
Abstract
Purpose
Adrenal incidentalomas (AIs) are incidentally discovered adrenal masses, during an imaging study undertaken for other reasons than the suspicion of adrenal disease. Their management is not a minor concern for patients and health-care related costs, since their increasing prevalence in the aging population. The exclusion of malignancy is the first question to attempt, then a careful evaluation of adrenal hormones is suggested. Surgery should be considered in case of overt secretion (primary aldosteronism, adrenal Cushing’s Syndrome or pheochromocytoma), however the management of subclinical secretion is still a matter of debate.
Methods
The aim of the present narrative review is to offer a practical guidance regarding the management of AI, by providing evidence-based answers to frequently asked questions.
Conclusion
The clinical experience is of utmost importance: a personalized diagnostic-therapeutic approach, based upon multidisciplinary discussion, is suggested.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adrenal incidentalomas (AIs) are incidentally-found lesions during radiological investigations unrelated to adrenal-related disorders. The availability of imaging techniques, especially computed tomography (CT) and magnetic resonance (MR), enabled the increased detection of AIs over the years. In most cases, AIs are non-functioning benign formations (cortical adenomas) that do not require treatment, however they represent an important challenge for differential diagnosis among several benign and malignant pathologies, with a wide spectrum of endocrine activity (adrenal secretion is a continuum from non-functioning to overt-secreting forms) [1,2,3,4].
The prevalence of AIs, derived from autopsy data, is variable according to age: < 1% in young people, 3% in middle-aged adults and > 15% in subjects over 70 years old [5, 6]. Considering radiological studies, the prevalence in middle-aged subjects is 2–4%, up to > 10% in patients aged 70 or over. It is slightly more frequent in women, with a moderate prevalence on the right side [3]. In 10–15% of cases AIs are bilateral. As far as size is concerned, most authors consider “incidentaloma” only a lesion > 1 cm in diameter [2].
Question 1: Is it malignant?
The first challenge for the endocrinologist is to promptly recognize a rare malignant form among the majority of benign lesions.
The adrenal glands have one of the greatest blood supply rates per gram of tissue [7]. Therefore, adrenal masses can be metastases of several primary tumors, especially if bilateral. Clinical history (positive for extra-adrenal malignancy) and patient’s presentation (age, rapid weight loss) should be ascertained before imaging. A recent population-based cohort study reported a 7.5% prevalence of metastasis in AIs, and that malignancy was 22 times more likely when the AI was discovered during cancer staging [8]. In patients with Colo-Rectal Cancer, the incidence of AIs was 10.5% (in 475 subjects); however, adrenal metastases could be ruled out in most cases (96%) with CT re-evaluation, follow-up imaging and multidisciplinary evaluation [9]. However, it is personal opinion of the authors that if an imaging study was performed during the evaluation of known extra-adrenal cancer, the AI does not fall into the definition of “incidentaloma”, because the imaging itself has been performed in a patient with an active malignancy, therefore the pre-test probability that the adrenal lesion represents a metastasis is high.
Morphological evaluation by CT and MR is of utmost importance to describe the shape and size of the lesion. Non-secreting cortical adenomas contain significant amounts of intracellular lipids. There is an inverse relationship between lipid concentration and the attenuation value obtained with un-enhanced CT: a signal intensity < 10 Hounsfield Units (HU) is suggestive of benign lipid-rich adenoma [2, 10, 11]. This approach fails to recognize up to 40% of benign adenomas (the so-called lipid-poor adenomas, with HU > 10) [12, 13]. A different un-enhanced CT attenuation value has been observed in patients with secreting adenoma: an overt cortisol secretion (defined with increased urinary cortisol excretion) may decrease the intracytoplasmic lipid droplets. 80% of cortisol-secreting adenomas have an un-enhanced attenuation value > 10 HU, and 65% ≥ 20 HU: doubling of the cortisol secretion is associated with a 4 HU increase [14]. In patients with subclinical cortisol secretion, unsuppressed serum cortisol after dexamethasone shows a positive correlation with adenoma diameters, morphologic parameters differ between secreting and non-secreting adenomas, either lipid-rich or lipid-poor [15]. In clinical practice, lipid-poor AIs are a heterogeneous group of tumors, and one third of those masses may have a borderline-malignant potential features, expressed by high Weiss or Lin-Weiss-Bisceglia score [16]. There is still debate whether the MR is better than the CT scan [2, 6, 17]. MR is used to characterize lipid-poor adenomas, especially when the appropriate methods (chemical shift among with in-phase and out-of-phase) are used [12].
In suspected cases, it is important to measure the wash-out time during enhanced CT: a wash-out > 60% in arterial phase or a relative washout > 40% in the delayed images (15 min for the venous phase) suggest a benign form with sensitivity of 82–97% and specificity of 92–100% [2, 18]. Azoury et al., in a retrospective study, considered patients who underwent unilateral adrenalectomy for an adrenal mass in 10 years (2005–2015): they achieved 100% specificity to predict adenomas considering benign features on preoperative CT (n = 143 with HU < 10, well-defined borders, homogeneous texture, absence of necrosis or calcifications, rapid washout on dynamic protocol) [19]. Recently, a European retrospective study collected attenuation values (HU) and absolute/relative washout in a cohort of patients with pheochromocytoma (PHEO). Only two out of 376 patients with PHEO presented a low attenuation value (< 10 HU), therefore Authors concluded that it seems reasonable to abstain from biochemical testing for PHEO in case of AI with an unenhanced attenuation value ≤ 10 HU [20].
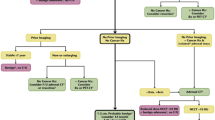
At CT, the adenoma appears rounded, homogeneous, rich in lipids and with well-defined margins. Malignant lesions are often > 4–6 cm in diameter, present with irregular margins and texture, calcifications, necrosis and in some cases with invasion of surrounding structures. Regarding MR, the malignant forms are hypo-intense in the T1 and hyper-intense in the T2 images, showing an intense enhancement and a delayed washout. A T2 signal hyper-intensity (the light-bulb sign) is common in PHEO [10]. Nevertheless, a wide spectrum of imaging is reported: PHEOs may mimic other benign or malignant adrenal lesions [21]. As indicated in Fig. 1, we suggest contrast-enhanced CT or MR in case of suspected AI.

Suggested flow-chart at baseline visit in patients with adrenal incidentaloma. CT computed tomography, HU Hounsfield Units, MR magnetic resonance, ACC adreno-cortical carcinoma, PET positron emission tomography
A 18–fluoro–2–deoxy–d–glucose (18F-FDG) positron emission tomography (PET) alone or combined with CT could be useful to characterize malignant forms. An adrenal mass is likely malignant when the uptake of 18-FDG is higher than that of the liver. However, false positives (sarcoidosis, tuberculosis, lipid-poor adenomas and PHEO) or negatives (necrotic-hemorrhagic areas present in malignant lesions) must be considered [22,23,24,25,26]. PET allowed to refine the diagnosis of pheochromocytoma by using 18F-meta-fluorobenzylguanidine, 18F-DOPA PET/CT, 18F-Fluorodopamine or 11C-hydroxyephedrine [27].
In addition to morphological diagnostic techniques, in case of indeterminate mass after an extensive biochemical and radiological assessment, a biopsy can be indicated in selected cases, after ruling out a PHEO. Nonetheless, adrenal biopsy presents low diagnostic value [28], a high-risk of cells spreading, and is not suggested in the study of AI [2].
In clinical practice, a close collaboration with an adrenal-dedicated radiologist is suggested, especially in a multidisciplinary team. In Fig. 2, we depicted a case discussed in the monthly multidisciplinary team dedicated to adrenal disease in Padova.

a Un-enhanced CT: right adrenal mass, 30 HU. b, c MR with chemical shift in-phase b and out-of-phase c without signal drop. d, e Enhanced MR with heterogeneous tissue and necrosis f 18-FDF PET with high uptake (SUV max 16). After surgery the histological examination was consistent with adrenal adenoma
Answer to question 1: Malignancy must be assessed at the baseline visit in patients with AI. Patient’s presentation (age and history of extra-adrenal malignancy) and radiological evaluation (attenuation value > 10 HU in non-secreting mass, delayed washout, increased 18-FDG PET uptake) are markers of suspicion. In all suspected cases, a multidisciplinary discussion is suggested.
Question 2: Is it hormonally active?
Considering AIs, non-secreting adenomas represent the largest part (70–80%), followed by glucocorticoid (CS), catecholamine (PHEO) and to a lesser extent mineralocorticoid secretion (Primary Aldosteronism, PA). Regardless of clinical aspect, the search for an increased endocrine production is generally carried out in all patients at baseline.
Considering cortisol excess, the hypothalamic–pituitary–adrenal (HPA) axis should be investigated in all patients presenting with an AI [2, 29,30,31,32]. Subclinical hypercortisolism does not present by definition any of the specific symptoms that characterize patients with overt Cushing’s syndrome (CS, as moon face, easy bruising, purple striae, cervical fat pad, and so on) [33]. Therefore, most authors preferred to use the term subclinical hypercortisolism, rather than subclinical CS or preclinical CS, because CS represents by definition a well-defined set of symptoms and the progression from non-secreting towards a clinical form is very rare [34]. These definitions have disappeared from the most recent Guidelines, replaced by autonomous cortisol secretion (ACS) [29]. The incidence of ACS is not well defined, because different diagnostic criteria have been adopted; it seems to be between 5 and 30% in most papers [35, 36], reaching even higher percentages in selected series [2, 3, 37, 38]. As a matter of fact, the lack of signs of CS or clinical features (hypertension, obesity, metabolic disorders, bone disease and so on) is not sufficient to define a non-functioning adenoma [37, 39].
