simplest and have been used successfully for template matching (Frangakis et al. 2002) and SPT (Nicastro et al. 2006; Schmid et al. 2006). These methods have the advantage of not requiring explicit identification of “missing voxels” in Fourier space. Other algorithms identify the missing wedge by presumed a priori knowledge of its exact location (Stölken et al. 2011) or by selection of a threshold value in Fourier space to constrain correlation (Bartesaghi et al. 2008; Förster et al. 2008; Schmid and Booth 2008). The rationale behind thresholding is that, for individual subtomograms, ~95% of the structural information is concentrated in approximately the top 1% strongest Fourier coefficients (Amat et al. 2010). Other proposed methods use an estimate of the true structure (Heumann et al. 2011) or fill in the missing information with data derived from the population average (Bostina et al. 2007). However, the latter methods assume structural homogeneity among particles. On the other hand, methods relying on thresholding assume that Fourier voxels corresponding to the missing wedge have small values and thus a threshold can be easily assigned to identify them. One caveat inherent to the thresholding method is that simple operations such as filtration and masking can alter the values of voxels inside or outside the missing wedge in different ways, leading to misidentification of missing wedge voxels and therefore increasing subtomogram alignment error. A dynamic, resolution-dependent, per-volume thresholding method was recently introduced (Galaz-Montoya et al. 2016a), which produces much more robust identification of the missing wedge than the use of a single threshold value, and better alignment results than adaptive low-pass filtering using estimates of the resolution (Hrabe et al. 2012) or FSC curves directly (Galaz-Montoya et al. 2015).
IMPROVING SINGLE PARTICLE TOMOGRAPHY
Measuring the defocus directly from experimental images, coupled with several other methodological improvements such as an optimized tilt-image acquisition scheme (Hagen et al. 2017) and damage compensation (Bammes and Jin 2014), also known as exposure filtering (Grant and Grigorieff 2015), as well as the usage of cutting-edge technology, are some of the latest improvements that have permitted cryoSPT to achieve ~4 Å resolution in the best case reported to date (Schur et al. 2016).
Ideally, one would like to produce a well-resolved 3D map with isotropic resolution for any specimen, be it particles extracted from cells, or macromolecules in solution. Covering the full 180° rotation about any single axis would yield a tiltseries without a missing wedge, but while this has been successfully implemented in material sciences, it is still not generally possible for biological specimens. Proof-of-concept methodologies that solve this problem have been proposed, such as combining FIB and SEM (Narayan et al. 2012; Saghi et al. 2016), or using a novel cylindrical specimen holder for cryoET (Palmer and Löwe 2014). Continued development of cylindrical holders or applying needle shaping of bulk specimens by FIB to cryoET could have a significant positive impact on cryoSPT, particularly for macromolecules in situ.
The ongoing development of tomographic reconstruction methods will also propel biological discoveries by cellular cryoSPT in the years to come. A current limiting factor is the reliable, automated identification of heterogeneous macromolecular complexes in crowded environments. Due to the missing wedge and the low SNR of cellular tomograms, automated particle picking is prone to bias and high false-positive rates (Yu and Frangakis 2014; Hrabe 2015; Kunz et al. 2015), though the use of neural networks has been proposed to reduce such issues (Yu and Frangakis 2011). Reliable automation of tiltseries reconstruction into tomograms will also increase the throughput of studies by cryoET (Mastronarde and Held 2017). Automated segmentation of features in tomograms remains poor and can be extremely subjective and time consuming when performed manually (Hecksel et al. 2016), particularly for thick frozen-hydrated specimens. The use of compressed sensing and other methodologies that improve the quality of tomographic reconstructions should facilitate more accurate particle picking and more objective, automated annotation of cellular tomograms.
Ongoing methodological developments and automation of algorithms on various fronts will also continue to improve image processing. For example, movie-mode data collection with relatively high contrast using DDDs now allows decoupling the decay in image quality due to radiation damage versus that stemming from beam-induced motions. Taking advantage of this, an “exposure filtering” method was recently proposed (Grant and Grigorieff 2015), which optimizes the SNR of every frame collected, thereby minimizing the deleterious effects of radiation damage. This idea was also proposed in an earlier study that filtered DDD frames based on cumulative exposure to compensate for radiation damage (Bammes and Jin 2014; Wang et al. 2014).
Since many subvolumes in random orientations are typically combined in SPT, there is seldom a need to use traditional small tilt steps of 1°–2° during data collection. As long as there are sufficient images to permit accurate alignment of the tiltseries, much larger tilt steps may be used in most cases (perhaps as high as 10°), depending on the size of the specimen. This can dramatically reduce data collection and processing times as well as data storage requirements, with no negative impact on final averages if enough subtomograms are averaged. Using a larger tilt step for cryoSPT also has the positive effect that each tilt image has a higher dose, permitting the application of some methods that require high contrast in each image of a tiltseries (Iwasaki et al. 2005; Bartesaghi et al. 2012; Zhang and Ren 2012). Unfortunately, to this day many cryoSPT studies continue to collect data using canonical parameters originally designed for cryoET of unique, non-repeating structures such as cells.
Several unconventional proposed improvements to general cryoEM have been reviewed in (Massover 2011) and might yield beneficial effects for cryoET and cryoSPT as well. For example, the development of specimen holders that are more stable and allow collecting data at slower exposure rates with “resting” periods in between exposures might make biological specimens more impervious to radiation damage (Chen et al. 2008; Karuppasamy et al. 2011).
Novel specimen grids (Rhinow and Kühlbrandt 2008; Pantelic et al. 2010; Yoshioka et al. 2010; Russo and Passmore 2016) made with materials that more readily dissipate charge and heat during imaging can minimize beam-induced specimen motions as well as the bubbling and distortions caused by radiation damage. The widespread adoption of such grids will also improve cryoET and cryoSPT and accelerate discoveries by these methodologies.
The problem of low-contrast at tolerable doses in cryoEM micrographs was first partially alleviated by the introduction of better illumination sources (field emission guns, opposed to metal filaments) (Tonomura et al. 1979) and the usage of large defocus settings for contrast enhancement (Erickson and Klug 1970). More recently, energy filters (Zhu et al. 1997) and phase plates (Danev and Nagayama 2001; Majorovits et al. 2007) have provided dramatic improvements in image contrast (Dai et al. 2013). Most significantly, the Volta phase plate (Danev et al. 2014) was recently used to demonstrate cryoSPT at subnanometer resolution (Khoshouei et al. 2016), averaging merely ~1400 ribosome subtomograms. Earlier phase plates caused strong ringing or “fringe” artifacts due to the cut-on frequency determined by the size of the central hole responsible for the phase shift, necessitating cumbersome computational correction (Kishchenko et al. 2015). Hole-less phase plates, such as the Volta phase plate, reduce such artifacts and are thus poised to revolutionize cryoET and cryoSPT. Unfortunately, a number of technical problems still remain that impede routine, straightforward use of this technology. The progressive change of phase with dose on Volta phase plates as well as the defocus gradient at high tilt both necessitate taking phase plates significantly out of focus. These factors greatly complicate CTF correction, particularly at high tilt (Sharp et al. 2017).
