
Abstract
The regulation of phosphate metabolism as an influence on bone homeostasis is profound. Recent advances in understanding the systemic control of fibroblast growth factor-23 (FGF23) has uncovered novel effectors of endocrine feedback loops for calcium, phosphate, and vitamin D balance that interact with ‘traditional’ feedback loops for mineral metabolism. Not only are these findings reshaping research studying phosphate handling and skeletal interactions, but they have also provided new therapeutic interventions. Emerging data support that the control of FGF23 production in bone and its circulating concentrations is a multilayered process, with some influences affecting FGF23 transcription and some posttranslational modification of the secreted, bioactive protein. Additionally, the actions of FGF23 on its target tissues via its coreceptor α-Klotho, are subject to regulatory events just coming to light. The recent findings of systemic influences on circulating FGF23 and the downstream manifestations on bone homeostasis will be reviewed herein.
Similar content being viewed by others

Introduction
Proper phosphate balance is required for normal bone development, growth, and function; therefore, blood phosphate is maintained within a fairly narrow range. Prolonged hypophosphatemia results in rickets in children through delayed endochondral ossification, and osteomalacia in children and adults. Hyperphosphatemia causes delayed bone mineralization, as well as precipitation of phosphate/calcium crystals resulting in ectopic and vascular calcification, with secondary effects on bone formation through indirect actions on calcitropic hormone production and activity. The bone-derived hormone fibroblast growth factor-23 (FGF23) is central to the maintenance of phosphate balance through its actions on its target organs including the kidney and parathyroid gland. The study of genetic and acquired diseases involving increased or decreased FGF23 bioactivity has revealed novel mechanisms controlling phosphate handling. FGF23 acts through its coreceptor α-Klotho (αKL), to control renal phosphate and vitamin D metabolism in kidney, the primary organ regulating shorter-term blood phosphate concentrations. This review will focus primarily upon recent findings describing novel regulators of phosphate metabolism and FGF23 that interact with “traditional” endocrine feedback loops, and their downstream influences on bone homeostasis. Finally, emerging therapeutic approaches targeting FGF23 that impact skeletal repair will be discussed.
FGF23 Actions in Renal Mineral Metabolism
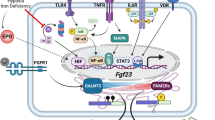
Systemic phosphate control is critical for hydroxyapatite formation during bone mineralization, and is the product of sensitive endocrine feedback loops involving the intestine, kidney, and the skeleton. FGF23 is produced in bone following an increase in serum phosphate or 1,25(OH)2 vitamin D (1,25D) [1, 2], and acts upon the kidney to decrease phosphate (Pi) reabsorption [3]. The type-II sodium phosphate cotransporters NPT2a and NPT2c are localized to the apical membrane of the renal proximal tubule and are the primary regulators of this process. Following FGF23 delivery, these transporters are downregulated fairly rapidly (within 1 hour) through internalization [3, 4]. FGF23 also has potent effects on renal 1,25D production and therefore calcium-phosphate balance, as FGF23 downregulates the renal 1α-hydroxylase (Cyp27b1) and stimulates the catabolic vitamin D 24-hydroxylase (Cyp24a1), inhibiting the synthesis of active 1,25D [2, 3]. Therefore, an increase in bioactive FGF23 (“intact” or “iFGF23”) results in reduced serum phosphate with “inappropriately” low or normal plasma 1,25D concentrations, with the converse biochemical profile for situations of reduced circulating FGF23. Reduced serum phosphate and 1,25D complete the feedback loop by inhibiting FGF23 production in bone (Fig. 1).

