
Abstract
Introduction
SB5 is an approved biosimilar of adalimumab, a monoclonal anti-tumor necrosis factor (anti-TNF) antibody. This study compared pharmacokinetics (PK), safety, tolerability, and immunogenicity between a new high-concentration, low-volume, and citrate-free formulation (40 mg/0.4 ml, SB5-HC) and the current low-concentration formulation with higher volume (40 mg/0.8 ml, SB5-LC) to evaluate the bioequivalence of the two formulations.
Methods
This study was a randomized, single-blind, two-arm, parallel-group, single-dose study in healthy male subjects. Subjects were randomized to receive either SB5-HC or SB5-LC via subcutaneous injection using a pre-filled syringe. Primary endpoints were the area under the curve of the concentration–time curve from zero to infinity (AUCinf) and maximum serum concentration (Cmax). Bioequivalence was achieved if the 90% confidence intervals (CIs) for the ratios of the geometric least squares mean (LSMean) of primary endpoints were within the pre-defined bioequivalence margins of 0.80–1.25. Secondary endpoints included safety, tolerability, and immunogenicity.
Results
Subjects (n = 188) were randomized to SB5-HC (n = 94) or SB5-LC (n = 94). Baseline characteristics were comparable between the two treatment groups. The mean values for AUCinf and Cmax were similar between the SB5-HC and SB5-LC groups. For the primary endpoints, the geometric LSMean ratios (90% CI) for AUCinf and Cmax were 0.920 (0.8262–1.0239) and 0.984 (0.9126–1.0604), respectively, placing the corresponding 90% CIs well within the pre-defined bioequivalence margin of 0.80–1.25. All treatment-emergent adverse events (TEAEs) were considered mild to moderate and were reported for 44.7% and 51.1% of subjects in the SB5-HC and SB5-LC groups, respectively. Immunogenicity assessed by frequency of occurrence of anti-drug antibodies (ADAs) and neutralizing antibodies (NAbs) was comparable between groups.
Conclusions
This bridging study demonstrated PK equivalence and comparable safety and tolerability of subcutaneous injection of SB5 via SB5-HC or SB5-LC.
Clinicaltrials.gov identifier
Similar content being viewed by others

Avoid common mistakes on your manuscript.
SB5 is a biosimilar of adalimumab, a monoclonal antibody used to treat autoimmune inflammatory diseases (rheumatoid arthritis, Crohn’s disease, ulcerative colitis, plaque psoriasis, etc.). |
This study investigated PK, safety, tolerability, and immunogenicity of a high-concentration, low-volume, citrate-free formulation of SB5 (SB5-HC) compared to a low-concentration, high-volume formulation of SB5 (SB5-LC). |
A total of 188 healthy male volunteers were randomly assigned to two groups that were either administered SB5-HC or SB5-LC and monitored for 57 days to assess PK, safety, tolerability, and immunogenicity. |
This study demonstrated the PK equivalence and comparable safety, tolerability, and immunogenicity of two formulations (SB5-HC and SB5-LC) in healthy male volunteers. |
Introduction
Adalimumab (ADL) is a recombinant, fully humanized monoclonal antibody that binds specifically to the inflammatory cytokine tumor necrosis factor (TNF), a major mediator of inflammation playing a pivotal role in autoimmune diseases [1]. Binding of ADL to TNF neutralizes the biological function of TNF. ADL is approved for the treatment of rheumatoid arthritis and several other inflammatory diseases [1,2,3].
SB5 is a biosimilar with ADL as the active substance [4]. It is approved in the European Union (EU) (Imraldi™) and the United States of America (US) (Hadlima™) for indications such as rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, axial spondylitis (ankylosing spondylitis [AS], axial spondyloarthritis without radiographic evidence of AS; in US, only AS is approved), Crohn’s disease, ulcerative colitis, and plaque psoriasis. In line with the reference product (Humira®, a registered trademark of AbbVie Inc., North Chicago, IL, USA), SB5 was developed as a 40 mg/0.8 ml formulation for subcutaneous injection (SB5-LC). A primary phase I clinical study in 189 healthy subjects established pharmacokinetic (PK) equivalence and comparable safety and immunogenicity profiles between single 40 mg doses of SB5-LC and its EU-sourced and US-sourced reference ADL [5]. A consecutive phase I study in 190 healthy subjects demonstrated bioequivalence, including comparable safety and tolerability, of single 40-mg doses of SB5-LC administered with an autoinjector and a pre-filled syringe, respectively [6].