DDDs are revolutionizing the field and also provide images with better contrast compared to CCDs by virtue of their improved DQE, allowing for the correction of beam-induced blurring at the whole-frame, or at the per-particle level in SPA cryoEM. A step further, a local, anisotropic motion correction method recently proposed for SPA cryoEM (Zheng et al. 2016) might find applications in effectively unblurring lower-SNR images of highly tilted, non-repeating specimens, which sometimes cannot be corrected with current methods (Fig. 6).
Lastly, the application of hybrid techniques such as combining FIB milling (Wang et al. 2012; Villa et al. 2013; Mahamid et al. 2016) and correlative light microscopy (Zhang 2013; Hampton et al. 2017) with cryoET and cryoSPT has opened the window for observing dynamic cellular processes in regions of thick mammalian cells at nanometer resolution. Further developments to make these hybrid methodologies accessible and routine are likely to lead the next generation of insights into the biochemistry occurring within cells.
Abbreviations
- CryoET:
-
Cryo-electron tomography
- CTF:
-
Contrast transfer function
- DDD:
-
Direct detection device
- SPT:
-
Single particle tomography
- STA:
-
Subtomogram averaging
References
Adrian M, Dubochet J, Lepault J, McDowall AW (1984) Cryo-electron microscopy of viruses. Nature 308(5954):32–36
Aganj I, Bartesaghi A, Borgnia M, Liao HY (2007) Regularization for inverting the radon transform with wedge consideration. In: 4th IEEE International Symposium on Biomedical Imaging: From Nano to Macro. pp. 217–220
Agulleiro JI, Vazquez F, Garzon EM, Fernandez JJ (2012) Hybrid computing: CPU + GPU co-processing and its application to tomographic reconstruction. Ultramicroscopy 115:109–114
Al-Amoudi A, Dubochet J, Gnaegi H, Lüthi W, Studer D (2003) An oscillating cryo-knife reduces cutting-induced deformation of vitreous ultrathin sections. J Microsc 212(1):26–33
Al-Amoudi A, Norlen LP, Dubochet J (2004) Cryo-electron microscopy of vitreous sections of native biological cells and tissues. J Struct Biol 148(1):131–135
Al-Amoudi A, Studer D, Dubochet J (2005) Cutting artefacts and cutting process in vitreous sections for cryo-electron microscopy. J Struct Biol 150(1):109–121
Amat F, Comolli LR, Moussavi F, Smit J, Downing KH, Horowitz M (2010) Subtomogram alignment by adaptive Fourier coefficient thresholding. J Struct Biol 171(3):332–344
Asano S, Fukuda Y, Beck F, Aufderheide A, Förster F, Danev R, Baumeister W (2015) A molecular census of 26S proteasomes in intact neurons. Science 347(6220):439–442
Asano S, Engel BD, Baumeister W (2016) In situ cryo-electron tomography: a post-reductionist approach to structural biology. J Mol Biol 428(2):332–343
Baker LA, Rubinstein JL (2010) Chapter fifteen—radiation damage in electron cryomicroscopy. Methods Enzymol 481:371–388
Bammes BE, Jin L (2014) Method of electron beam imaging of a specimen by combining images of an image sequence. Direct Electron, Lp. U.S. Patent 8,809,781
Bammes BE, Jakana J, Schmid MF, Chiu W (2010) Radiation damage effects at four specimen temperatures from 4 to 100 K. J Struct Biol 169(3):331–341
Bartesaghi A, Sprechmann P, Liu J, Randall G, Sapiro G, Subramaniam S (2008) Classification and 3D averaging with missing wedge correction in biological electron tomography. J Struct Biol 162(3):436–450
Bartesaghi A, Lecumberry F, Sapiro G, Subramaniam S (2012) Protein secondary structure determination by constrained single-particle cryo-electron tomography. Structure 20(12):2003–2013
Batenburg KJ, Sijbers J (2009) Adaptive thresholding of tomograms by projection distance minimization. Pattern Recogn 42(10):2297–2305
Baumeister W (2002) Electron tomography: towards visualizing the molecular organization of the cytoplasm. Curr Opin Struct Biol 12(5):679–684
Belnap DM (2015) Electron microscopy and image processing: essential tools for structural analysis of macromolecules. Curr Protocols Protein Sci 17(17):2
Bharat TA, Russo CJ, Löwe J, Passmore LA, Scheres SH (2015) Advances in single-particle electron cryomicroscopy structure determination applied to sub-tomogram averaging. Structure 23(9):1743–1753
Booy FP, Pawley JB (1993) Cryo-crinkling: what happens to carbon films on copper grids at low temperature. Ultramicroscopy 48(3):273–280
Bostina M, Bubeck D, Schwartz C, Nicastro D, Filman DJ, Hogle JM (2007) Single particle cryoelectron tomography characterization of the structure and structural variability of poliovirus–receptor–membrane complex at 30 Å resolution. J Struct Biol 160(2):200–210
Brandt S, Heikkonen J, Engelhardt P (2001) Automatic alignment of transmission electron microscope tilt series without fiducial markers. J Struct Biol 136(3):201–213
Brandt F, Carlson L-A, Hartl FU, Baumeister W, Grünewald K (2010) The three-dimensional organization of polyribosomes in intact human cells. Mol Cell 39(4):560–569
Briegel A, Ortega DR, Mann P, Kjær A, Ringgaard S, Jensen GJ (2016) Chemotaxis cluster 1 proteins form cytoplasmic arrays in vibrio cholerae and are stabilized by a double signaling domain receptor DosM. Proc Natl Acad Sci USA 113(37):10412–10417
Brilot AF, Chen JZ, Cheng A, Pan J, Harrison SC, Potter CS, Carragher B, Henderson R, Grigorieff N (2012) Beam-induced motion of vitrified specimen on holey carbon film. J Struct Biol 177(3):630–637
Brink J, Sherman MB, Berriman J, Chiu W (1998) Evaluation of charging on macromolecules in electron cryomicroscopy. Ultramicroscopy 72(1):41–52
Campbell MG, Cheng A, Brilot AF, Moeller A, Lyumkis D, Veesler D, Pan J, Harrison SC, Potter CS, Carragher B, Grigorieff N (2012) Movies of ice-embedded particles enhance resolution in electron cryo-microscopy. Structure 20(11):1823–1828