The diagnosis of overt hypercortisolism (CS) is based upon first-line screening tests: urinary-free cortisol (UFC), dexamethasone suppression test (DST) and late-night salivary cortisol (LNSC) [33, 40]. To reduce the risks of false positives, and in agreement with many other authors, we consider a high likelihood of overt CS the alteration of at least two screening tests [2, 3, 29]. In case of overt cortisol secretion, low-suppressed ACTH levels are used to confirm adrenal origin [40, 41].
In normal subjects, a supra-physiological dose of glucocorticoids prompts the suppression of cortisol secretion: the DST explores the HPA axis negative feedback [42]. The overnight 1-mg DST is considered the most sensitive screening test in AI, even if there is still no complete agreement regarding cortisol cut-off. The Endocrine Society guidelines suggest to use the 50 nmol/L (1.8 μg/dL) cut-off to exclude overt hypercortisolism [31]. NIH and other scientific societies recommend the 138 nmol/L (5 μg/dL) to avoid false positives [2, 30, 32], also intermediate thresholds have been proposed (Chiodini et al. suggests 3 μg/dL as the best compromise [37]). The European Society of Endocrinology (ESE) Guidelines in collaboration with the Working Group of the European Network for the Study of Adrenal Tumors (ENS@T) consider ACS, amenable of surgical management in case of comorbidities, if cortisol after 1-mg DST is > 138 nmol/L, and possible ACS in the grey-zone 50–138 nmol/L (in such cases further biochemical tests to confirm cortisol secretion might be required, as UFC, LNSC or ACTH) [29]. At 50 nmol/L the sensitivity for CS is high (up to 100%), at the expense of a low specificity (< 85–90% [43, 44]). To add further insights, even patients with cortisol < 50 nmol/L after 1-mg DST might present some cortisol-related comorbidities, as increased insulin resistance and endothelial dysfunction [45]. The DST provide false positive results if cortisol binding globulin (CBG) levels are increased (as in case of estrogen therapy); on the other hand, falsely negative in case of hypoalbuminemia and low levels of CBG are reported. Moreover, inaccurate tests are not so rare, due to the interference of drugs that can accelerate or reduce the liver metabolism of dexamethasone [46]. To avoid these diagnostic pitfalls, combined cortisol and dexamethasone measurement after DST have been proposed [47,48,49].
ACTH, LNSC and UFC are used to assess HPA axis, however, they are conditioned by methodological issues. UFC is accurate in diagnosing overt CS, especially when measured with liquid chromatography tandem-mass spectrometry (LC–MS) [50, 51], however, it can be increased in physiologic/non-neoplastic hypercortisolism (obesity, depression, diabetes, pseudo-CS state [52,53,54,55]) and sometimes normal also in overt CS [50]. A poor diagnostic accuracy of LNSC to detect ACS has been documented, even if LC–MS methods are used [56,57,58]. Loss of circadian cortisol rhythm is a peculiar feature of overt CS, and LNSC should not be used in AI. ACTH measurement requires careful pre-analytical sampling and analytical procedures: commercially-available ACTH immunoassays are imprecise in the normal-low range (< 20 pg/mL) [59]: in selected cases, a human CRH test might unmask the ACTH responsiveness of ACTH-dependent hypercortisolism [60]. Moreover, also sensitivity of glucocorticoid receptor can affect the interpretation of first-line screening tests [61, 62]. Cortisol secretion is a continuum: no cut-off can unequivocally distinguish the physiological HPA axis from its abnormal activation (pseudo-CS) or overt CS. A post-surgical adrenal insufficiency may occur also in patients with “non-functioning” adrenal adenomas, because first line-screening tests can fail to recognize a mild cortisol secretion [63, 64]. The adrenal scintigraphy with 131I-19 norcholesterol should be considered to identify those patients who could benefit from adrenalectomy; however, it is of limited availability [65].
PA represents the most frequent form of endocrine hypertension. It affects 6% of the hypertensive population (up to 20% in selected cohorts). Patients with PA presents with suppressed renin, normal-elevated aldosterone concentration and hypokalemia in some cases [66]. Despite the relatively high prevalence of PA in the hypertensive population, its finding among AI is relatively low, ranging from 1.5 to 7% [2, 67]. Adrenal lesions of patients investigated with abdominal imaging in search of the causes of secondary hypertension should not considered “incidentalomas” [68]. Moreover, some guidelines recommend exploring mineralocorticoid secretion only in hypertensive patients with AI [2, 30], thus excluding those with normal/borderline blood pressure, in which an autonomous secretion of aldosterone could not be excluded a priori [69, 70]. It has been reported that forms of subclinical hyperaldosteronism are not so rare in patients with AI, especially in those with increased diastolic blood pressure levels [71]. Recent acquisitions have radically changed the classic division of the adrenal cortex into zones: clusters of aldosterone-producing cells are found in normal adrenals and close to the aldosterone-secreting tumors [72].
Aldosterone to renin ratio (ARR) is recommended by the Endocrine Society guidelines as the most reliable test for PA screening [66]. Serum potassium levels should be normalized and interfering drugs should be discontinued before ARR: all diuretics must be suspended for 4–6 weeks; angiotensin converting enzyme inhibitors, angiotensin receptor blockers and beta blockers require 2–4 weeks of withdrawal [66]. Recently, it has been reported that an early mineralcorticoid-receptor antagonists treatment did not seem to endanger the accuracy of ARR [73]. After the diagnosis of PA, confirmatory tests and PA subtyping (monolateral vs bilateral disease) are suggested [66], because a non-functioning AI may coexist with bilateral PA, in order to avoid an unnecessary adrenalectomy [74, 75].
PHEO represents about 7% of AIs as reported by the ESE-ENS@T guidelines [29], with an even higher prevalence for other authors [76], and a modern presentation of PHEO (despite the classic triad of headaches, sweating and palpitation) has been suggested in patients with AI [77]. With the currently available tests (metanephrines and accurate imaging), a diagnosis of PHEO should be rule-out at diagnosis in all patients with AI, especially before surgery [78].
Lenders et al. reported that 30% of PHEOs are incidental, and that the prevalence of the tumor is increasing over time [79]; these data are confirmed also in other series [80, 81], especially in large AIs. The clinical history reports hypertensive episodes only in a modest percentage of cases, therefore all guidelines suggest to test for PHEO all patients with AI [2, 29, 30, 32]. Assessment of plasma or urinary metanephrines levels are the screening test in a patient with suspected PHEO [82]. The diagnostic accuracy of metanephrines is higher than that of catecholamines, with reduced false positives, especially in the elderly. In this regard, recent data from Eisenhofer et al. offer specific age-related reference intervals for both plasma and urinary metanephrines [83]. The biochemical diagnosis of PHEO could be conditioned by numerous interfering factors: stress, increased sympathetic activity (myocardial infarction, stroke, hypoxia), the use of antidepressants, antipsychotics, beta blockers, antibiotics and glucocorticoid [82]. Moreover, also some foods (bananas, cheeses, coffee, yogurt, cured meats, soya sauce, red wine, fish, chocolate, figs, fava beans) should be avoided for 3–5 days before urinary collection [68].
The dosage of adrenal androgens, its precursors and metabolites in patients with AIs is commonly reserved for suspected cases of Congenital Adrenal Hyperplasia (often associated with normal-low cortisol and normal-high ACTH levels) [84, 85]. On the other hand, the androgens and adrenal sex hormones should be considered in case of rapid-onset hirsutism in female, because adrenocortical cancer (ACC) could be a rare but reasonable diagnosis [18].
An emerging approach is the measurement of urinary steroid metabolites: 24-h urine steroid excretion allow the identification of different steroid patterns, especially if measured with mass spectrometry [86]. In patients with PA, a glucocorticoid co-secretion is frequently found, contributing to metabolic risk [87] and ventricular hypertrophy [88]. The combination of CT and a panel of 15-steroid profile had the same accuracy of vein sampling to differentiate unilateral PA (amenable of surgical remission) from bilateral adrenal hyperplasia [89]. In case of ACTH-dependent CS there is a lower androgen precursors’ and a higher 11-dexoycorticosterone secretion than ACTH-dependent forms of hypercortisolism [90]. In patients with AIs, 21-deoxycortisol and 11-deoxycorticosterone after ACTH test were increased in ACS [91], and cortisol and corticosterone after 1-mg DST were associated with higher prevalence of severe-resistant hypertension and higher incidence of cardiovascular events [92]. An immature steroidogenesis is able to identify ACC with a specific fingerprint [93].
Answer to question 2: According to available literature, urinary metanephrines, ARR (after appropriate drug washout) and serum cortisol after 1-mg DST are suggested in all patients with an AI.
Question 3: Is surgery an appropriate treatment in patients with ACS?
Surgery is the suggested therapy for AI in patients with secreting adenoma, suspicion of malignancy or in case of ACS and cortisol-related comorbidities (hypertension, impaired glucose metabolism, obesity, dyslipidemia, osteoporosis) in the ESE-ENS@T Guidelines [18]. It seems a clear and easy-to-manage indication; however, if the diagnosis of overt CS (with the full-blown complete clinical picture) could be plain, the appropriate detection of ACS and its cortisol-related comorbidities requires extensive clinical practice. There is a greater prevalence of cortisol-related comorbidities in patients with AI (either non-functioning or ACS), compared to normal control subjects [94,95,96,97,98,99]. Therefore, the reversal of cortisol-related comorbidities through the surgical management of ACS seems a reasonable choice.