Schematic of FGF23 pathways affecting systemic actions of FGF23 and bone homeostasis. FGF23 is produced in osteoblasts and osteocytes (left) and controls phosphate (Pi) metabolism in the kidney and parathyroid glands, sites of αKL expression (green lines indicate stimulatory pathways, red lines inhibitory). In kidney, FGF23 reduces phosphate reabsorption through downregulation of NPT2a and NPT2c and decreases 1,25D production through control of 1,25D metabolizing enzymes. The resulting decreases in serum phosphate from decreased 1,25D-dependent intestinal Pi absorption reduce FGF23 production in bone through a negative feedback loop. Evidence supports PTH stimulation of FGF23 in bone, and FGF23 acts in the parathyroid glands to reduce PTH. Other regulators such as calcium (Ca2+) and iron (Fe3+) deficiency may act directly on osteoblasts/osteocytes to increase FGF23 mRNA. The intracellular factors FAM20c, GALNT3, and furin are dynamically controlled to influence the production of either bioactive, intact FGF23 (iFGF23), or inactive FGF23 fragments as another important mode to regulate iFGF23 concentrations. Factors such as cKL and FGF2/FGFR1 likely act in bone to control circulating FGF23. Iron (II) sulfate therapy and FGF23 neutralizing antibodies reduce FGF23 production and bioactivity, respectively
Through genetic and molecular experiments, it was demonstrated that FGF23 requires the coreceptor α-Klotho (αKL) to elicit high-affinity bioactivity in its target tissues, primarily kidney and parathyroid gland [5]. The mature αKL protein is produced as several isoforms: the “membrane”-bound (“mKL”) form is a 130 kD single-pass transmembrane protein with a large extracellular domain that interacts with FGF23, as well as a short intracellular region that does not appear to be capable of independent signaling [6]. At the cell surface, the cleavage of mKL occurs near the transmembrane domain by membrane-bound secretases including those from the ADAM and BACE families [7], thereby releasing a circulating form of αKL referred to as “cut-” or “cleaved-KL” (‘cKL’). In vitro, mKL recruitment of canonical FGF receptors (FGFRs) forms a complex permitting FGF23 signaling [8]. Initial findings supported specific interactions among FGF23, FGFR1c, and αKL [5]; however, multiple “c” isoforms of FGFRs (inclusion of exon 9 vs exon 8 near the transmembrane domain) appear to mediate FGF23-αKL signaling in vitro [17]. Consistent with FGFR signaling, renal FGF23 bioactivity is primarily mediated through mitogen-activated protein kinase (MAPK) cascades [9]; however, whether a single or multiple FGFRs permit FGF23-αKL signaling in vivo, and the cell-specificity of these interactions, is currently unclear.
To begin identifying the FGFRs involved in FGF23 activity, a study demonstrated that individual deletion of FGFR3 and −4 on the Hyp genetic background (mouse model of X-linked hypophosphatemia (XLH) with 10-fold elevated FGF23) did not correct the hypophosphatemia in this model. However, compound deletion of FGFR3/4 partially reversed the Hyp biochemical defects [10]. In a more recent study, breeding a kidney conditional FGFR1-KO using the metanephric mesenchyme Pax3-cre recombinase (global FGFR1-KO is lethal) on a global FGFR4-KO background produced double mutant mice with significantly elevated serum FGF23 (approximately 45-fold) [11•]. The compound mutants also had elevated serum phosphate despite the high serum FGF23, as well as increased brush border membrane phosphate transport [11•] and decreased pERK1/2 activity in response to FGF23 injections, supporting FGF23 resistance with FGFR deletion. These studies were consistent with FGFR1, and perhaps FGFR4, as playing key roles in renal FGF23 bioactivity.
To examine the role of FGFR-dependent signaling in renal phosphate metabolism, wild-type (WT) and NPT2a-KO mice were provided high and low phosphate-containing feed, as well as acute switches between the two diets. Interestingly, during a switch from high phosphate diet to low phosphate, over an 8-h time course serum FGF23 concentrations remained steady; however, brush border membrane Pi transport activity and NPT2a, but not NPT2c and Pit-2, abundance acutely increased with this change [12]. The adapter protein FRS2α was downregulated with diet switch [12], supporting the idea that changes in the expression of signaling proteins may control FGF23 bioactivity through FGFRs and αKL in kidney, providing a “brake” when FGF23 is inappropriately elevated or during metabolic adaptation.