Carazo JM, Carrascosa JL (1987) Information recovery in missing angular data cases: an approach by the convex projections method in three dimensions. J Microsc 145(1):23–43
Carbajal-González BI, Heuser T, Fu X, Lin J, Smith BW, Mitchell DR, Nicastro D (2013) Conserved structural motifs in the central pair complex of eukaryotic flagella. Cytoskeleton 70(2):101–120
Cardone G, Grünewald K, Steven AC (2005) A resolution criterion for electron tomography based on cross-validation. J Struct Biol 151(2):117–129
Cassidy CK, Himes BA, Alvarez FJ, Ma J, Zhao G, Perilla JR, Schulten K, Zhang P (2015) CryoEM and computer simulations reveal a novel kinase conformational switch in bacterial chemotaxis signaling. eLife 4:e08419
Castaño-Díez D, Al-Amoudi A, Glynn A-M, Seybert A, Frangakis AS (2007) Fiducial-less alignment of cryo-sections. J Struct Biol 159(3):413–423
Castaño-Díez D, Kudryashev M, Arheit M, Stahlberg H (2012) Dynamo: a flexible, user-friendly development tool for subtomogram averaging of cryo-EM data in high-performance computing environments. J Struct Biol 178(2):139–151
Chen Y, Förster F (2014) Iterative reconstruction of cryo-electron tomograms using nonuniform fast Fourier transforms. J Struct Biol 185(3):309–316
Chen JZ, Sachse C, Xu C, Mielke T, Spahn CM, Grigorieff N (2008) A dose-rate effect in single-particle electron microscopy. J Struct Biol 161(1):92–100
Chen D, Goris B, Bleichrodt F, Mezerji HH, Bals S, Batenburg KJ, de With G, Friedrich H (2014) The properties of SIRT, TVM, and DART for 3D imaging of tubular domains in nanocomposite thin-films and sections. Ultramicroscopy 147:137–148
Chen Y, Zhang Y, Zhang K, Deng Y, Wang S, Zhang F, Sun F (2016) FIRT: filtered iterative reconstruction technique with information restoration. J Struct Biol 195(1):49–61
Cheng N, Wu W, Steven AC, Thomas J, Black L (2012) Bubblegrams reveal the inner body of bacteriophage phiKZ. Microsc Microanal 18(S2):112–113
Comolli LR, Downing KH (2005) Dose tolerance at helium and nitrogen temperatures for whole cell electron tomography. J Struct Biol 152(3):149–156
Cosslett VE (1978) Radiation damage in the high resolution electron microscopy of biological materials: a review. J Microsc 113(2):113–129
Crowther RA (1971) Procedures for three-dimensional reconstruction of spherical viruses by Fourier synthesis from electron micrographs. Philos Trans R Soc Lond B 261(837):221–230
Crowther RA (2010) From envelopes to atoms: the remarkable progress of biological electron microscopy. Adv Protein Chem Struct Biol 81:1–32
Crowther RA, DeRosier DJ, Klug A (1970) The reconstruction of a three-dimensional structure from projections and its application to electron microscopy. Proc R Soc Lond A 317(1530):319–340
Dai W, Fu C, Raytcheva D, Flanagan J, Khant HA, Liu X, Rochat RH, Haase-Pettingell C, Piret J, Ludtke SJ (2013) Visualizing virus assembly intermediates inside marine cyanobacteria. Nature 502(7473):707–710
Dalm D, Galaz-Montoya JG, Miller JL, Grushin K, Villalobos A, Koyfman AY, Schmid MF, Stoilova-McPhie S (2015) Dimeric organization of blood coagulation factor VIII bound to lipid nanotubes. Sci Rep 5:11212
Danev R, Nagayama K (2001) Transmission electron microscopy with Zernike phase plate. Ultramicroscopy 88(4):243–252
Danev R, Buijsse B, Khoshouei M, Plitzko JM, Baumeister W (2014) Volta potential phase plate for in-focus phase contrast transmission electron microscopy. Proc Natl Acad Sci USA 111(44):15635–15640
Darrow MC, Sergeeva OA, Isas JM, Galaz-Montoya JG, King JA, Langen R, Schmid MF, Chiu W (2015) Structural mechanisms of mutant huntingtin aggregation suppression by the synthetic chaperonin-like CCT5 complex explained by cryoelectron tomography. J Biol Chem 290(28):17451–17461
Davies KM, Strauss M, Daum B, Kief JH, Osiewacz HD, Rycovska A, Zickermann V, Kühlbrandt W (2011) Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc Natl Acad Sci USA 108(34):14121–14126
De Rosier DJ, Klug A (1968) Reconstruction of three dimensional structures from electron micrographs. Nature 217(5124):130–134
Deng Y, Chen Y, Zhang Y, Wang S, Zhang F, Sun F (2016) ICON: 3D reconstruction with ‘missing-information’ restoration in biological electron tomography. J Struct Biol 195(1):100–112
Díez DC, Mueller H, Frangakis AS (2007) Implementation and performance evaluation of reconstruction algorithms on graphics processors. J Struct Biol 157(1):288–295
Donoho DL (2006) Compressed sensing. IEEE Trans Inf Theory 52(4):1289–1306
Dubochet J, Booy FP, Freeman R, Jones AV, Walter CA (1981) Low temperature electron microscopy. Annu Rev Biophys Bioeng 10(1):133–149
Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, Schultz P (1988) Cryoelectron microscopy of vitrified specimens. Q rev biophys 21(2):129–228
Eibauer M, Hoffmann C, Plitzko JM, Baumeister W, Nickell S, Engelhardt H (2012) Unraveling the structure of membrane proteins in situ by transfer function corrected cryo-electron tomography. J Struct Biol 180(3):488–496
Eibauer M, Pellanda M, Turgay Y, Dubrovsky A, Wild A, Medalia O (2015) Structure and gating of the nuclear pore complex. Nat Commun 6:7532
Eisenstein M (2016) The field that came in from the cold. Nat Methods 13(1):19–22
Erickson HP, Klug A (1970) The Fourier transform of an electron micrograph: effects of defocussing and aberrations, and implications for the use of underfocus contrast enhancement. Ber Bunsenges Phys Chem 74(11):1129–1137
Erickson HP, Klug A (1971) Measurement and compensation of defocusing and aberrations by Fourier processing of electron micrographs. Philos Trans R Soc Lond B 261(837):105–118
Fernandez J-J (2012) Computational methods for electron tomography. Micron 43(10):1010–1030