A phase III clinical study in 544 patients with moderate-to-severe active rheumatoid arthritis despite methotrexate therapy demonstrated equivalent efficacy between SB5-LC and the reference ADL (both 40 mg subcutaneously every other week) with regard to the primary endpoint (American College of Rheumatology 20% improvement criteria, ACR20) and other efficacy endpoints [7]. ACR20 response rates at week 24 were 72.4% and 72.2% in SB5-LC and the reference ADL, respectively. Efficacy, safety, and immunogenicity remained comparable up to week 52 [8]. Also, among patients who switched from reference ADL to SB5-LC after week 24 or continued to receive reference ADL, the ACR20, ACR50 and ACR70 response rates, as well as safety profiles and the incidence of anti-drug antibodies (ADAs), were comparable up to week 52.
A new formulation of the reference ADL has been approved with a reduced volume and higher concentration (40 mg/0.4 ml) than the prior formulation (40 mg/0.8 ml) and six excipients including citric acid being removed [2, 3]. Accordingly, a high-concentration, low-volume, citrate-free formulation of SB5 (SB5-HC) has been newly developed as a part of life cycle management to accommodate the reference ADL formulation. The current study aimed to demonstrate the PK equivalence and comparable safety, tolerability, and immunogenicity of subcutaneous SB5-HC (40 mg/0.4 ml) and SB5-LC (40 mg/0.8 ml) formulation in healthy male subjects.
Methods
Study Population
The study population comprised healthy male subjects. Key inclusion criteria were age of 18–55 years, body weight of 65.0–90.0 kg, and a body mass index (BMI) of 20.0–29.9 kg/m2 at screening and baseline (day 1). In addition, eligible subjects were to show no clinically relevant abnormalities in 12-lead electrocardiogram (ECG), vital signs, physical examination, and clinical laboratory tests at screening and baseline. Key exclusion criteria were a diagnosis of active or latent tuberculosis, previous treatment with ADL, and known or suspected clinically relevant drug hypersensitivity to ADL or to any of the excipients.
Study Design
This was a randomized, single-blind, two-arm, parallel-group, single-dose study performed at Parexel Early Phase Clinical Unit, Berlin, Germany, between August 2020 and May 2021 (clinicaltrials.gov identifier NCT04514796). The final study protocol was approved by the local Independent Ethics Committee (IEC; Ethics committee of the state of Berlin, State Office of Health and Social Affairs, Berlin, Germany, reference number 20/0178). This study was conducted in accordance with the Declaration of Helsinki (1996) and is consistent with the International Council for Harmonization Good Clinical Practice guidelines and applicable local regulatory requirements and laws. Written informed consent was obtained from each subject before enrolment. The consent form was reviewed and approved by the IEC prior to use.
Eligible subjects were randomized in a 1:1 ratio to receive a single 40-mg dose of either SB5-HC or SB5-LC, subcutaneously injected with a pre-filled syringe in the left or right upper abdominal quadrant of the periumbilical area on day 1. Subjects were observed for 57 days during which PK, safety, tolerability, and immunogenicity measurements were performed. Primary endpoints were the area under the concentration–time curve from time zero to infinity (AUCinf) and maximum serum concentration (Cmax). Secondary PK endpoints included area under the concentration–time curve from time zero to the last quantifiable concentration (AUClast), time to Cmax (Tmax), apparent volume of distribution during the terminal phase (Vz/F), terminal rate constant (λz), terminal half-life (t1/2), apparent total body clearance (CL/F), and percentage of AUCinf due to extrapolation from time of last measurable concentration (Tlast) to infinity (%AUCextrap). The safety endpoints were adverse events (AEs) and serious AEs (SAEs), clinical laboratory values, 12-lead ECG, vital signs, physical examination, and injection site assessment. Immunogenicity endpoints were the incidence of ADAs and neutralizing antibodies (NAbs) to ADL.
Pharmacokinetic Evaluation
Blood samples for PK analysis were collected at 0 (pre-dose), 24, 48, 96, 120, 144, 168, 216, 264, 336, 408, 504, 600, 696, 840, 1008, and 1344 hours (h) post-dose. Subjects were discharged on day 2 with the rest of study period consisting of outpatient visits. Serum concentrations of ADL were measured using a validated enzyme-linked immunosorbent assay (ELISA) specific for the detection and quantification of ADL. The lower limit of quantitation was 0.05 μg/ml, and the upper limit of quantitation was 1.50 μg/ml. Intra- and inter-assay precision and accuracy displayed a percent coefficient of variation (CV%) of 6.6% and 14.8%, respectively. The primary endpoint AUCinf was calculated as AUClast plus last observed concentration (Ct) divided by λz. Linear regression after log-transformation using the last three (or more) non-zero concentrations was used to calculate λz. Terminal half-life (t1/2) was calculated by ln(2)/λz.