Beneficial effects of adrenalectomy on cardiovascular risk in patients with ACS are not evidence-based, yet. From a clinical perspective, most patients are aged and have longstanding comorbidities, thus with limited improvement after surgery. On the other hand, the beneficial effect of surgery is based upon uncontrolled retrospective studies, where a selection-bias (versus conservative management in old or worst-prognosis patients) could not be ruled-out. Toniato et al. in one of the first prospective study described an improvement in metabolic and cardiovascular parameters after surgery [100], confirmed by other authors [101, 102]. In a systematic review, Iacobone et al. selected six retrospective and one prospective studies, including 230 patients: surgical treatment was able to normalize hypercortisolism, improving blood pressure, glucose metabolism and obesity in 72, 46 and 39% of patients, respectively, when compared with the conservative-treatment group [103]. Petramala et al. evaluated 70 patients with ACS: only those treated by unilateral adrenalectomy (n = 26) showed a significant decrease in blood pressure levels [104].
A metanalysis of 26 papers (584 patients) comparing surgery and conservative management in patients with ACS, reported that adrenalectomy improves arterial hypertension, diabetes, dyslipidemia and obesity [105]. However, the collection of heterogeneous studies resulted in a low-to-moderate level of evidence. In the meta-analyses, as in clinical practice, the criteria to define ACS were different. Moreover, not only the confidence intervals were large, suggesting a significant individual variability, but also inconsistent definitions of comorbidities and the degrees of improvement limited the final accuracy of the outcome. Even patients with non-functioning AI showed an improvement in blood pressure after adrenalectomy [106]. Most of the non-functioning AI remain asymptomatic for life, and the natural history of non-functioning or subclinical forms is still poorly understood and follow-up too limited [107, 108].
Taking into account that the main consequences of subclinical hypercortisolism are arterial hypertension, metabolic disorders and diabetes, the New Zealand group coordinated by Goh et al. suggests to evaluate case by case before engaging in too many investigations and wasting unnecessary resources [109]. We must keep in mind that in ACS the cortisol-related comorbidities, such as cardiovascular risk factors, impaired glucose or lipid metabolism, bone disease and the impaired quality of life [62, 110,111,112,113,114,115], are related to modest changes of cortisol secretion.
In a follow-up study (average of 7.5 years), Di Dalmazi et al. observed that in patients with stable/intermediate phenotype of hypercortisolism and in those with ACS the overall survival (considering all causes of death) was lower than in non-secreting AIs [96]. Similar results have been reported by De Bono et al.: mortality from cardiovascular and infectious diseases was increased in cases of ACS [97]. In light of these literature data, which have not been adequately considered by the latest ESE-ENS@T guidelines, Morelli et al. underline the need to follow all incidentalomas with adequate clinical and endocrine follow-up associated with a careful assessment of possible cardiovascular risks [116].
At present, there are no prospective randomized studies on cases of patients with functioning incidentaloma to undergo surgery or medical therapy (the drugs available are those reserved for overt CS), to define which is the best treatment.
To conclude, several factors must be considered before surgery in a patient with ACS (age, cortisol-related comorbidities [117, 118]) in a multidisciplinary team discussion, in order to identify those patients that would be most likely to benefit from surgery [18].
Answer to question 3: Surgery should be considered for patients with ACS and cortisol-related comorbidities. However, a comprehensive multidisciplinary discussion is suggested, in order to balance the benefit and the risk of surgery.
Question 4: Can a non-functioning lesion become ACS and a benign adenoma become malignant?
The natural history of AI is not fully reported: there is few evidence that non-functional and benign cortical adenomas can become functional, or even malignant [4, 119].
In a retrospective study, Pantalone et al. showed that an absolute increase of 0.8 cm, an annual growth of 0.64 cm or an increase in size > 25% are indicative of malignancy [120]. However, malignancy is reported also in cases without any significant growth in 3–16 months of follow-up [120]. In a meta-analysis of over 1000 patients with benign AI, two cases of malignancy were reported after 2–4 years [6]. Similar results can be found in a Turkish study: only one malignant node was found among 277 patients followed for 2 years (the adrenal mass increased from 24 to 84 mm in diameter in 6 months [121]). In a 4-year follow-up study of 77 patients, an increase > 0.5 cm has been observed in 30% and > 1 cm in ten cases; malignancy was not observed, four increased masses become functioning; a reduction was reported in six adenomas [122]. No evidence of malignancy or hyper-function has been reported in a Swedish 2-year follow-up study involving 126 patients [123]. Several studies reported the development of an overt secretion in up to 11% of cases [1, 121, 122], especially in masses > 3 cm [124]. Morelli V. et al.in a retrospective study of 206 patients concluded that masses > 24 mm in diameter present high-risk to develop ACS [125].
The meta-analysis of Cawood et al., including data collected between 1980 and 2008, reports an increase in size in 15% of AIs and a new-onset of hyper-function in 1% [4]. According to these data, it was subsequently suggested to limit the follow-up. Therefore, unlike the previous guidelines [30] which suggested a radiological control every 6–12 months for 1–2 years and then annually for 5 years, the ESE-ENS@T guidelines do not foresee any follow-up for smaller incidentalomas (< 4 cm), with benign features (< HU at CT) and non-functioning behavior at the baseline evaluation [18, 126].
A group of Finnish researchers, hypothesizing that a lipid-rich adenoma (HU < 10) requires a radiological follow-up after 5 years, described 54 patients with AI (< 4 cm) and with normal functional tests (two cases of hyperfunction). After 5 years the size of the lesion remained unchanged (from 19 ± 6 mm to 20 ± 7 mm), as well as the function. Only one case showed 8 mm growth, surgery confirmed a cortical adenoma [127]. A group of Korean researchers retrospectively tested 1149 patients (mean age 54 years) collected between 2000 and 2013 in a single center [119], according to the ESE-ENS@T guidelines 68% had non-functioning AI. To distinguish between benign and malignant masses it was calculated that the mass cut-off should have a diameter of 3.4 cm (with a sensitivity of 100% and specificity of 95%) and a HU value at CT of 19.9 (with sensitivity of 100% and specificity of 67.4%). Most of the non-functioning masses showed no change in size during 4 years of follow-up [119]. Considering the cut-off of 50 nmol/L after 1 mg DST, 28% of non-functioning AI developed an ACS during follow-up, no progression to an overt CS was observed.
Finally, in a recent study from New Zealand, 101 patients were followed for 3-years. Growth was observed in 5% of cases, malignancy was not documented. At the beginning of the study nine cases showed signs of cortisol excess. At 3 years, five of the nine cases of hyperfunction normalized, while the other five showed a subclinical cortisol secretion (5%) [109].
Since no cases of hyperaldosteronism have been reported during the numerous follow-up studies, it is quite common practice not to control the mineralocorticoid function after baseline evaluation. Also anecdotal cases of PHEO have been reported in previous studies with less accurate diagnostic tests [120,121,122, 124].
Answer to question 4: The progression to a malignant form is anecdotal. In some patients a non-functioning AI could evolve in ACS, therefore 1-mg DST is suggested in the follow-up.
Question 5: Is perioperative period a matter of concern?
Surgery of functioning adrenal tumors remains at a higher risk for comorbidities, also in patients with ACS, despite continuous evolution, technical improvements and reduction of perioperative risks. There are no evidence-based protocols, probably due to the heterogeneity of the cases and the limited number of patients reported.
Laparoscopic surgery, well-tolerated and accompanied by less morbidity, is effective in normalizing cortisol levels and reducing cortisol-related morbidity [128, 129]. Perioperative management is critical, not only for overt CS [130]: patients with ACS may present cortisol-related comorbidities that could increase surgical risk [110, 131, 132]. After adrenalectomy, replacement therapy with hydrocortisone is mandatory in CS, and should be considered also in patients with ACS, to avoid post-surgical adrenal insufficiency [64, 133]. HPA axis inhibition must be taken into account even in case of ACS or non-functioning incidentalomas, therefore blood pressure, serum glucose and sodium levels must be assessed in the perioperative period, as suggested in Fig. 3. The correct diagnosis of HPA axis before surgery is able to predict post-surgical adrenal insufficiency: 90% of patients diagnosed as cortisol-secreting according to strict criteria (DST + 2 screening tests) need replacement therapy against only 50% of cases diagnosed with DST alone [134]. Cortisol levels > 83 nmol/L after DST, increased UFC and ACTH < 10 pg/mL are the best parameters for predicting post-surgical hypoadrenalism [135].

Suggested management of adrenal incidentaloma. ACC adrenocortical cancer, PA primary aldosteronism, AVS adrenal vein sampling, CS Cushing’s syndrome, ARR aldosterone to renin ratio, PHEO pheochromocytoma, DST dexamethasone suppression test, CT computed tomography, MR magnetic resonance
In patients with PA, the normalization of blood pressure and potassium levels should be considered before surgery [136, 137]. Post-intervention data show changes in renal sodium reabsorption, vasodilatation of the arterioles, structural changes of the renal parenchyma and, in case of long-lasting PA, suppression of mineralocorticoids of the residual adrenal gland [138]. A subset of patients with PA have cortisol co-secretion [87], thus requiring glucocorticoid treatment.