Fernández JJ, Li S, Crowther RA (2006) CTF determination and correction in electron cryotomography. Ultramicroscopy 106(7):587–596
Fontana J, Jurado KA, Cheng N, Engelman A, Steven AC (2015) Exploiting the susceptibility of HIV-1 nucleocapsid protein to radiation damage in tomo-bubblegram imaging. Microsc Microanal 21(S3):545–546
Förster F, Hegerl R (2007) Structure determination in situ by averaging of tomograms. Methods Cell Biol 79:741–767
Förster F, Medalia O, Zauberman N, Baumeister W, Fass D (2005) Retrovirus envelope protein complex structure in situ studied by cryo-electron tomography. Proc Natl Acad Sci USA 102(13):4729–4734
Förster F, Pruggnaller S, Seybert A, Frangakis AS (2008) Classification of cryo-electron sub-tomograms using constrained correlation. J Struct Biol 161(3):276–286
Frangakis AS, Böhm J, Förster F, Nickell S, Nicastro D, Typke D, Hegerl R, Baumeister W (2002) Identification of macromolecular complexes in cryoelectron tomograms of phantom cells. Proc Natl Acad Sci USA 99(22):14153–14158
Frank J, Verschoor A, Boublik M (1982) Multivariate statistical analysis of ribosome electron micrographs: L and R lateral views of the 40 S subunit from HeLa cells. J Mol Biol 161(1):107–133
Galaz JG, Flanagan JF, Schmid MF, Chiu W, Ludtke S (2012) Single particle tomography in EMAN2. Microsc Microanal 18(S2):552
Galaz-Montoya JG, Flanagan J, Schmid MF, Ludtke SJ (2015) Single particle tomography in EMAN2. J Struct Biol 190(3):279–290
Galaz-Montoya JG, Hecksel CW, Baldwin PR, Wang E, Weaver SC, Schmid MF, Ludtke SJ, Chiu W (2016a) Alignment algorithms and per-particle CTF correction for single particle cryo-electron tomography. J Struct Biol 194(3):383–394
Galaz-Montoya JG, Hecksel CW, Chin J, Wang R, Lewis CW, Haemmerle M, Schmid MF, Ludtke SJ, Sood AK, Chiu W (2016b) Computational tools to improve visualization by cryo-electron tomography. Biophys J 110:159a
Gilbert P (1972) Iterative methods for the three-dimensional reconstruction of an object from projections. J Theor Biol 36(1):105–117
Glaeser RM (1971) Limitations to significant information in biological electron microscopy as a result of radiation damage. J Ultrastruct Res 36(3):466–482
Glaeser RM, Taylor KA (1978) Radiation damage relative to transmission electron microscopy of biological specimens at low temperature: a review. J Microsc 112(1):127–138
Goris B, Van den Broek W, Batenburg KJ, Mezerji HH, Bals S (2012) Electron tomography based on a total variation minimization reconstruction technique. Ultramicroscopy 113:120–130
Goris B, Roelandts T, Batenburg KJ, Mezerji HH, Bals S (2013) Advanced reconstruction algorithms for electron tomography: from comparison to combination. Ultramicroscopy 127:40–47
Grant T, Grigorieff N (2015) Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. Elife 4:e06980
Grange M, Vasishtan D, Grünewald K (2017) Cellular electron cryo tomography and in situ sub-volume averaging reveal the context of microtubule-based processes. J Struct Biol 197(2):181–190
Grubb DT (1974) Radiation damage and electron microscopy of organic polymers. J Mater Sci 9(10):1715–1736
Grünewald K, Medalia O, Gross A, Steven AC, Baumeister W (2002) Prospects of electron cryotomography to visualize macromolecular complexes inside cellular compartments: implications of crowding. Biophys Chem 100(1):577–591
Hagen WJ, Wan W, Briggs JA (2017) Implementation of a cryo-electron tomography tilt-scheme optimized for high resolution subtomogram averaging. J Struct Biol 197(2):191–198
Hampton CM, Strauss JD, Ke Z, Dillard RS, Hammonds JE, Alonas E, Desai TM, Marin M, Storms RE, Leon F, Melikyan GB, Santangelo PJ, Spearman PW, Wright ER (2017) Correlated fluorescence microscopy and cryo-electron tomography of virus-infected or transfected mammalian cells. Nat Protoc 12(1):150–167
Han R, Wang L, Liu Z, Sun F, Zhang F (2015) A novel fully automatic scheme for fiducial marker-based alignment in electron tomography. J Struct Biol 192(3):403–417
Harris A, Cardone G, Winkler DC, Heymann JB, Brecher M, White JM, Steven AC (2006) Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc Natl Acad Sci USA 103(50):19123–19127
Harris AK, Meyerson JR, Matsuoka Y, Kuybeda O, Moran A, Bliss D, Das SR, Yewdell JW, Sapiro G, Subbarao K (2013) Structure and accessibility of HA trimers on intact 2009 H1N1 pandemic influenza virus to stem region-specific neutralizing antibodies. Proc Natl Acad Sci USA 110(12):4592–4597
Hart RG (1968) Electron microscopy of unstained biological material: the polytropic montage. Science 159(3822):1464–1467
Hawkes PW (2007) The electron microscope as a structure projector. In: Frank J (ed) Electron tomography. Springer, New York, pp 83–111
Hecksel CW, Darrow MC, Dai W, Galaz-Montoya JG, Chin JA, Mitchell PG, Chen S, Jakana J, Schmid MF, Chiu W (2016) Quantifying variability of manual annotation in cryo-electron tomograms. Microsc Microanal 22:1–10
Henderson R, Chen S, Chen JZ, Grigorieff N, Passmore LA, Ciccarelli L, Rubinstein JL, Crowther RA, Stewart PL, Rosenthal PB (2011) Tilt-pair analysis of images from a range of different specimens in single-particle electron cryomicroscopy. J Mol Biol 413(5):1028–1046
Henderson R, Sali A, Baker ML, Carragher B, Devkota B, Downing KH, Egelman EH, Feng Z, Frank J, Grigorieff N (2012) Outcome of the first electron microscopy validation task force meeting. Structure 20(2):205–214
Heumann JM, Hoenger A, Mastronarde DN (2011) Clustering and variance maps for cryo-electron tomography using wedge-masked differences. J Struct Biol 175(3):288–299
Heymann JB, Belnap DM (2007) Bsoft: image processing and molecular modeling for electron microscopy. J Struct Biol 157(1):3–18