Safety Evaluation
All reported terms for AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA®) version 23.0. Specifically, injection sites were assessed for redness, bruising, swelling, itching, and pain using a numeric rating scale from 0 to 3 (0 = none, 1 = mild, 2 = moderate, 3 = severe). The total score was the sum of these points, ranging from 0 to 15. If an injection site reaction reached a total score sum ≥ 2, it was recorded as an AE. The injection site assessment was conducted at six time points (pre-dose, immediately post-dose, 15 minutes (min) post-dose, 24 h, 48 h, and 96 h post-dose). For the pain evaluation during injection site assessment, a visual analogue scale (VAS; 0 mm = no pain; 100 mm = intolerable pain) was used and the readouts were categorized as no pain (scale < 10 mm), mild pain (scale ≥ 10 to < 50 mm), moderate pain (scale ≥ 50 to < 70 mm), and severe pain (scale ≥ 70 mm) with pain defined to interfere with life if VAS was ≥ 50 mm [9,10,11].
Immunogenicity Evaluation
Blood samples for immunogenicity were collected on day 1 (pre-dose), day 26, and day 57 to detect ADAs and NAbs to ADL. The samples for immunogenicity were analyzed using the Meso Scale Discovery® platform (Rockville, MD, USA) with acid dissociation to release any ADAs complexed with free drug.
Statistical Methods
The original sample size of 232 subjects to achieve 90% power at a 5% dropout rate was reduced to 188 subjects to achieve 85% power at a 1% dropout rate. This change was implemented as a protocol amendment during the COVID-19 pandemic in accordance with local conditions. A sample size of 93 completing subjects per treatment group provided 85% power to detect a 20% difference in PK between the test and reference investigational product based on the assumption of 5% difference in true geometric means between test and reference group and inter-subject percent coefficient of variation (CV%) of 46%. Considering AUCinf and Cmax as the primary endpoints for this clinical study, 41% inter-subject CV% was used as a conservative approach, based on known variability of PK parameters in previous studies involving SB5-LC [5, 6]. An additional 5% uncertainty was added to account for the different concentration of the new formulation SB5-HC. Sample size was calculated in respect to two one-sided t tests, each at 5% significance level.
The safety set consisted of all subjects who received the study drug, and the PK analysis set consisted of all subjects who were included in the safety set and had at least one PK sample analyzed without any major protocol deviation that has an impact on PK analysis.
Statistical analyses of primary endpoints were based on an analysis of variance (ANOVA) model with treatment as fixed effect. The difference in geometric least squares means (LSMeans) of primary endpoints between the SB5-HC and SB5-LC group and the associated 90% confidence intervals (CIs) for the ratio of the geometric LSMeans were determined. Back transformation provided the ratio of geometric LSMeans and 90% CIs for these ratios. Equivalence of the primary endpoints was determined if the 90% CI for the ratio of geometric LSMeans of the SB5-HC to SB5-LC group was within the acceptance interval of 0.80–1.25. A subgroup analysis was conducted to investigate impact of post-dose ADA status on PK parameters. The post-dose ADA status was defined as positive for subjects with at least one ADA positive test post-baseline, and defined as negative for subjects without any ADA positive test post-baseline. PK parameters were calculated using Phoenix® WinNonlin® version 8.2 (Certara, Palo Alto, CA, USA).
Results
Subject Disposition and Baseline Characteristics
A total of 411 subjects were screened, of whom 188 received a single dose of study drug (SB5-LC, n = 94; SB5-HC, n = 94) (Fig. 1). Mean age was 38.4 years (range, 18–55 years) and baseline characteristics were comparable between the treatment groups (Table 1). All randomized subjects completed the study. One subject in the SB5-HC group was excluded from the PK analysis set for major protocol deviation (i.e., not being withdrawn in the event of confirmed COVID-19).