Local FGFR/FGF Regulation of Phosphate Metabolism Through FGF23
Fibroblast Growth Factor Receptor-1 (FGFR1) Activity
It has come to light that FGF23 production in bone relies not only upon systemic signals but perhaps upon more local signals known to be important for bone cell growth and homeostasis, including those elicited from the autocrine/paracrine FGFs. To test the role of bone FGFR1 activity on FGF23 expression, the receptor was conditionally deleted with the osteocyte-targeted Dentin matrix protein-1 (DMP1)-cre [13•]. Additionally, to examine the role of FGFR1 in XLH, Hyp mice were generated with osteocyte-deleted FGFR1. Dmp1-cre/FGFR1−/− mice had significantly reduced serum intact FGF23 concentrations versus the prevailing elevated levels in Hyp mice, but no alterations in serum phosphate, vitamin D homeostasis, or discernable effects on the skeleton. Interestingly, compound Hyp/FGFR1−/− mice had improvement of the rickets and osteomalacia phenotypes as well as a significant reduction in serum FGF23 and increased serum phosphate compared to Hyp [13•]. These results were corroborated in vitro with the demonstration that FGF23 promoter activity could be stimulated with FGFR1 agonists and was inhibited by a dominant negative FGFR1 construct as well as PLC and MAPK inhibitors. In addition, delivery of a monoclonal anti-FGFR1 activating antibody “R1Mab” to normal mice resulted in increased FGF23 and a mild hypophosphatemia [14], and treatment of primary cultures of differentiated rat osteoblasts induced FGF23 mRNA and FGF23 secretion. Interestingly, treatment of a kidney cell line with R1Mab was FGF23-mimetic and FGFR1 knockdown experiments inhibited these effects [14]. Thus, bone and kidney FGFR1 expression may be required for maintaining normal circulating concentrations of FGF23.
FGF2 Isoforms
Studies have also tested the cognate ligands for FGFR-mediated regulation of FGF23 production in bone. Low molecular weight (18 kD) FGF2 activates cell surface FGFRs, but high molecular weight (HMW)-FGF2 isoforms interact with intranuclear FGFR1 to activate integrative nuclear FGFR1 signaling (INFS). Interestingly, overexpression of nuclear HMW-FGF2 in bone increased FGF23 production and induced a hypophosphatemic rickets phenotype [15]. Bone marrow stromal cell cultures (BMSCs) from HMW-FGF2 transgenic mice had elevated FGF23 and an intrinsic defect in mineralization which was rescued by an FGF23 neutralizing antibody, an MAPK inhibitor and an FGFR tyrosine kinase inhibitor [15]. Conversely, global deletion of HMW-FGF2 resulted in mice with high BMD versus control littermates with increased osteoblastic activity [16•]. Although there was a reduction of FGF23 mRNA in the HMWFGF2-KO mice, normal serum phosphate and parathyroid hormone(PTH) were observed [16•]. Taken together, these studies suggest that HMW-FGF2 may influence bone through controlling FGF23 production (Fig. 1), and in situations of very high levels, FGF23 may act on bone cells through FGFRs. The extent of the role of HMW-FGF2 and whether control of this factor affects FGF23 in disorders of phosphate handling remain to be determined.
To identify the functional differences between FGF2 isoforms (HMW and low molecular weight or “LMW”), studies were also undertaken in isolated osteoblast cell lines. It was shown that LMW-FGF2 induced NFAT and Ets1 binding and activation of the FGF23 promoter through the PLCγ/calcineurin/NFAT and MAPK pathways [17]. In contrast, HMWFGF2 demonstrated cAMP-dependent binding of FGFR1 and cAMP-response element-binding protein (CREB) to a conserved cAMP response element (CRE) in the FGF23 promoter adjacent to the NFAT binding site [17]. These findings are consistent with the above studies in which deletion of FGFR1 in normal and Hyp osteocytes reduced FGF23 and implicate FGF2 as a possible FGFR1 ligand playing a role in normal FGF23 expression.
Circulating α-Klotho and Interactions with Bone
The role of skeletal FGFRs in the control of FGF23 may also be associated with systemic regulators of phosphate homeostasis including cKL. A patient with severe hypophosphatemia and harboring a balanced chromosomal translocation (t9:13) in proximity to the α-KLOTHO gene was identified [18]. In addition, this patient had elevated FGF23, inappropriately normal 1,25D, as well as hyperparathyroidism. Upon testing the patient’s serum, it was determined that the translocation caused elevated cKL [18], supporting that the excess cKL drove the expression of FGF23 under prevailing hypophosphatemic conditions (Fig. 1), typically suppressive for FGF23 production. In an effort to determine the molecular mechanisms underlying the role of cKL in FGF23 expression, using adeno-associated virus (AAV-cKL), this isoform was delivered to mice for 4–8 weeks [19•]. Similar to the translocation patient, the cKL-treated mice had highly elevated serum intact FGF23 with osteomalacia, as well as increased bone FGF23 mRNA despite hypophosphatemia. In vitro, treatment of FGFR1-stably expressing cells with cKL resulted in increased p-ERK1/2 activity, supporting interactions between cKL and FGFRs upstream of FGF23 production [19•]. The regulation of cKL cleavage from mKL in terms of phosphate handling is not well understood; however, the major site of αKL expression is kidney, thus molecular interactions between cKL and FGFRs in bone could potentially act to fine-tune circulating FGF23.