The treatment of choice for PHEO is adrenalectomy [82, 139]. Surgery can be complicated by the sudden release of catecholamines due to anesthesia induction or manipulation of the adrenal mass, moreover, the tumor can be silent [81]. All patients with PHEO needs adequate preparation before surgery with α-adrenergic receptor blockers (2 weeks) and hydration (immediately before surgery), reducing the perioperative morbidity from 40 to 7% [140].
Answer to question 5: A close cooperation between endocrinologists and surgeons is mandatory: endocrine management of perioperative period is of utmost importance. Glucocorticoid substitutive treatment must be considered in case of cortisol secretion (either overt CS or ACS).
Question 6: Is a different management required for bilateral adrenal incidentaloma?
The usual presentation of bilateral adrenal incidentalomas (BAI, up to 9–17% of AI) is that of two discrete cortical adenomas, one on each adrenal; in rare cases, both adrenals may be massively enlarged by the presence of multiple macronodules (> 1 cm), termed Primary Macronodular Adrenal Hyperplasia (PMAH). In BAI with PMAH, the concept of ACTH-independent cortisol secretion was abandoned after the demonstration of paracrine and autocrine adrenal ACTH secretion [141].
The various etiologies of unilateral or bilateral AI vary depending on whether recruitment was performed in an endocrine or surgical setting: non-secreting AI are prevalent in the former, while ACC and secreting tumors are over-represented in the latter [3, 142, 143]. Adrenal metastases are often bilateral, on the contrary bilateral ACC is a rare condition [144]. In clinical practice, bilateral masses do not necessarily represent the same entity on both adrenals: the coexistence of different adrenal diseases is not uncommon, and in selected cases nodular adrenal hyperplasia could be ACTH-dependent, as in Cushing’s Disease [145] or Congenital Adrenal Hyperplasia [84].
The initial evaluation of BAI should aim to assess malignancy and functional status. Regarding imaging, each lesion should be assessed separately [29]. Benign lesions are lipid-rich (attenuation value < 10 HU), in case of lipid-poor adenomas an enhanced CT or a MR is able to further characterize the mass.
The endocrine assessment is similar to that of unilateral AI. The most common endocrine disease is cortisol excess (ACS), which is more prevalent in BAI. The prevalence of metabolic cortisol-related comorbidities (obesity, diabetes, hypertension, dyslipidemia) is similar to those patients with unilateral AI [146, 147]; however, only fracture risk seems increased in BAI [148].
Bilateral non-adrenal malignancies (metastasis, adrenal lymphoma [149]) may lead to adrenal insufficiency due to neoplastic infiltration of the adrenal glands: in selected cases, especially if imaging is not conclusive for lipid-rich AI and ACTH levels are increased, an insufficient cortisol secretion should be reasonably suspected.
In patients with PMAH and cortisol secretion, a special relevance is the study of illegitimate membrane G-protein coupled receptors (GPCRs). The activation of these GPCRs activate the cyclic-AMP/protein kinase A (PKA) signaling, leading to the transcription of steroidogenic factors, acting as ACTH [142]. These receptors could be ectopic or eutopic (over-expressed), and are studied applying various stimuli with dynamic tests (as meal test, upright posture test, luteinizing hormone-releasing hormone or metoclopramide test), to document a paradoxical cortisol secretion [150]. In case of aberrant receptors, an attempt to control cortisol hypersecretion could be performed with octreotide, propranolol, long-acting gonadotropin-releasing hormone agonist or other selected receptor agonists or antagonists [144, 151].
Bilateral adrenal masses, along with cortisol secretion and the documentation of familial cases, suggests a genetic predisposition. Recently, Armadillo repeat containing 5 (ARMC5) mutations have been described in familial cases and in sporadic patients with CS [152, 153]. Although ARMC5 mutations reduce the steroid secretory capacity of each cell, the overall cortisol secretion is increased due to massive enlargement of the adrenals [154,155,156]. In 2015, the association between ARMC5 and the presence of meningiomas has been described [157].
In patients with PMAH and cortisol secretion, the treatment of choice (surgery or medical therapy with steroidogenesis inhibitors) is not evidence-based, therefore a multidisciplinary evaluation in a referral center is suggested.
Answer to question 6: The assessment of malignancy and functional status in bilateral AI is similar to that of unilateral lesion. However, PMAH and ARMC5 mutations should be considered in case of overt cortisol secretion.
Conclusions
Several issues, summarized in Table 1, are still debated in patients with AI.
As a matter of fact, is difficult to define a rigid protocol in the absence of prospective studies in patients with AI, as well as lacking of prognostic factors to predict morpho-functional modifications of the mass. Without these elements, the hormonal studies, the type of imaging, the plan of follow-up and its duration must be based on the characteristics of the lesion (size, morphology, unilateral, bilateral), the age and gender of the patient and the presence of comorbidities (active cancer, cardiovascular diseases, high blood pressure, diabetes, osteoporosis etc.).
The clinical experience is of utmost importance: a personalized diagnostic-therapeutic approach, based upon multidisciplinary discussion, is suggested.
Data availability
Not applicable in a review paper.
References
Barzon L, Sonino N, Fallo F, Palu G, Boscaro M (2003) Prevalence and natural history of adrenal incidentalomas. Eur J Endocrinol. https://doi.org/10.1530/eje.0.1490273
Terzolo M, Stigliano A, Chiodini I et al (2011) AME position statement on adrenal incidentaloma. Eur J Endocrinol 164(6):851–870. https://doi.org/10.1530/EJE-10-1147
Mantero F, Terzolo M, Arnaldi G et al (2000) A survey on adrenal incidentaloma in Italy. Study group on adrenal tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab 85(2):637–644. https://doi.org/10.1210/jcem.85.2.6372
Cawood TJ, Hunt PJ, O’Shea D, Cole D, Soule S (2009) Recommended evaluation of adrenal incidentalomas is costly, has high false-positive rates and confers a risk of fatal cancer that is similar to the risk of the adrenal lesion becoming malignant; time for a rethink? Eur J Endocrinol 161(4):513–527. https://doi.org/10.1530/EJE-09-0234
Kloos RT, Gross MD, Francis IR, Korobkin M, Shapiro B (1995) Incidentally discovered adrenal masses. Endocr Rev 16(4):460–484. https://doi.org/10.1210/edrv-16-4-460
Grumbach MM, Biller BMK, Braunstein GD et al (2003) Management of the clinically inapparent adrenal mass (“incidentaloma”). Ann Intern Med 138(5):424–429. https://doi.org/10.7326/0003-4819-138-5-200303040-00013
Jaffe RB, Mesiano S, Smith R, Coulter CL, Spencer SJ, Chakravorty A (1998) The regulation and role of fetal adrenal development in human pregnancy. Endocr Res 24(3–4):919–926. https://doi.org/10.3109/07435809809032707
Ebbehoj A, Li D, Kaur RJ et al (2020) Epidemiology of adrenal tumours in Olmsted County, Minnesota, USA: a population-based cohort study. Lancet Diabetes Endocrinol 8(11):894–902. https://doi.org/10.1016/S2213-8587(20)30314-4
van den Broek J, Geenen R, Heijnen L, Kobus C, Schreurs H (2018) Adrenal incidentalomas during diagnostic work-up of colorectal cancer patients: what is the risk of metastases? Ann Surg Oncol 25(7):1986–1991. https://doi.org/10.1245/s10434-018-6501-y
Blake MA, Cronin CG, Boland GW (2010) Adrenal imaging. AJR Am J Roentgenol 194(6):1450–1460. https://doi.org/10.2214/AJR.10.4547
Blake MA, Kalra MK, Sweeney AT et al (2006) Distinguishing benign from malignant adrenal masses: multi-detector row CT protocol with 10-minute delay. Radiology 238(2):578–585. https://doi.org/10.1148/radiol.2382041514
McCarthy CJ, McDermott S, Blake MA (2016) Adrenal imaging: magnetic resonance imaging and computed tomography. Front Horm Res 45:55–69. https://doi.org/10.1159/000442313
Wang F, Liu J, Zhang R et al (2018) CT and MRI of adrenal gland pathologies. Quant Imaging Med Surg 8(8):853–875. https://doi.org/10.21037/qims.2018.09.13