CONSORT Subject disposition. One subject in the SB5-HC group was excluded from the PK analysis set for a major protocol deviation (i.e., for not being withdrawn in the event of confirmed COVID-19). PK = pharmacokinetic; SB5-HC = 40 mg/0.4 ml SB5 in a pre-filled syringe; SB5-LC = 40 mg/0.8 ml SB5 in a pre-filled syringe
Pharmacokinetic Evaluation
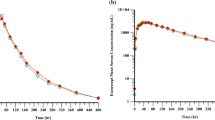
The mean serum concentration–time profiles were superimposable between SB5-LC and SB5-HC (Fig. 2) and the mean values and ranges of PK parameters were comparable between treatments (Table 2). Maximum serum concentrations (Cmax) were reached between 24 and 410 h after injection. The 90% CIs of the geometric LSMean ratios for AUCinf, Cmax, and AUClast were fully contained within the pre-defined bioequivalence margin of 0.80–1.25 (Table 3). The geometric LSMean ratios (90% CI) of the primary endpoints AUCinf and Cmax for SB5-HC compared with SB5-LC were 0.920 (0.8262–1.0239) and 0.984 (0.9126–1.0604), respectively.

Mean adalimumab serum concentrations versus nominal times on a linear and a semi-logarithmic scale (PK analysis set). Mean serum concentrations versus nominal times on linear (top graph) and semi-logarithmic scale (bottom graph) of SB5-HC (40 mg/0.4 ml) and SB5-LC (40 mg/0.8 ml)
For one subject in the SB5-LC group, only Cmax, Tmax, and AUClast were included in the PK analysis as the regression slope, necessary to determine the remaining parameters, could not be reliably estimated because most of the measured serum concentrations were below the lower limit of quantification during the elimination phase.
Safety Evaluation
The proportion of subjects who experienced a treatment-emergent adverse event (TEAE) was comparable between the SB5-HC and SB5-LC groups (42 subjects [44.7%] and 48 subjects [51.1%], respectively; Table 4). All TEAEs were mild or moderate in severity and no TEAE led to study discontinuation. There were no SAEs reported. The most frequently reported TEAE in both treatment groups was headache (10.6% in the SB5-HC and 12.8% in the SB5-LC group, Table 4). The proportion of subjects who experienced TEAEs considered to be related to the study drug was 18.1% in the SB5-HC and 29.8% in the SB5-LC group. The most frequently reported TEAEs in any treatment group considered to be related to the study drug were injection site reaction (score of ≥ 2 according to the rating scale), headache, and nausea. The incidence of injection site reactions in the SB5-HC group was lower than that in the SB5-LC group (four subjects [4.3%] vs. ten subjects [10.6%]). The mean VAS scores immediately after injection were 3.1 ± 4.98 mm in the SB5-HC group and 9.2 ± 12.98 mm in the SB5-LC group. The mean VAS scores 15 min after injection were 0.5 ± 1.34 mm in the SB5-HC group and 1.7 ± 3.55 mm in the SB5-LC group (Supplementary Table 1). Laboratory data, vital signs, and ECG parameters did not show clinically relevant changes that might be considered related to the study drug over time.
The overall incidence of subjects with post-dose ADAs to ADL was comparable between the two groups (Table 5), 93.6% of subjects in the SB5-HC group and 94.7% of subjects in the SB5-LC group tested positive for the presence of ADAs post-dose. The overall incidence of subjects with post-dose NAbs to ADL among those who had a positive ADA result was comparable between the SB5-HC and SB5-LC formulations (75.0 and 67.4%, respectively; Table 5). A subgroup analysis by post-dose ADA status showed no impact of ADA positivity on the equivalence of SB5-HC and SB5-LC. PK parameters such as AUCinf, AUClast, Cmax, CL/F and t1/2 were comparable between the two treatment groups within each ADA-positive and ADA-negative subgroups (Supplementary Table 2).
Discussion
This randomized, single-blinded, two-arm, parallel-group bridging study was designed to demonstrate PK equivalence between SB5-HC and SB5-LC in healthy male subjects. All participants received a single subcutaneous injection of 40 mg ADL as either higher concentration (40 mg/0.4 ml, SB5-HC) or lower concentration (40 mg/0.8 ml, SB5-LC) formulation of ADL. The study results demonstrated that the two formulations showed comparable PK characteristics. In addition, safety and immunogenicity profiles were also comparable between both groups.
For the primary endpoints AUCinf and Cmax, the 90% CI for the ratio of geometric LSMean of SB5-HC and SB5-LC were within the pre-defined bioequivalence margins of 0.80 to 1.25, indicating that the two treatments were bioequivalent. All remaining PK parameters were comparable between both treatment groups, consistent with the previous phase I PK studies of SB5-LC which demonstrated PK equivalence of SB5-LC to both EU-sourced and US-sourced reference ADL [5].