Hoppe W, Langer R, Knesch G, Poppe C (1968) Protein-kristallstrukturanalyse mit elektronenstrahlen. Naturwissenschaften 55(7):333–336
Hoppe W, Gassmann J, Hunsmann N, Schramm HJ, Sturm M (1974) Three-dimensional reconstruction of individual negatively stained yeast fatty-acid synthetase molecules from tilt series in the electron microscope. Hoppe-Seyler’s Z Phys Chem 355(11):1483–1487
Hrabe T (2015) Localize. pytom: a modern webserver for cryo-electron tomography. Nucleic Acids Res 43(W1):W231–W236
Hrabe T, Chen Y, Pfeffer S, Kuhn Cuellar L, Mangold A-V, Förster F (2012) PyTom: a python-based toolbox for localization of macromolecules in cryo-electron tomograms and subtomogram analysis. J Struct Biol 178(2):177–188
Hu B, Margolin W, Molineux IJ, Liu J (2013) The bacteriophage t7 virion undergoes extensive structural remodeling during infection. Science 339(6119):576–579
Huiskonen JT, Hepojoki J, Laurinmäki P, Vaheri A, Lankinen H, Butcher SJ, Grünewald K (2010) Electron cryotomography of Tula hantavirus suggests a unique assembly paradigm for enveloped viruses. J Virol 84(10):4889–4897
Iancu CV, Wright ER, Heymann JB, Jensen GJ (2006) A comparison of liquid nitrogen and liquid helium as cryogens for electron cryotomography. J Struct Biol 153(3):231–240
Iwasaki K, Mitsuoka K, Fujiyoshi Y, Fujisawa Y, Kikuchi M, Sekiguchi K, Yamada T (2005) Electron tomography reveals diverse conformations of integrin αIIbβ3 in the active state. J Struct Biol 150(3):259–267
Jensen GJ, Kornberg RD (2000) Defocus-gradient corrected back-projection. Ultramicroscopy 84(1):57–64
Karuppasamy M, Karimi Nejadasl F, Vulovic M, Koster AJ, Ravelli RB (2011) Radiation damage in single-particle cryo-electron microscopy: effects of dose and dose rate. J Synchrotron Radiat 18(3):398–412
Kazantsev IG, Klukowska J, Herman GT, Cernetic L (2010) Fully three-dimensional defocus-gradient corrected backprojection in cryoelectron microscopy. Ultramicroscopy 110(9):1128–1142
Khoshouei M, Pfeffer S, Baumeister W, Förster F, Danev R (2016) Subtomogram analysis using the Volta phase plate. J Struct Biol 197(2):94–101
Kishchenko GP, Danev R, Fisher R, He J, Hsieh C, Marko M, Sui H (2015) Effect of fringe-artifact correction on sub-tomogram averaging from Zernike phase-plate cryo-TEM. J Struct Biol 191(3):299–305
Knauer V, Hegerl R, Hoppe W (1983) Three-dimensional reconstruction and averaging of 30S ribosomal subunits of Escherichia coli from electron micrographs. J Mol Biol 163(3):409–430
Kosinski J, Mosalaganti S, von Appen A, Teimer R, DiGuilio AL, Wan W, Bui KH, Hagen WJ, Briggs JA, Glavy JS (2016) Molecular architecture of the inner ring scaffold of the human nuclear pore complex. Science 352(6283):363–365
Koster AJ, Grimm R, Typke D, Hegerl R, Stoschek A, Walz J, Baumeister W (1997) Perspectives of molecular and cellular electron tomography. J Struct Biol 120(3):276–308
Koyfman AY, Schmid MF, Gheiratmand L, Fu CJ, Khant HA, Huang D, He CY, Chiu W (2011) Structure of Trypanosoma brucei flagellum accounts for its bihelical motion. Proc Natl Acad Sci USA 108(27):11105–11108
Kremer JR, Mastronarde DN, McIntosh JR (1996) Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116(1):71–76
Kudryashev M, Castaño-Díez D, Stahlberg H (2012) Limiting factors in single particle cryo electron tomography. Comput Struct Biotechnol J 1(2):1–6
Kudryashev M, Castaño-Díez D, Deluz C, Hassaine G, Grasso L, Graf-Meyer A, Vogel H, Stahlberg H (2016) The structure of the mouse serotonin 5-HT 3 receptor in lipid vesicles. Structure 24(1):165–170
Kunz M, Frangakis AS (2014) Super-sampling SART with ordered subsets. J Struct Biol 188(2):107–115
Kunz M, Frangakis AS (2016) Three-dimensional CTF correction improves the resolution of electron tomograms. J Struct Biol 197(2):114–122
Kunz M, Yu Z, Frangakis AS (2015) M-free: mask-independent scoring of the reference bias. J Struct Biol 192(2):307–311
Lanzavecchia S, Cantele F, Bellon PL, Zampighi L, Kreman M, Wright E, Zampighi GA (2005) Conical tomography of freeze-fracture replicas: a method for the study of integral membrane proteins inserted in phospholipid bilayers. J Struct Biol 149(1):87–98
Le Gros MA, McDermott G, Larabell CA (2005) X-ray tomography of whole cells. Curr Opin Struct Biol 15(5):593–600
Leary R, Saghi Z, Midgley PA, Holland DJ (2013) Compressed sensing electron tomography. Ultramicroscopy 131:70–91
Leschziner AE, Nogales E (2006) The orthogonal tilt reconstruction method: an approach to generating single-class volumes with no missing cone for ab initio reconstruction of asymmetric particles. J Struct Biol 153(3):284–299
Li S, Sun Z, Pryce R, Parsy ML, Fehling SK, Schlie K, Siebert CA, Garten W, Bowden TA, Strecker T, Huiskonen JT (2016) Acidic pH-induced conformations and LAMP1 binding of the Lassa virus glycoprotein spike. PLoS Pathog 12(2):e1005418
Liu Y, Penczek PA, McEwen BF, Frank J (1995) A marker-free alignment method for electron tomography. Ultramicroscopy 58(3):393–402
Ludtke SJ, Baldwin PR, Chiu W (1999) EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128(1):82–97
Mahamid J, Pfeffer S, Schaffer M, Villa E, Danev R, Cuellar LK, Förster F, Hyman AA, Plitzko JM, Baumeister W (2016) Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science 351(6276):969–972
Maimon T, Elad N, Dahan I, Medalia O (2012) The human nuclear pore complex as revealed by cryo-electron tomography. Structure 20(6):998–1006
Maiorca M, Millet C, Hanssen E, Abbey B, Kazmierczak E, Tilley L (2014) Local regularization of tilt projections reduces artifacts in electron tomography. J Struct Biol 186(1):28–37
Majorovits E, Barton B, Schultheiss K, Perez-Willard F, Gerthsen D, Schröder RR (2007) Optimizing phase contrast in transmission electron microscopy with an electrostatic (Boersch) phase plate. Ultramicroscopy 107(2):213–226