Chambre C, McMurray E, Baudry C et al (2015) The 10 Hounsfield units unenhanced computed tomography attenuation threshold does not apply to cortisol secreting adrenocortical adenomas. Eur J Endocrinol 173(3):325–332. https://doi.org/10.1530/EJE-15-0036
Mosconi C, Vicennati V, Papadopoulos D et al (2017) Can imaging predict subclinical cortisol secretion in patients with adrenal adenomas? A CT predictive score. Am J Roentgenol 209(1):122–129. https://doi.org/10.2214/AJR.16.16965
De Leo A, Mosconi C, Zavatta G et al (2020) Radiologically defined lipid-poor adrenal adenomas: histopathological characteristics. J Endocrinol Invest 43(9):1197–1204. https://doi.org/10.1007/s40618-020-01198-5
Fassnacht M, Kenn W, Allolio B (2004) Adrenal tumors: how to establish malignancy ? J Endocrinol Invest 27(4):387–399. https://doi.org/10.1007/BF03351068
Fassnacht M, Dekkers OM, Else T et al (2018) European society of endocrinology clinical practice guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the study of adrenal tumors. Eur J Endocrinol 179(4):G1–G46. https://doi.org/10.1530/EJE-18-0608
Azoury SC, Nagarajan N, Young A et al (2017) Computed tomography in the management of adrenal tumors. J Comput Assist Tomogr 41(4):628–632. https://doi.org/10.1097/RCT.0000000000000578
Canu L, Van Hemert JAW, Kerstens MN et al (2019) CT characteristics of pheochromocytoma: relevance for the evaluation of adrenal incidentaloma. J Clin Endocrinol Metab 104(2):312–318. https://doi.org/10.1210/jc.2018-01532
Blake MA, Kalra MK, Maher MM et al (2004) Pheochromocytoma: an imaging chameleon. Radiographics 24(Suppl 1):S87-99. https://doi.org/10.1148/rg.24si045506
Erasmus JJ, Patz EF, McAdams HP et al (1997) Evaluation of adrenal masses in patients with bronchogenic carcinoma using 18F-fluorodeoxyglucose positron emission tomography. AJR Am J Roentgenol 168(5):1357–1360. https://doi.org/10.2214/ajr.168.5.9129444
Maurea S, Mainolfi C, Bazzicalupo L et al (1999) Imaging of adrenal tumors using FDG PET: comparison of benign and malignant lesions. AJR Am J Roentgenol 173(1):25–29. https://doi.org/10.2214/ajr.173.1.10397094
Yun M, Kim W, Alnafisi N, Lacorte L, Jang S, Alavi A (2001) 18F-FDG PET in characterizing adrenal lesions detected on CT or MRI. J Nucl Med 42(12):1795–1799. http://www.ncbi.nlm.nih.gov/pubmed/11752075
Tenenbaum F, Groussin L, Foehrenbach H et al (2004) 18F-fluorodeoxyglucose positron emission tomography as a diagnostic tool for malignancy of adrenocortical tumours? Preliminary results in 13 consecutive patients. Eur J Endocrinol 150(6):789–792. https://doi.org/10.1530/eje.0.1500789
Groussin L, Bonardel G, Silvéra S et al (2009) 18F-Fluorodeoxyglucose positron emission tomography for the diagnosis of adrenocortical tumors: a prospective study in 77 operated patients. J Clin Endocrinol Metab 94(5):1713–1722. https://doi.org/10.1210/jc.2008-2302
Taïeb D, Pacak K (2018) Molecular imaging and theranostic approaches in pheochromocytoma and paraganglioma. Cell Tissue Res 372(2):393–401. https://doi.org/10.1007/s00441-018-2791-4
Tirabassi G, Kola B, Ferretti M et al (2012) Fine-needle aspiration cytology of adrenal masses: a re-assessment with histological confirmation. J Endocrinol Invest 35(6):590–594. https://doi.org/10.3275/8010
Fassnacht M, Arlt W, Bancos I et al (2016) Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the study of adrenal tumors. Eur J Endocrinol 175(2):G34. https://doi.org/10.1530/EJE-16-0467
Zeiger MA, Thompson GB, Duh Q-Y et al (2009) American association of clinical endocrinologists and American association of endocrine surgeons medical guidelines for the management of adrenal incidentalomas: executive summary of recommendations. Endocr Pract 15(5):450–453. https://doi.org/10.4158/EP.15.5.450
Nieman LK, Biller BMK, Findling JW et al (2008) The diagnosis of Cushing’s syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 93(5):1526–1540. https://doi.org/10.1210/jc.2008-0125
Tabarin A, Bardet S, Bertherat J et al (2008) Exploration and management of adrenal incidentalomas. French society of endocrinology consensus. Ann Endocrinol (Paris) 69(6):487–500. https://doi.org/10.1016/j.ando.2008.09.003
Boscaro M, Arnaldi G (2009) Approach to the patient with possible Cushing’s syndrome. J Clin Endocrinol Metab 94(9):3121–3131. https://doi.org/10.1210/jc.2009-0612
Tabarin A (2018) Do the diagnostic criteria for subclinical hypercortisolism exist? Ann Endocrinol (Paris) 79(3):146–148. https://doi.org/10.1016/j.ando.2018.03.013
Reincke M (2000) Subclinical Cushing’s syndrome. Endocrinol Metab Clin North Am 29(1):43–56. https://doi.org/10.1016/s0889-8529(05)70115-8
Chiodini I, Morelli V (2016) Subclinical hypercortisolism: how to deal with it? Front Horm Res 46:28–38. https://doi.org/10.1159/000443862
Chiodini I (2011) Clinical review: diagnosis and treatment of subclinical hypercortisolism. J Clin Endocrinol Metab 96(5):1223–1236. https://doi.org/10.1210/jc.2010-2722
Stewart PM (2010) Is subclinical Cushing’s syndrome an entity or a statistical fallout from diagnostic testing? Consensus surrounding the diagnosis is required before optimal treatment can be defined. J Clin Endocrinol Metab 95(6):2618–2620. https://doi.org/10.1210/jc.2010-0633
Morelli V, Donadio F, Eller-Vainicher C et al (2010) Role of glucocorticoid receptor polymorphism in adrenal incidentalomas. Eur J Clin Invest 40(9):803–811. https://doi.org/10.1111/j.1365-2362.2010.02330.x
Arnaldi G, Angeli A, Atkinson AB et al (2003) Diagnosis and complications of Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab 88(12):5593. http://www.ncbi.nlm.nih.gov/pubmed/14671138. Accessed 17 Feb 2021
(2002) NIH state-of-the-science statement on management of the clinically inapparent adrenal mass (“incidentaloma”). NIH Consens State Sci Statements 19(2):1–25. http://www.ncbi.nlm.nih.gov/pubmed/14768652
Ceccato F, Boscaro M (2016) Cushing’s syndrome: screening and diagnosis. High Blood Press Cardiovasc Prev 23(3):209–215. https://doi.org/10.1007/s40292-016-0153-4
Wood PJ, Barth JH, Freedman DB, Perry L, Sheridan B (1997) Evidence for the low dose dexamethasone suppression test to screen for Cushing’s syndrome–recommendations for a protocol for biochemistry laboratories. Ann Clin Biochem 34(Pt 3):222–229. https://doi.org/10.1177/000456329703400302
Ceccato F, Antonelli G, Frigo AC et al (2017) First-line screening tests for Cushing’s syndrome in patients with adrenal incidentaloma: the role of urinary free cortisol measured by LC-MS/MS. J Endocrinol Invest. https://doi.org/10.1007/s40618-017-0644-8
Androulakis II, Kaltsas GA, Kollias GE et al (2014) Patients with apparently nonfunctioning adrenal incidentalomas may be at increased cardiovascular risk due to excessive cortisol secretion. J Clin Endocrinol Metab 99(8):2754–2762. https://doi.org/10.1210/jc.2013-4064
Valassi E, Swearingen B, Lee H et al (2009) Concomitant medication use can confound interpretation of the combined dexamethasone-corticotropin releasing hormone test in Cushing’s syndrome. J Clin Endocrinol Metab 94(12):4851–4859. https://doi.org/10.1210/jc.2009-1500
Meikle AW (1982) Dexamethasone suppression tests: usefulness of simultaneous measurement of plasma cortisol and dexamethasone. Clin Endocrinol (Oxf) 16(4):401–408. http://www.ncbi.nlm.nih.gov/pubmed/7094363
Ceccato F, Artusi C, Barbot M et al (2020) Dexamethasone measurement during low-dose suppression test for suspected hypercortisolism: threshold development with and validation. J Endocrinol Invest 43(8):1105–1113. https://doi.org/10.1007/s40618-020-01197-6
de Graaf AJ, Mulder AL, Krabbe JG (2019) Retrospective analysis of repeated dexamethasone suppression tests—the added value of measuring dexamethasone. Ann Clin Biochem Int J Lab Med 56(6):708–710. https://doi.org/10.1177/0004563219870834
Ceccato F, Antonelli G, Barbot M et al (2014) The diagnostic performance of urinary free cortisol is better than the cortisol:cortisone ratio in detecting de novo Cushing’s syndrome: the use of a LC–MS/MS method in routine clinical practice. Eur J Endocrinol 171(1):1–7. https://doi.org/10.1530/EJE-14-0061
Ceccato F, Barbot M, Zilio M et al (2015) Screening tests for Cushing’s syndrome: urinary free cortisol role measured by LC-MS/MS. J Clin Endocrinol Metab. https://doi.org/10.1210/jc.2015-2507
Scaroni C, Albiger NM, Palmieri S et al (2020) Approach to patients with pseudo-Cushing’s states. Endocr Connect 9(1):R1–R13. https://doi.org/10.1530/EC-19-0435