PTH, Calcium Balance, and FGF23 Interactions
In addition to local control of FGF23 in bone, key findings in the systemic regulation of FGF23 have revealed potentially novel regulatory steps in phosphate homeostasis. PTH is secreted following a reduction in serum Ca2+ concentrations and acts upon the renal distal nephron to increase Ca2+ reabsorption, as well as downregulates NPT2a in proximal tubule cells to reduce phosphate reabsorption [20]. PTH also increases the renal 1α-hydroxylase (Cyp27b1), thus stimulating the synthesis of active 1,25D, the reciprocal effect of FGF23. The elevation in plasma 1,25D promotes calcium and phosphate absorption in the intestine, and through negative feedback, reduces PTH production in the parathyroid glands (Fig. 1). The relationships between PTH and FGF23 in vivo appears to be more complex than those between 1,25D and FGF23, where 1,25D has been shown to directly stimulate FGF23 production through VDR sites on the FGF23 promoter in vitro [21], supporting in vivo studies with similar effects on FGF23 following 1,25D injections into rodents [2]. Results from ex vivo studies demonstrated that FGF23 suppressed PTH in cultured bovine parathyroid cells [22] and during direct incubation of the exposed parathyroid gland in live rats [23]. The parathyroid glands are known to express αKL [22, 23], and the inhibition of PTH by FGF23 is in agreement with a potential negative feedback loop where PTH increased FGF23 in primary cultures of differentiated osteoblasts/osteocytes [24].
To identify the mechanisms underlying the influence of FGF23 on PTH, conditional genetic studies have been performed in mice. In this regard, parathyroid-specific deletion of αKL with a PTH-promoter Cre (producing PTH-KL−/− mice) reduced MAPK/ERK signaling [25•], thought to be the primary pathway for FGF23-αKL-FGFR bioactivity [26]. In mice devoid of parathyroid αKL, PTHR expression, serum PTH concentrations, as well as responses to FGF23 challenge were however not different from control mice [25•]. In contrast, serum 1,25D concentrations in PTH-KL−/− mice were double those of control littermates. The nuclear factor of activated T cells (NFAT) cascade was then tested as an alternative FGFR-mediated signaling pathway in parathyroid glands [25•]. Interestingly, the PTH-KL−/− mice had constitutively active calcineurin-NFAT signaling, as tested by increased MCIP1 and nuclear localization of NFATC2. Treatment of the PTH-KL−/− mice with the calcineurin-inhibitor cyclosporine A abolished FGF23-mediated PTH suppression whereas wild-type mice remained responsive to FGF23 administration. Similar results were observed in thyro-parathyroid explants, supporting FGF23 actions through an FGFR and NFAT signaling [25•]; however, the FGFR receptor(s) guiding these pathways have yet to be identified. Taken together, these studies support the concept that FGF23 may signal through mechanisms in addition to αKL-FGFR MAPK/ERK cascades in the parathyroid glands. Additionally, future studies on patients receiving kidney transplants and treated with immunosuppressive therapies, such as cyclosporine A, could be an important research area as this patient population may be susceptible to enhanced PTH secretion.
To test the mechanisms underlying the effects of PTH-stimulated FGF23 expression in bone as the closing actions in a negative feedback loop between the parathyroids and bone, the nuclear orphan receptor Nurr1 was examined as downstream of PTH actions [27]. Results using the UMR-106 osteosarcoma cell line demonstrated that Nurr1 was required for the FGF23 mRNA increases following PTH treatment. In addition, luciferase and ChIP assays supported direct interactions between Nurr1 and the FGF23 promoter [27]. In a rat CKD model, the calcimimetic R586 decreased Nurr1 expression which coincided with decreases in FGF23 mRNA. Thus, the presence of a feedback loop between bone and parathyroid through FGF23 may be important for maintaining skeletal homeostasis through phosphate and calcium balance.