Marabini R, Herman GT, Carazo JM (1998) 3D reconstruction in electron microscopy using ART with smooth spherically symmetric volume elements (blobs). Ultramicroscopy 72(1):53–65
Marabini R, Carragher B, Chen S, Chen J, Cheng A, Downing KH, Frank J, Grassucci RA, Heymann JB, Jiang W, Jonic S (2015) CTF challenge: result summary. J Struct Biol 190(3):348–359
Marko M, Hsieh C, Schalek R, Frank J, Mannella C (2007) Focused-ion-beam thinning of frozen-hydrated biological specimens for cryo-electron microscopy. Nat Methods 4(3):215–217
Massover WH (2011) New and unconventional approaches for advancing resolution in biological transmission electron microscopy by improving macromolecular specimen preparation and preservation. Micron 42(2):141–151
Mastronarde DN (1997) Dual-axis tomography: an approach with alignment methods that preserve resolution. J Struct Biol 120(3):343–352
Mastronarde DN, Held SR (2017) Automated tilt series alignment and tomographic reconstruction in IMOD. J Struct Biol 197(2):102–113
Mattei S, Glass B, Hagen WJ, Kräusslich HG, Briggs JA (2016) The structure and flexibility of conical HIV-1 capsids determined within intact virions. Science 354(6318):1434–1437
McEwen BF, Downing KH, Glaeser RM (1995) The relevance of dose-fractionation in tomography of radiation-sensitive specimens. Ultramicroscopy 60(3):357–373
Medalia O, Typke D, Hegerl R, Angenitzki M, Sperling J, Sperling R (2002a) Cryoelectron microscopy and cryoelectron tomography of the nuclear pre-mRNA processing machine. J Struct Biol 138(1):74–84
Medalia O, Weber I, Frangakis AS, Nicastro D, Gerisch G, Baumeister W (2002b) Macromolecular architecture in eukaryotic cells visualized by cryoelectron tomography. Science 298(5596):1209–1213
Messaoudi C, Loubresse NG, Boudier T, Dupuis-Williams P, Marco S (2006) Multiple-axis tomography: applications to basal bodies from Paramecium tetraurelia. Biol Cell 98(7):415–425
Milazzo A-C, Leblanc P, Duttweiler F, Jin L, Bouwer JC, Peltier S, Ellisman M, Bieser F, Matis HS, Wieman H (2005) Active pixel sensor array as a detector for electron microscopy. Ultramicroscopy 104(2):152–159
Milazzo A-C, Moldovan G, Lanman J, Jin L, Bouwer JC, Klienfelder S, Peltier ST, Ellisman MH, Kirkland AI, Xuong N-H (2010) Characterization of a direct detection device imaging camera for transmission electron microscopy. Ultramicroscopy 110(7):741–744
Miller MK, Kelly TF, Rajan K, Ringer SP (2012) The future of atom probe tomography. Mater Today 15(4):158–165
Mindell JA, Grigorieff N (2003) Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 142(3):334–347
Murata K, Zhang Q, Galaz-Montoya JG, Fu C, Coleman ML, Osburne MS, Schmid MF, Sullivan MB, Chisholm SW, Chiu W (2017) Visualizing adsorption of cyanophage P-SSP7 onto marine Prochlorococcus. Sci Rep 7:44176
Murray SC, Galaz-Montoya JG, Tang G, Flanagan JF, Ludtke SJ (2014) EMAN2. 1-a new generation of software for validated single particle analysis and single particle tomography. Microsc Microanal 20(S3):832–833
Nans A, Kudryashev M, Saibil HR, Hayward RD (2015) Structure of a bacterial type III secretion system in contact with a host membrane in situ. Nat Commun 6:10114
Narayan K, Prosa TJ, Fu J, Kelly TF, Subramaniam S (2012) Chemical mapping of mammalian cells by atom probe tomography. J Struct Biol 178(2):98–107
Nicastro D, Schwartz C, Pierson J, Gaudette R, Porter ME, McIntosh JR (2006) The molecular architecture of axonemes revealed by cryoelectron tomography. Science 313(5789):944–948
Noble AJ, Stagg SM (2015) Automated batch fiducial-less tilt-series alignment in Appion using Protomo. J Struct Biol 192(2):270–278
Nogales E (2016) The development of cryo-EM into a mainstream structural biology technique. Nat Methods 13(1):24–27
Oettl H, Hegerl R, Hoppe W (1983) Three-dimensional reconstruction and averaging of 50S ribosomal subunits of Escherichia coli from electron micrographs. J Mol Biol 163(3):431–450
Ortiz JO, Förster F, Kürner J, Linaroudis AA, Baumeister W (2006) Mapping 70S ribosomes in intact cells by cryoelectron tomography and pattern recognition. J Struct Biol 156(2):334–341
Ortiz JO, Brandt F, Matias VR, Sennels L, Rappsilber J, Scheres SH, Eibauer M, Hartl FU, Baumeister W (2010) Structure of hibernating ribosomes studied by cryoelectron tomography in vitro and in situ. J Cell Biol 190(4):613–621
Palmer CM, Löwe J (2014) A cylindrical specimen holder for electron cryo-tomography. Ultramicroscopy 137:20–29
Pantelic RS, Meyer JC, Kaiser U, Baumeister W, Plitzko JM (2010) Graphene oxide: a substrate for optimizing preparations of frozen-hydrated samples. J Struct Biol 170(1):152–156
Pascual-Montano A, Taylor KA, Winkler H, Pascual-Marqui RD, Carazo J-M (2002) Quantitative self-organizing maps for clustering electron tomograms. J Struct Biol 138(1):114–122
Penczek PA (2010) Chapter one-fundamentals of three-dimensional reconstruction from projections. Methods Enzymol 482:1–33
Penczek P, Marko M, Buttle K, Frank J (1995) Double-tilt electron tomography. Ultramicroscopy 60(3):393–410
Penczek PA, Renka R, Schomberg H (2004) Gridding-based direct Fourier inversion of the three-dimensional ray transform. JOSA A 21(4):499–509
Peralta B, Gil-Carton D, Castaño-Díez D, Bertin A, Boulogne C, Oksanen HM, Bamford DH, Abrescia NG (2013) Mechanism of membranous tunnelling nanotube formation in viral genome delivery. PLoS Biol 11(9):e1001667
Pfeffer S, Brandt F, Hrabe T, Lang S, Eibauer M, Zimmermann R, Förster F (2012) Structure and 3D arrangement of endoplasmic reticulum membrane-associated ribosomes. Structure 20(9):1508–1518