Ceccato F, Lizzul L, Barbot M, Scaroni C (2020) Pituitary-adrenal axis and peripheral cortisol metabolism in obese patients. Endocrine 69(2):386–392. https://doi.org/10.1007/s12020-020-02392-4
Findling JW, Raff H (2017) Diagnosis of endocrine disease: differentiation of pathologic/neoplastic hypercortisolism (Cushing’s syndrome) from physiologic/non-neoplastic hypercortisolism (formerly known as pseudo-Cushing’s syndrome). Eur J Endocrinol 176(5):R205–R216. https://doi.org/10.1530/EJE-16-0946
Ceccato F, Marcelli G, Martino M et al (2019) The diagnostic accuracy of increased late night salivary cortisol for Cushing’s syndrome: a real-life prospective study. J Endocrinol Invest 42(3):327–335. https://doi.org/10.1007/s40618-018-0921-1
Ceccato F, Barbot M, Zilio M et al (2013) Performance of salivary cortisol in the diagnosis of Cushing’s syndrome, adrenal incidentaloma, and adrenal insufficiency. Eur J Endocrinol 169(1):31–36. https://doi.org/10.1530/EJE-13-0159
Masserini B, Morelli V, Bergamaschi S et al (2009) The limited role of midnight salivary cortisol levels in the diagnosis of subclinical hypercortisolism in patients with adrenal incidentaloma. Eur J Endocrinol 160(1):87–92. https://doi.org/10.1530/EJE-08-0485
Ceccato F, Barbot M, Albiger N et al (2017) Daily salivary cortisol and cortisone rhythm in patients with adrenal incidentaloma. Endocrine. https://doi.org/10.1007/s12020-017-1421-3
Pecori Giraldi F, Saccani A, Cavagnini F (2011) Study group on the hypothalamo-pituitary-adrenal axis of the Italian Society of Endocrinology. Assessment of ACTH assay variability: a multicenter study. Eur J Endocrinol 164(4):505–512. https://doi.org/10.1530/EJE-10-0962
Ceccato F, Tizianel I, Vedolin CK, Boscaro M, Barbot M, Scaroni C (2020) Human corticotropin-releasing hormone tests: 10 years of real-life experience in pituitary and adrenal disease. J Clin Endocrinol Metab. https://doi.org/10.1210/clinem/dgaa564
Trementino L, Appolloni G, Concettoni C, Cardinaletti M, Boscaro M, Arnaldi G (2012) Association of glucocorticoid receptor polymorphism A3669G with decreased risk of developing diabetes in patients with Cushing’s syndrome. Eur J Endocrinol 166(1):35–42. https://doi.org/10.1530/EJE-11-0722
Trementino L, Appolloni G, Ceccoli L et al (2014) Bone complications in patients with Cushing’s syndrome: looking for clinical, biochemical, and genetic determinants. Osteoporos Int 25(3):913–921. https://doi.org/10.1007/s00198-013-2520-5
Eller-Vainicher C, Morelli V, Salcuni AS et al (2010) Post-surgical hypocortisolism after removal of an adrenal incidentaloma: is it predictable by an accurate endocrinological work-up before surgery? Eur J Endocrinol 162(1):91–99. https://doi.org/10.1530/EJE-09-0775
Di Dalmazi G, Berr CM, Fassnacht M, Beuschlein F, Reincke M (2014) Adrenal function after adrenalectomy for subclinical hypercortisolism and Cushing’s syndrome: a systematic review of the literature. J Clin Endocrinol Metab 99(8):2637–2645. https://doi.org/10.1210/jc.2014-1401
Ricciato MP, Di Donna V, Perotti G, Pontecorvi A, Bellantone R, Corsello SM (2014) The role of adrenal scintigraphy in the diagnosis of subclinical Cushing’s syndrome and the prediction of post-surgical hypoadrenalism. World J Surg 38(6):1328–1335. https://doi.org/10.1007/s00268-014-2482-6
Funder JW, Carey RM, Mantero F et al (2016) The management of primary aldosteronism: case detection, diagnosis, and treatment: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 101(5):1889–1916. https://doi.org/10.1210/jc.2015-4061
Young WF (2007) Clinical practice. The incidentally discovered adrenal mass. N Engl J Med 356(6):601–610. https://doi.org/10.1056/NEJMcp065470
Grasso M, Boscaro M, Scaroni C, Ceccato F (2018) Secondary arterial hypertension: from routine clinical practice to evidence in patients with adrenal tumor. High Blood Press Cardiovasc Prev 25(4):345–354. https://doi.org/10.1007/s40292-018-0288-6
Médeau V, Moreau F, Trinquart L et al (2008) Clinical and biochemical characteristics of normotensive patients with primary aldosteronism: a comparison with hypertensive cases. Clin Endocrinol (Oxf) 69(1):20–28. https://doi.org/10.1111/j.1365-2265.2008.03213.x
Ito Y, Takeda R, Karashima S, Yamamoto Y, Yoneda T, Takeda Y (2011) Prevalence of primary aldosteronism among prehypertensive and stage 1 hypertensive subjects. Hypertens Res 34(1):98–102. https://doi.org/10.1038/hr.2010.166
Piaditis GP, Kaltsas GA, Androulakis II et al (2009) High prevalence of autonomous cortisol and aldosterone secretion from adrenal adenomas. Clin Endocrinol (Oxf) 71(6):772–778. https://doi.org/10.1111/j.1365-2265.2009.03551.x
Nishimoto K, Seki T, Hayashi Y et al (2016) Human adrenocortical remodeling leading to aldosterone-producing cell cluster generation. Int J Endocrinol 2016:7834356. https://doi.org/10.1155/2016/7834356
Rossi GP, Ceolotto G, Rossitto G, Maiolino G, Cesari M, Seccia TM (2020) Effects of mineralocorticoid and AT1 receptor antagonism on the aldosterone-renin ratio in primary aldosteronism-the EMIRA study. J Clin Endocrinol Metab. https://doi.org/10.1210/clinem/dgaa080
Fallo F, Barzon L, Boscaro M, Sonino N (1997) Coexistence of aldosteronoma and contralateral nonfunctioning adrenal adenoma in primary aldosteronism. Am J Hypertens 10(4 Pt 1):476–478. https://doi.org/10.1016/s0895-7061(96)00506-7
Sarlon-Bartoli G, Michel N, Taieb D et al (2011) Adrenal venous sampling is crucial before an adrenalectomy whatever the adrenal-nodule size on computed tomography. J Hypertens 29(6):1196–1202. https://doi.org/10.1097/HJH.0b013e32834666af
Kim HY, Kim SG, Lee KW et al (2005) Clinical study of adrenal incidentaloma in Korea. Korean J Intern Med 20(4):303–309. https://doi.org/10.3904/kjim.2005.20.4.303
Falhammar H, Kjellman M, Calissendorff J (2018) Initial clinical presentation and spectrum of pheochromocytoma: a study of 94 cases from a single center. Endocr Connect 7(1):186–192. https://doi.org/10.1530/EC-17-0321
Oshmyansky AR, Mahammedi A, Dackiw A et al (2013) Serendipity in the diagnosis of pheochromocytoma. J Comput Assist Tomogr 37(5):820–823. https://doi.org/10.1097/RCT.0b013e31829cbecf
Lenders JWM, Eisenhofer G, Mannelli M, Pacak K (2005) Phaeochromocytoma. Lancet (London, England) 366(9486):665–675. https://doi.org/10.1016/S0140-6736(05)67139-5
Mannelli M, Ianni L, Cilotti A, Conti A (1999) Pheochromocytoma in Italy: a multicentric retrospective study. Eur J Endocrinol 141(6):619–624. https://doi.org/10.1530/eje.0.1410619
Kopetschke R, Slisko M, Kilisli A et al (2009) Frequent incidental discovery of phaeochromocytoma: data from a German cohort of 201 phaeochromocytoma. Eur J Endocrinol 161(2):355–361. https://doi.org/10.1530/EJE-09-0384
Lenders JWM, Duh Q-Y, Eisenhofer G et al (2014) Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 99(6):1915–1942. https://doi.org/10.1210/jc.2014-1498
Eisenhofer G, Peitzsch M, Kaden D et al (2019) Reference intervals for LC-MS/MS measurements of plasma free, urinary free and urinary acid-hydrolyzed deconjugated normetanephrine, metanephrine and methoxytyramine. Clin Chim Acta 490:46–54. https://doi.org/10.1016/j.cca.2018.12.019
Speiser PW, Azziz R, Baskin LS et al (2010) Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 95(9):4133–4160. https://doi.org/10.1210/jc.2009-2631
Ceccato F, Mantero F (2019) Monogenic forms of hypertension. Endocrinol Metab Clin North Am 48(4):795–810. https://doi.org/10.1016/j.ecl.2019.08.009
Fanelli F, Di Dalmazi G (2019) Serum steroid profiling by mass spectrometry in adrenocortical tumors: diagnostic implications. Curr Opin Endocrinol Diabetes Obes 26(3):160–165. https://doi.org/10.1097/MED.0000000000000475
Arlt W, Lang K, Sitch AJ et al (2017) Steroid metabolome analysis reveals prevalent glucocorticoid excess in primary aldosteronism. JCI Insight. https://doi.org/10.1172/jci.insight.93136
Adolf C, Köhler A, Franke A et al (2018) Cortisol excess in patients with primary aldosteronism impacts left ventricular hypertrophy. J Clin Endocrinol Metab 103(12):4543–4552. https://doi.org/10.1210/jc.2018-00617
Yang Y, Burrello J, Burrello A et al (2019) Classification of microadenomas in patients with primary aldosteronism by steroid profiling. J Steroid Biochem Mol Biol 189:274–282. https://doi.org/10.1016/j.jsbmb.2019.01.008
Eisenhofer G, Masjkur J, Peitzsch M et al (2018) Plasma steroid metabolome profiling for diagnosis and subtyping patients with Cushing syndrome. Clin Chem 64(3):586–596. https://doi.org/10.1373/clinchem.2017.282582