In this study, a single dose of SB5-HC or SB5-LC administered to healthy male subjects showed comparable safety and tolerability between the treatment groups. The proportion of subjects who experienced TEAEs was comparable between the SB5-HC and SB5-LC groups. The most frequently reported TEAEs considered to be related to the study drug were injection site reaction, headache, and nausea, which all resolved spontaneously by the end of the study visit. No severe TEAEs were reported and no subjects discontinued because of a TEAE. Furthermore, no safety concerns were found based on clinical laboratory evaluations, vital signs, and 12-lead ECGs.
The incidence and severity of injection site reactions were low in both the SB5-HC and SB5-LC group throughout the study while lower incidence was found in the SB5-HC compared to the SB5-LC group. In terms of pain VAS intensity, the largest difference was found immediately after injection when the highest intensity was measured in both groups. Those reactions mostly diminished in both groups after 15 min (Supplement 1). Factors known to contribute to subcutaneous injection site reactions can be grouped into product-related and patient-related factors (e.g., low body weight or needle phobia) [12]. The product-related factors include the formulation (ingredients, pH, or buffers), injection volume, needle gauge size, or type of device. Although the amount of the active ingredient is the same (i.e., 40 mg ADL), SB5-HC is different from SB5-LC by means of 50% reduced injection volume (i.e., 40 vs. 80 ml) and excipients (e.g., citrate-free). With high injection volume and certain excipients being potential triggers of injection site pain [13,14,15], the lower volume and difference in the formulation may be the reason for the reduction of injection site related reactions.
The reduction of injection-site reactions and pain may positively affect treatment adherence in patients with chronic conditions who require repeated dosing [16]. Currently, the recommended subcutaneous dosage of ADL for adult patients with rheumatoid arthritis, psoriatic arthritis, or ankylosing spondylitis is 40 mg administered every other week [2, 3]. As previous reports showed that reduced injection site pain, achieved by low-volume or citrate-free formulations and other means, significantly increased patients’ compliance [17, 18], a newly developed, low-volume and citrate-free formulation of SB5 (SB5-HC) may contribute to a better patient adherence to treatment.
Limitations of the present study include factors related to the study population. Only healthy male subjects were selected because they were considered to be a more homogeneous and represent a sensitive population to compare PK characteristics than patients with various disease-related factors [19].
Conclusions
A newly developed, high-concentration, low-volume, citrate-free formulation of SB5 (SB5-HC) demonstrated PK bioequivalence with comparable safety and immunogenicity characteristics to the low-concentration, high-volume formulation of SB5 (SB5-LC) in healthy male subjects.
References
Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol. 2016;12(1):49–62.
EMA. Humira®: EPAR—Product Information; Annex I, Summary of product characteristics. EMA; 2021.
FDA. Humira®: Prescribing Information. FDA; 2021.
Frampton JE. SB5: an adalimumab biosimilar. BioDrugs. 2018;32(5):507–10.
Shin D, Lee Y, Kim H, Kornicke T, Fuhr R. A randomized phase I comparative pharmacokinetic study comparing SB5 with reference adalimumab in healthy volunteers. J Clin Pharm Ther. 2017;42(6):672–8.
Shin D, Lee Y, Jeong D, Ellis-Pegler R. Comparative pharmacokinetics of an adalimumab biosimilar SB5 administered via autoinjector or prefilled syringe in healthy subjects. Drug Des Devel Ther. 2018;12:3799–805.
Weinblatt ME, Baranauskaite A, Niebrzydowski J, Dokoupilova E, Zielinska A, Jaworski J, et al. Phase III randomized study of SB5, an adalimumab biosimilar, versus reference adalimumab in patients with moderate-to-severe rheumatoid arthritis. Arthritis Rheumatol. 2018;70(1):40–8.
Weinblatt ME, Baranauskaite A, Dokoupilova E, Zielinska A, Jaworski J, Racewicz A, et al. Switching from reference adalimumab to SB5 (Adalimumab Biosimilar) in patients with rheumatoid arthritis. Arthritis Rheumatol. 2018;70(6):832–40.
Jensen MP, Smith DG, Ehde DM, Robinsin LR. Pain site and the effects of amputation pain: further clarification of the meaning of mild, moderate, and severe pain. Pain. 2001;91(3):317–22.
Palos GR, Mendoza TR, Mobley GM, Cantor SB, Cleeland CS. Asking the community about cutpoints used to describe mild, moderate, and severe pain. J Pain. 2006;7(1):49–56.