Pfeffer S, Woellhaf MW, Herrmann JM, Förster F (2015a) Organization of the mitochondrial translation machinery studied in situ by cryoelectron tomography. Nat Commun 6:6019
Pfeffer S, Burbaum L, Unverdorben P, Pech M, Chen Y, Zimmermann R, Beckmann R, Förster F (2015b) Structure of the native Sec61 protein-conducting channel. Nat Commun 6:6019
Radermacher M (1988) Three-dimensional reconstruction of single particles from random and nonrandom tilt series. J Electron Microsc Tech 9(4):359–394
Radermacher M (2007) Electron tomography. Springer, New York
Radermacher M, Wagenknecht T, Verschoor A, Frank J (1986) A new 3-d reconstruction scheme applied to the 50 s ribosomal subunit of E. coli. J Microsc 141(1):1–2
Radon J (1917) Uber die Bestimmung von Funktionen durch ihre Integralwerte langs gewissez Mannigfaltigheiten, Ber. Verh. Sachs. Akad. Wiss. Leipzig, Math Phys Klass, 69
Renken C, McEwen B (2003) Markerless alignment: bridging the gap between theory and practice. Microsc Microanal 9(S02):1170–1171
Rhinow D, Kühlbrandt W (2008) Electron cryo-microscopy of biological specimens on conductive titanium–silicon metal glass films. Ultramicroscopy 108(7):698–705
Riedel C, Vasishtan D, Siebert CA, Whittle C, Lehmann MJ, Mothes W, Grünewald K (2017) Native structure of a retroviral envelope protein and its conformational change upon interaction with the target cell. J Struct Biol 197(2):172–180
Russo CJ, Passmore LA (2016) Ultrastable gold substrates: properties of a support for high-resolution electron cryomicroscopy of biological specimens. J Struct Biol 193(1):33–44
Saghi Z, Holland DJ, Leary R, Falqui A, Bertoni G, Sederman AJ, Gladden LF, Midgley PA (2011) Three-dimensional morphology of iron oxide nanoparticles with reactive concave surfaces. A compressed sensing-electron tomography (CS-ET) approach. Nano Lett 11(11):4666–4673
Saghi Z, Divitini G, Winter B, Leary R, Spiecker E, Ducati C, Midgley PA (2016) Compressed sensing electron tomography of needle-shaped biological specimens–Potential for improved reconstruction fidelity with reduced dose. Ultramicroscopy 160:230–238
Schaller RR (1997) Moore’s law: past, present and future. IEEE Spectr 34(6):52–59
Scheres SH (2014) Beam-induced motion correction for sub-megadalton cryo-EM particles. elife 3:e03665
Schmid MF (2011) Single-particle electron cryotomography (cryoET). Adv Protein Chem Struct Biol 82:37–65
Schmid MF, Booth CR (2008) Methods for aligning and for averaging 3D volumes with missing data. J Struct Biol 161(3):243–248
Schmid MF, Paredes AM, Khant HA, Soyer F, Aldrich HC, Chiu W, Shively JM (2006) Structure of halothiobacillus neapolitanus carboxysomes by cryo-electron tomography. J Mol Biol 364(3):526–535
Schmid MF, Hecksel CW, Rochat RH, Bhella D, Chiu W, Rixon FJ (2012) A tail-like assembly at the portal vertex in intact herpes simplex type-1 virions. PLoS Pathog 8(10):e1002961
Schur FK, Hagen W, de Marco A, Briggs JA (2013) Determination of protein structure at 8.5 Å resolution using cryo-electron tomography and subtomogram averaging. J Struct Biol 184(3):394–400
Schur FK, Hagen WJ, Rumlová M, Ruml T, Müller B, Kräusslich H-G, Briggs JA (2015a) Structure of the immature HIV-1 capsid in intact virus particles at 8.8 A resolution. Nature 517(7535):505–508
Schur FK, Dick RA, Hagen WJ, Vogt VM, Briggs JA (2015b) The structure of immature virus-like Rous sarcoma virus Gag particles reveals a structural role for the p10 domain in assembly. J Virol 89(20):10294–10302
Schur FK, Obr M, Hagen WJ, Wan W, Jakobi AJ, Kirkpatrick JM, Sachse C, Kräusslich HG, Briggs JA (2016) An atomic model of HIV-1 capsid-SP1 reveals structures regulating assembly and maturation. Science 353(6298):506–508
Schwartz CL, Heumann JM, Dawson SC, Hoenger A (2012) A detailed, hierarchical study of Giardia lamblia’s ventral disc reveals novel microtubule-associated protein complexes. PLoS ONE 7(9):e43783
Shahmoradian SH, Galaz-Montoya JG, Schmid MF, Cong Y, Ma B, Spiess C, Frydman J, Ludtke SJ, Chiu W (2013) TRiCs tricks inhibit huntingtin aggregation. eLife 2:e00710
Sharp T, Koster A, Gros P (2016) Heterogeneous MAC initiator and pore structures in a lipid bilayer by phase-plate cryo-electron tomography. Cell Rep 15(1):1–8
Sharp TH, Faas FG, Koster AJ, Gros P (2017) Imaging complement by phase-plate cryo-electron tomography from initiation to pore formation. J Struct Biol 197(2):155–162
Song K, Comolli LR, Horowitz M (2012) Removing high contrast artifacts via digital inpainting in cryo-electron tomography: an application of compressed sensing. J Struct Biol 178(2):108–120
Stoffler D, Feja B, Fahrenkrog B, Walz J, Typke D, Aebi U (2003) Cryo-electron tomography provides novel insights into nuclear pore architecture: implications for nucleocytoplasmic transport. J Mol Biol 328(1):119–130
Stölken M, Beck F, Haller T, Hegerl R, Gutsche I, Carazo J-M, Baumeister W, Scheres SH, Nickell S (2011) Maximum likelihood based classification of electron tomographic data. J Struct Biol 173(1):77–85
Sun L, Young LN, Zhang X, Boudko SP, Fokine A, Zbornik E, Roznowski AP, Molineux IJ, Rossmann MG, Fane BA (2014) Icosahedral bacteriophage [Phi] X174 forms a tail for DNA transport during infection. Nature 505(7483):432–435
Taylor KA, Glaeser RM (1974) Electron diffraction of frozen, hydrated protein crystals. Science 186(4168):1036–1037
Tong J, Arslan I, Midgley P (2006) A novel dual-axis iterative algorithm for electron tomography. J Struct Biol 153(1):55–63
Tonomura A, Matsuda T, Todokoro H, Komoda T (1979) Development of a field emission electron microscope. J Electron Microsc 28(1):1–11
Toyoshima C, Unwin N (1988) Contrast transfer for frozen-hydrated specimens: determination from pairs of defocused images. Ultramicroscopy 25(4):279–291