Di Dalmazi G, Fanelli F, Mezzullo M et al (2015) Steroid profiling by LC-MS/MS in nonsecreting and subclinical cortisol-secreting adrenocortical adenomas. J Clin Endocrinol Metab. https://doi.org/10.1210/JC.2015-1992
Di Dalmazi G, Fanelli F, Zavatta G et al (2019) The steroid profile of adrenal incidentalomas: subtyping subjects with high cardiovascular risk. J Clin Endocrinol Metab 104(11):5519–5528. https://doi.org/10.1210/jc.2019-00365
Arlt W, Biehl M, Taylor AE et al (2011) Urine steroid metabolomics as a biomarker tool for detecting malignancy in adrenal tumors. J Clin Endocrinol Metab 96(12):3775–3784. https://doi.org/10.1210/jc.2011-1565
Garrapa GG, Pantanetti P, Arnaldi G, Mantero F, Faloia E (2001) Body composition and metabolic features in women with adrenal incidentaloma or Cushing’s syndrome. J Clin Endocrinol Metab 86(11):5301–5306. https://doi.org/10.1210/jcem.86.11.8059
Giordano R, Marinazzo E, Berardelli R et al (2010) Long-term morphological, hormonal, and clinical follow-up in a single unit on 118 patients with adrenal incidentalomas. Eur J Endocrinol 162(4):779–785. https://doi.org/10.1530/EJE-09-0957
Di Dalmazi G, Vicennati V, Garelli S et al (2014) Cardiovascular events and mortality in patients with adrenal incidentalomas that are either non-secreting or associated with intermediate phenotype or subclinical Cushing’s syndrome: a 15-year retrospective study. Lancet Diabetes Endocrinol 2(5):396–405. https://doi.org/10.1016/S2213-8587(13)70211-0
Debono M, Bradburn M, Bull M, Harrison B, Ross RJ, Newell-Price J (2014) Cortisol as a marker for increased mortality in patients with incidental adrenocortical adenomas. J Clin Endocrinol Metab 99(12):4462–4470. https://doi.org/10.1210/jc.2014-3007
Di Dalmazi G, Vicennati V, Pizzi C et al (2020) Prevalence and incidence of atrial fibrillation in a large cohort of adrenal incidentalomas: a long-term study. J Clin Endocrinol Metab. https://doi.org/10.1210/clinem/dgaa270
Peppa M, Koliaki C, Raptis SA (2010) Adrenal incidentalomas and cardiometabolic morbidity: an emerging association with serious clinical implications. J Intern Med 268(6):555–566. https://doi.org/10.1111/j.1365-2796.2010.02291.x
Toniato A, Merante-Boschin I, Opocher G, Pelizzo MR, Schiavi F, Ballotta E (2009) Surgical versus conservative management for subclinical Cushing syndrome in adrenal incidentalomas: a prospective randomized study. Ann Surg 249(3):388. http://www.ncbi.nlm.nih.gov/pubmed/19247023
Chiodini I, Morelli V, Salcuni AS et al (2010) Beneficial metabolic effects of prompt surgical treatment in patients with an adrenal incidentaloma causing biochemical hypercortisolism. J Clin Endocrinol Metab 95(6):2736–2745. https://doi.org/10.1210/jc.2009-2387
Guerrieri M, Campagnacci R, Patrizi A, Romiti C, Arnaldi G, Boscaro M (2010) Primary adrenal hypercortisolism: minimally invasive surgical treatment or medical therapy? A retrospective study with long-term follow-up evaluation. Surg Endosc 24(10):2542–2546. https://doi.org/10.1007/s00464-010-1000-7
Iacobone M, Citton M, Scarpa M, Viel G, Boscaro M, Nitti D (2015) Systematic review of surgical treatment of subclinical Cushing’s syndrome. Br J Surg 102(4):318–330. https://doi.org/10.1002/bjs.9742
Petramala L, Cavallaro G, Galassi M et al (2017) Clinical benefits of unilateral adrenalectomy in patients with subclinical hypercortisolism due to adrenal incidentaloma: results from a single center. High Blood Press Cardiovasc Prev 24(1):69–75. https://doi.org/10.1007/s40292-017-0182-7
Bancos I, Alahdab F, Crowley RK et al (2016) Therapy of endocrine disease: improvement of cardiovascular risk factors after adrenalectomy in patients with adrenal tumors and subclinical Cushing’s syndrome: a systematic review and meta-analysis. Eur J Endocrinol 175(6):R283–R295. https://doi.org/10.1530/EJE-16-0465
Rossi R, Tauchmanova L, Luciano A et al (2000) Subclinical Cushing’s syndrome in patients with adrenal incidentaloma: clinical and biochemical features. J Clin Endocrinol Metab 85(4):1440–1448. https://doi.org/10.1210/jcem.85.4.6515
Tsagarakis S, Vassiliadi D, Thalassinos N (2006) Endogenous subclinical hypercortisolism: diagnostic uncertainties and clinical implications. J Endocrinol Invest 29(5):471–482. https://doi.org/10.1007/BF03344133
Mansmann G, Lau J, Balk E, Rothberg M, Miyachi Y, Bornstein SR (2004) The clinically inapparent adrenal mass: update in diagnosis and management. Endocr Rev 25(2):309–340. https://doi.org/10.1210/er.2002-0031
Goh Z, Phillips I, Hunt PJ, Soule S, Cawood TJ (2020) Three-year follow up of adrenal incidentalomas in a New Zealand centre. Intern Med J 50(3):350–356. https://doi.org/10.1111/imj.14332
Tauchmanovà L, Rossi R, Biondi B et al (2002) Patients with subclinical Cushing’s syndrome due to adrenal adenoma have increased cardiovascular risk. J Clin Endocrinol Metab 87(11):4872–4878. https://doi.org/10.1210/jc.2001-011766
Swiatkowska-Stodulska R, Kaniuka-Jakubowska S, Wisniewski P, Skibowska-Bielinska A, Sworczak K (2011) The estimation of selected endogenous anticoagulation system parameters in patients with subclinical Cushing’s syndrome. Eur J Endocrinol 165(6):865–871. https://doi.org/10.1530/EJE-11-0535
Giordano R, Guaraldi F, Berardelli R et al (2012) Glucose metabolism in patients with subclinical Cushing’s syndrome. Endocrine 41(3):415–423. https://doi.org/10.1007/s12020-012-9628-9
Scaroni C, Zilio M, Foti M, Boscaro M (2017) Glucose metabolism abnormalities in Cushing syndrome: from molecular basis to clinical management. Endocr Rev 38(3):189–219. https://doi.org/10.1210/er.2016-1105
Arnaldi G, Scandali VM, Trementino L, Cardinaletti M, Appolloni G, Boscaro M (2010) Pathophysiology of dyslipidemia in Cushing’s syndrome. Neuroendocrinology 92(1):86–90. https://doi.org/10.1159/000314213
Kastelan D, Dzubur F, Dusek T et al (2011) Health-related quality of life and fatigue in patients with adrenal incidentaloma. Endocrine 40(1):84–89. https://doi.org/10.1007/s12020-011-9456-3
Morelli V, Arosio M, Chiodini I (2018) Cardiovascular mortality in patients with subclinical Cushing. Ann Endocrinol (Paris) 79(3):149–152. https://doi.org/10.1016/j.ando.2018.03.005
Eller-Vainicher C, Morelli V, Salcuni AS et al (2010) Accuracy of several parameters of hypothalamic-pituitary-adrenal axis activity in predicting before surgery the metabolic effects of the removal of an adrenal incidentaloma. Eur J Endocrinol 163(6):925–935. https://doi.org/10.1530/EJE-10-0602
Chiodini I, Albani A, Ambrogio AG et al (2016) Six controversial issues on subclinical Cushing’s syndrome. Endocrine 56(2):262–266. https://doi.org/10.1007/s12020-016-1017-3
Hong AR, Kim JH, Park KS et al (2017) Optimal follow-up strategies for adrenal incidentalomas: reappraisal of the 2016 ESE-ENSAT guidelines in real clinical practice. Eur J Endocrinol 177(6):475–483. https://doi.org/10.1530/EJE-17-0372
Pantalone KM, Gopan T, Remer EM et al (2010) Change in adrenal mass size as a predictor of a malignant tumor. Endocr Pract 16(4):577–587. https://doi.org/10.4158/EP09351.OR
Yener S, Ertilav S, Secil M et al (2010) Prospective evaluation of tumor size and hormonal status in adrenal incidentalomas. J Endocrinol Invest 33(1):32–36. https://doi.org/10.1007/BF03346546
Vassilatou E, Vryonidou A, Michalopoulou S et al (2009) Hormonal activity of adrenal incidentalomas: results from a long-term follow-up study. Clin Endocrinol (Oxf) 70(5):674–679. https://doi.org/10.1111/j.1365-2265.2008.03492.x
Bülow B, Jansson S, Juhlin C et al (2006) Adrenal incidentaloma—follow-up results from a Swedish prospective study. Eur J Endocrinol 154(3):419–423. https://doi.org/10.1530/eje.1.02110
Barzon L, Scaroni C, Sonino N, Fallo F, Paoletta A, Boscaro M (1999) Risk factors and long-term follow-up of adrenal incidentalomas. J Clin Endocrinol Metab 84(2):520–526. https://doi.org/10.1210/jcem.84.2.5444
Morelli V, Reimondo G, Giordano R et al (2014) Long-term follow-up in adrenal incidentalomas: an Italian multicenter study. J Clin Endocrinol Metab 99(3):827–834. https://doi.org/10.1210/jc.2013-3527
Dinnes J, Bancos I, di Ruffano LF et al (2016) Management of endocrine disease: imaging for the diagnosis of malignancy in incidentally discovered adrenal masses: a systematic review and meta-analysis. Eur J Endocrinol 175(2):R51–R64. https://doi.org/10.1530/EJE-16-0461