Turner JA, Franklin G, Heagerty PJ, Wu R, Egan K, Fulton-Kehoe D, et al. The association between pain and disability. Pain. 2004;112(3):307–14.
St Clair-Jones A, Prignano F, Goncalves J, Paul M, Sewerin P. Understanding and minimising injection-site pain following subcutaneous administration of biologics: a narrative review. Rheumatol Ther. 2020;7(4):741–57.
Anderson G, Meyer D, Herrman CE, Sheppard C, Murray R, Fox EJ, et al. Tolerability and safety of novel half milliliter formulation of glatiramer acetate for subcutaneous injection: an open-label, multicenter, randomized comparative study. J Neurol. 2010;257(11):1917–23.
Nash P, Vanhoof J, Hall S, Arulmani U, Tarzynski-Potempa R, Unnebrink K, et al. Randomized crossover comparison of injection site pain with 40 mg/0.4 or 0.8 ml formulations of adalimumab in patients with rheumatoid arthritis. Rheumatol Ther. 2016;3(2):257–70.
Taub KJ, Blake PG, Langlois S, Jindal KK. A double-blind, randomized, crossover study of the local tolerability of erythropoietin alfa formulations in dialysis patients. Can J Hosp Pharm. 1999;52:24–9.
Beer K, Muller M, Hew-Winzeler AM, Bont A, Maire P, You X, et al. The prevalence of injection-site reactions with disease-modifying therapies and their effect on adherence in patients with multiple sclerosis: an observational study. BMC Neurol. 2011;11:144.
Bergman M, Patel P, Chen N, Jing Y, Saffore CD. Evaluation of adherence and persistence differences between adalimumab citrate-free and citrate formulations for patients with immune-mediated diseases in the United States. Rheumatol Ther. 2021;8(1):109–18.
Salaffi F, Di Carlo M, Farah S, Carotti M. Adherence to subcutaneous anti-TNFα agents in patients with rheumatoid arthritis is largely influenced by pain and skin sensations at the injection site. Int J Rheum Dis. 2020;23(4):480–7.
EMA. Guideline on similar biological medicinal products containing monoclonal antibodies—non-clinical and clinical issues. EMA; 2012.
Acknowledgements
Funding
Planning, conduct, and analysis of the study as well as the journal’s Rapid Service Fee were funded by Samsung Bioepis Co., Ltd.
Medical Writing, Editorial, and Other Assistance
Medical writing support was provided by SFL Regulatory Affairs & Scientific Communications, Switzerland, and funded by Samsung Bioepis Co., Ltd.
Author Contributions
SS and MK were responsible for study concepts and design; YM and SH were responsible for data acquisition; SS, MK, YM were responsible for data analysis and interpretation; SS and MK were responsible for manuscript preparation; all authors were responsible for manuscript review and approval.
Disclosures
So-shin Ahn, Minkyung Lee, Yumin Baek, and Sukho Lee are employees of Samsung Bioepis Co., Ltd. There are no other relationships or activities that could appear to influence the submitted work. The results of this study have been submitted to EULAR 2022 and accepted as a poster presentation (POS0641).
Compliance with Ethics Guidelines
The final study protocol was approved by the Independent Ethics Committee (Ethics committee of the state of Berlin, State Office of Health and Social Affairs Berlin, Germany). This study was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki (1996) and that are consistent with the latest International Council for Harmonization Good Clinical Practice guidelines (ICH E6) and applicable local regulatory requirements and laws. The nature and purpose of the study was fully explained to each subject and written informed consent was obtained from each subject before the subject was entered into the study. The consent documents for the study were reviewed and approved by the appropriate IEC prior to use.
Data Availability
Upon request, and subject to certain criteria, conditions, and exceptions, Samsung Bioepis will provide access to individual de-identified participant data to researchers whose proposals meet the research criteria and other conditions and for which an exception does not apply. Proposals should be directed to the corresponding author. For access, data requestors must enter into a data access agreement with Samsung Bioepis.
Thanking Volunteer Subjects
The authors are grateful to the volunteers for participating in the clinical trial.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Ahn, Ss., Lee, M., Baek, Y. et al. A Randomized Pharmacokinetic Study in Healthy Male Subjects Comparing a High-concentration, Citrate-free SB5 Formulation (40 mg/0.4 ml) and Prior SB5 (Adalimumab Biosimilar). Rheumatol Ther 9, 1157–1169 (2022). https://doi.org/10.1007/s40744-022-00471-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-022-00471-8