Turoňová B, Marsalek L, Davidovič T, Slusallek P (2015) Progressive stochastic reconstruction technique (PSRT) for cryo electron tomography. J Struct Biol 189(3):195–206
Van Heel M (1984) Multivariate statistical classification of noisy images (randomly oriented biological macromolecules). Ultramicroscopy 13(1):165–183
Van Heel M (1987) Angular reconstitution: a posteriori assignment of projection directions for 3D reconstruction. Ultramicroscopy 21(2):111–123
Van Heel M, Frank J (1981) Use of multivariate statistics in analysing the images of biological macromolecules. Ultramicroscopy 6(2):187–194
Villa E, Schaffer M, Plitzko JM, Baumeister W (2013) Opening windows into the cell: focused-ion-beam milling for cryo-electron tomography. Curr Opin Struct Biol 23(5):771–777
Voortman LM, Stallinga S, Schoenmakers RH, van Vliet LJ, Rieger B (2011) A fast algorithm for computing and correcting the CTF for tilted, thick specimens in TEM. Ultramicroscopy 111(8):1029–1036
Wan W, Briggs JAG (2016) Chapter thirteen-cryo-electron tomography and subtomogram averaging. Methods Enzymol 579:329–367
Wan X, Zhang F, Chu Q, Zhang K, Sun F, Yuan B, Liu Z (2011) Three-dimensional reconstruction using an adaptive simultaneous algebraic reconstruction technique in electron tomography. J Struct Biol 175(3):277–287
Wang Y, Jacobsen C, Maser J, Osanna A (2000) Soft X-ray microscopy with a cryo scanning transmission X-ray microscope: II. Tomography. J Microsc 197(Pt 1):80–93
Wang K, Strunk K, Zhao G, Gray JL, Zhang P (2012) 3D structure determination of native mammalian cells using cryo-FIB and cryo-electron tomography. J Struct Biol 180(2):318–326
Wang Z, Hryc CF, Bammes B, Afonine PV, Jakana J, Chen DH, Liu X, Baker ML, Kao C, Ludtke SJ, Schmid MF (2014) An atomic model of brome mosaic virus using direct electron detection and real-space optimization. Nat Commun 5:4808
Winkler H (2007) 3D reconstruction and processing of volumetric data in cryo-electron tomography. J Struct Biol 157(1):126–137
Winkler H, Taylor KA (1999) Multivariate statistical analysis of three-dimensional cross-bridge motifs in insect flight muscle. Ultramicroscopy 77(3):141–152
Winkler H, Taylor KA (2003) Focus gradient correction applied to tilt series image data used in electron tomography. J Struct Biol 143(1):24–32
Wu W, Thomas JA, Cheng N, Black LW, Steven AC (2012) Bubblegrams reveal the inner body of bacteriophage φKZ. Science 335(6065):182
Xiong Q, Morphew MK, Schwartz CL, Hoenger AH, Mastronarde DN (2009) CTF determination and correction for low dose tomographic tilt series. J Struct Biol 168(3):378–387
Xu P, Donaldson LA, Gergely ZR, Staehelin LA (2007) Dual-axis electron tomography: a new approach for investigating the spatial organization of wood cellulose microfibrils. Wood Sci Technol 41(2):101–116
Yoshioka C, Carragher B, Potter CS (2010) Cryomesh: a new substrate for cryo-electron microscopy. Microsc Microanal 16(01):43–53
Yu Z, Frangakis AS (2011) Classification of electron sub-tomograms with neural networks and its application to template-matching. J Struct Biol 174(3):494–504
Yu Z, Frangakis AS (2014) M-free: Scoring the reference bias in sub-tomogram averaging and template matching. J Struct Biol 187(1):10–19
Yu L, Snapp RR, Ruiz T, Radermacher M (2010) Probabilistic principal component analysis with expectation maximization (PPCA-EM) facilitates volume classification and estimates the missing data. J Struct Biol 171(1):18–30
Yu L, Snapp RR, Ruiz T, Radermacher M (2013) Projection-based volume alignment. J Struct Biol 182(2):93–105
Zanetti G, Riches JD, Fuller SD, Briggs JA (2009) Contrast transfer function correction applied to cryo-electron tomography and sub-tomogram averaging. J Struct Biol 168(2):305–312
Zhang P (2013) Correlative cryo-electron tomography and optical microscopy of cells. Curr Opin Struct Biol 23(5):763–770
Zhang L, Ren G (2012) IPET and FETR: experimental approach for studying molecular structure dynamics by cryo-electron tomography of a single-molecule structure. PLoS ONE 7(1):e30249
Zhao X, Zhang K, Boquoi T, Hu B, Motaleb MA, Miller KA, James ME, Charon NW, Manson MD, Norris SJ (2013) Cryoelectron tomography reveals the sequential assembly of bacterial flagella in Borrelia burgdorferi. Proc Natl Acad Sci USA 110(35):14390–14395
Zheng SQ, Palovcak E, Armache JP, Cheng Y, Agard DA (2016) Anisotropic correction of beam-induced motion for improved single-particle electron cryo-microscopy. BioArxiv. doi:10.1101/061960
Zhou Z, Li Y, Zhang F, Wan X (2015) FASART: an iterative reconstruction algorithm with inter-iteration adaptive NAD filter. Bio-Med Mater Eng 26(s1):S1409–S1415
Zhu J, Penczek PA, Schröder R, Frank J (1997) Three-dimensional reconstruction with contrast transfer function correction from energy-filtered cryoelectron micrographs: procedure and application to the 70s Escherichia coli ribosome. J Struct Biol 118(3):197–219
Acknowledgements
We thank Boxue Ma from Dr. Wah Chiu’s lab at the NCMI, Baylor College of Medicine (Houston, TX, USA), for collecting the platelet tiltseries, and Dr. Anil Sood’s lab at MD Anderson Cancer Center (Houston, TX, USA) for providing the platelet specimen.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Jesús G. Galaz-Montoya and Steven J. Ludtke declare that they have no conflicts of interest.
Human and animal rights and informed consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Galaz-Montoya, J.G., Ludtke, S.J. The advent of structural biology in situ by single particle cryo-electron tomography. Biophys Rep 3, 17–35 (2017). https://doi.org/10.1007/s41048-017-0040-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41048-017-0040-0