Schalin-Jäntti C, Raade M, Hämäläinen E, Sane T (2015) A 5-year prospective follow-up study of lipid-rich adrenal incidentalomas: no tumor growth or development of hormonal hypersecretion. Endocrinol Metab (Seoul, Korea) 30(4):481–487. https://doi.org/10.3803/EnM.2015.30.4.481
Iacobone M, Citton M, Viel G et al (2012) Adrenalectomy may improve cardiovascular and metabolic impairment and ameliorate quality of life in patients with adrenal incidentalomas and subclinical Cushing’s syndrome. Surgery 152(6):991–997. https://doi.org/10.1016/j.surg.2012.08.054
Perysinakis I, Marakaki C, Avlonitis S et al (2013) Laparoscopic adrenalectomy in patients with subclinical Cushing syndrome. Surg Endosc 27(6):2145–2148. https://doi.org/10.1007/s00464-012-2730-5
Barbot M, Ceccato F, Lizzul L et al (2019) Perioperative multidisciplinary management of endoscopic transsphenoidal surgery for sellar lesions: practical suggestions from the Padova model. Neurosurg Rev. https://doi.org/10.1007/s10143-019-01132-1
Iacobellis G, Petramala L, Barbaro G et al (2013) Epicardial fat thickness and left ventricular mass in subjects with adrenal incidentaloma. Endocrine 44(2):532–536. https://doi.org/10.1007/s12020-013-9902-5
Sbardella E, Minnetti M, D’Aluisio D et al (2018) Cardiovascular features of possible autonomous cortisol secretion in patients with adrenal incidentalomas. Eur J Endocrinol 178(5):501–511. https://doi.org/10.1530/EJE-17-0986
Alesina PF, Hommeltenberg S, Meier B et al (2010) Posterior retroperitoneoscopic adrenalectomy for clinical and subclinical Cushing’s syndrome. World J Surg 34(6):1391–1397. https://doi.org/10.1007/s00268-010-0453-0
Berr CM, Di Dalmazi G, Osswald A et al (2015) Time to recovery of adrenal function after curative surgery for Cushing’s syndrome depends on etiology. J Clin Endocrinol Metab 100(4):1300–1308. https://doi.org/10.1210/jc.2014-3632
Morelli V, Minelli L, Eller-Vainicher C et al (2018) Predictability of hypoadrenalism occurrence and duration after adrenalectomy for ACTH-independent hypercortisolism. J Endocrinol Invest 41(4):485–493. https://doi.org/10.1007/s40618-017-0788-6
Steichen O, Amar L, Chaffanjon P, Kraimps J-L, Ménégaux F, Zinzindohoue F (2016) SFE/SFHTA/AFCE consensus on primary aldosteronism, part 6: adrenal surgery. Ann Endocrinol (Paris) 77(3):220–225. https://doi.org/10.1016/j.ando.2016.01.009
Choi SH, Kwon TG, Kim T-H (2012) Active potassium supplementation might be mandatory during laparoscopic adrenalectomy for primary hyperaldosteronism. J Endourol 26(6):666–669. https://doi.org/10.1089/end.2011.0566
Fischer E, Hanslik G, Pallauf A et al (2012) Prolonged zona glomerulosa insufficiency causing hyperkalemia in primary aldosteronism after adrenalectomy. J Clin Endocrinol Metab 97(11):3965–3973. https://doi.org/10.1210/jc.2012-2234
Iacobone M, Citton M, Viel G, Schiavone D, Torresan F (2017) Surgical approaches in hereditary endocrine tumors. Updates Surg 69(2):181–191. https://doi.org/10.1007/s13304-017-0451-y
Livingstone M, Duttchen K, Thompson J et al (2015) Hemodynamic stability during pheochromocytoma resection: lessons learned over the last two decades. Ann Surg Oncol 22(13):4175–4180. https://doi.org/10.1245/s10434-015-4519-y
Louiset E, Duparc C, Young J et al (2013) Intraadrenal corticotropin in bilateral macronodular adrenal hyperplasia. N Engl J Med 369(22):2115–2125. https://doi.org/10.1056/NEJMoa1215245
Bourdeau I, El Ghorayeb N, Gagnon N, Lacroix A (2018) Management of endocrine disease: differential diagnosis, investigation and therapy of bilateral adrenal incidentalomas. Eur J Endocrinol 179(2):R57–R67. https://doi.org/10.1530/EJE-18-0296
Barzon L, Scaroni C, Sonino N et al (1998) Incidentally discovered adrenal tumors: endocrine and scintigraphic correlates. J Clin Endocrinol Metab 83(1):55–62. https://doi.org/10.1210/jcem.83.1.4501
Vassiliadi DA, Partsalaki E, Tsagarakis S (2020) Approach to patients with bilateral adrenal incidentalomas. Curr Opin Endocrinol Diabetes Obes 27(3):125–131. https://doi.org/10.1097/MED.0000000000000536
Albiger NM, Occhi G, Sanguin F et al (2011) Adrenal nodules in patients with Cushing’s disease: prevalence, clinical significance and follow-up. J Endocrinol Invest 34(8):e204–e209. https://doi.org/10.3275/7349
Vassilatou E, Vryonidou A, Ioannidis D, Paschou SA, Panagou M, Tzavara I (2014) Bilateral adrenal incidentalomas differ from unilateral adrenal incidentalomas in subclinical cortisol hypersecretion but not in potential clinical implications. Eur J Endocrinol 171(1):37–45. https://doi.org/10.1530/EJE-13-0848
Paschou SA, Kandaraki E, Dimitropoulou F, Goulis DG, Vryonidou A (2016) Subclinical Cushing’s syndrome in patients with bilateral compared to unilateral adrenal incidentalomas: a systematic review and meta-analysis. Endocrine 51(2):225. https://doi.org/10.1007/s12020-015-0776-6
Morelli V, Palmieri S, Salcuni AS et al (2013) Bilateral and unilateral adrenal incidentalomas: biochemical and clinical characteristics. Eur J Endocrinol 168(2):235–241. https://doi.org/10.1530/EJE-12-0777
Majidi F, Martino S, Kondakci M et al (2020) Clinical spectrum of primary adrenal lymphoma: results of a multicenter cohort study. Eur J Endocrinol 183(4):453–462. https://doi.org/10.1530/EJE-19-0506
Regazzo D, Barbot M, Scaroni C, Albiger N, Occhi G (2020) The pathogenic role of the GIP/GIPR axis in human endocrine tumors: emerging clinical mechanisms beyond diabetes. Rev Endocr Metab Disord 21(1):165–183. https://doi.org/10.1007/s11154-019-09536-6
Albiger NM, Occhi G, Mariniello B et al (2007) Food-dependent Cushing’s syndrome: from molecular characterization to therapeutical results. Eur J Endocrinol 157(6):771–778. https://doi.org/10.1530/EJE-07-0253
Alencar GA, Lerario AM, Nishi MY et al (2014) ARMC5 mutations are a frequent cause of primary macronodular adrenal hyperplasia. J Clin Endocrinol Metab 99(8):E1501–E1509. https://doi.org/10.1210/jc.2013-4237
Vassiliadi DA, Tsagarakis S (2019) Diagnosis and management of primary bilateral macronodular adrenal hyperplasia. Endocr Relat Cancer 26(10):R567–R581. https://doi.org/10.1530/ERC-19-0240
Assié G, Libé R, Espiard S et al (2013) ARMC5 mutations in macronodular adrenal hyperplasia with cushing’s syndrome. N Engl J Med 369(22):2105–2114. https://doi.org/10.1056/NEJMoa1304603
Cavalcante IP, Nishi M, Zerbini MCN et al (2018) The role of ARMC5 in human cell cultures from nodules of primary macronodular adrenocortical hyperplasia (PMAH). Mol Cell Endocrinol 460:36–46. https://doi.org/10.1016/j.mce.2017.06.027
da Conceição BB, Cavalcante IP, Kremer JL et al (2020) ARMC5 mutations are associated with high levels of proliferating cell nuclear antigen and the presence of the serotonin receptor 5HT4R in PMAH nodules. Arch Endocrinol Metab 64(4):390–401. https://doi.org/10.20945/2359-3997000000236
Elbelt U, Trovato A, Kloth M et al (2015) Molecular and clinical evidence for an ARMC5 tumor syndrome: concurrent inactivating germline and somatic mutations are associated with both primary macronodular adrenal hyperplasia and meningioma. J Clin Endocrinol Metab 100(1):E119–E128. https://doi.org/10.1210/jc.2014-2648
Funding
Open access funding provided by Università degli Studi di Padova within the CRUI-CARE Agreement. This study did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Author information
Authors and Affiliations
Contributions
All Authors gave an equal contribution to the review of literature, in drafting the manuscript and in the final approval of the paper.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflicts of interest that might be perceived as influencing the impartiality of the reported research.
Ethical approval
All the procedures performed in our study involving human participants were in accordance with the ethical standards of the institutional Ethic Committee (Comitato Etico per la Sperimentazione Scientifica, CESC of Padova), and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Research involving human participants and patient consent
Informed consent has been obtained in the manuscript reported.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ceccato, F., Barbot, M., Scaroni, C. et al. Frequently asked questions and answers (if any) in patients with adrenal incidentaloma. J Endocrinol Invest 44, 2749–2763 (2021). https://doi.org/10.1007/s40618-021-01615-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-021-01615-3