Calcium and FGF23
FGF23 activity in the kidney can affect not only phosphate handling but also calcium metabolism through its actions on 1,25D and PTH production. However, whether calcium plays a direct role in FGF23 induction within a novel feedback loop is unclear. Indeed, when FGF23 suppresses renal 1,25D production, dietary uptake of phosphate and calcium is reduced. PTH, as stated above, counters the FGF23 effects on kidney 1,25D production; therefore, it was postulated that excess calcium stimulates FGF23 in a feedback regulatory loop similar to phosphate. Treating mice for 4 weeks with PTH, 1,25D, or placing mice on a high calcium (2 %)/high phosphate (1.2 %) diet for 9 weeks showed a significant positive correlation between serum calcium and iFGF23 although FGF23 is known to be stimulated by PTH, 1,25D, and high phosphate [28•]. Increased blood calcium represses serum PTH within 5 min after infusion [29]. In contrast, response of FGF23 was delayed with a modest induction of serum intact levels at 5 h after calcium injection and continued in an upward trend to 12 h posttreatment [28•]. Genetic ablation of PTH or the calcium-sensing receptor (CaSR) in vivo did not prevent calcium from stimulating FGF23 protein [30]. Conversely, Cyp27b1-KO mice which are hypophosphatemic due to a lack of 1,25D production, showed no increase in FGF23 when given a bolus injection of calcium [28•, 31]. This suggests, in parallel to other in vivo studies, low phosphate levels may block the ability of calcium to activate FGF23 (Fig. 1). In corollary in vitro studies, addition of exogenous calcium modestly increased FGF23 promoter activity and treatment of cells with the calcium channel blocker nefidipine ablated the promoter response to calcium. Additionally, in concordance with in vivo studies, cells cultured in low calcium conditions elicited no upregulation of FGF23 in the presence of known activators 1,25D or phosphate [28•, 31]. Although the molecular mechanisms behind this regulation remain unknown, direct interactions of calcium and phosphate at the bone cell level may be instrumental for the regulation of FGF23.
Dynamic Posttranslational Processing of FGF23 and Systemic Impact
Circulating FGF23 increases in response to a positive phosphate balance in mice and humans [32, 33], but to date, a phosphate-sensor system that controls FGF23 production in bone (or through indirect actions of other tissues) has not been identified. In addition to effects on FGF23 mRNA levels, recent evidence suggests that the control of iFGF23 secretion may arise through posttranslational regulation via a process involving stepwise modifications and minute to minute “decision-making” on the status of FGF23 protein by osteoblasts and osteocytes. This cell system has activity that results in either iFGF23 secretion (residues 25–251 comprises the full-length protein) or secretion as at least two inactive proteolytic cleavage products (N-terminal residues 25–179) and C-terminal residues 180–251). There are several ELISAs available to measure serum FGF23. ELISAs that recognize FGF23 antigens “C-terminal” (“cFGF23”) to the FGF23 176RXXR179/S180 SPC cleavage site (from Immutopics, Int’l.; [34]) have been developed for human and rodent FGF23. This assay binds bioactive iFGF23, as well as C-terminal proteolytic fragments that arise from cleavage of the mature hormone. The cFGF23 assays therefore may act as an approximate serum “read out” for bone FGF23 mRNA concentrations. An ELISA that uses conformation-specific monoclonal antibodies that biochemically react with epitopes N- and C-terminal to the FGF23 176RXXR179/S180 SPC site detects iFGF23 across a fairly wide species range (from Kainos, Inc.; [35]). More recently, a rodent-specific iFGF23 assay has been developed that reacts with rat and mouse FGF23 but not human (from Immutopics, Int’l.). As described below, the assays have been useful in determining the ratios of iFGF23/cFGF23, which are diagnostic for changes in the balance of circulating FGF23 isoforms under systemic challenge, and in some FGF23-related diseases where patients have inappropriate “over” or “under” degradation of FGF23 protein.
Mature FGF23 protein is O-glycosylated on T178 by the GALNT3 GalNAc-transferase within the FGF23 RHT178R/S subtilisin-like proprotein convertase protease (SPC) motif, and this glycosylation is necessary for intact hormone stability [36, 37]. Clues to the concept of regulated FGF23 protein processing to control active blood levels arose from the disorder GALNT3-hfTC (hyperphosphatemic familial tumoral calcinosis) and the hfTC variant hyperostosis-hyperphosphatemia syndrome (HHS). In these conditions, loss of function mutations in GALNT3 leads to destabilization of iFGF23 from lack of glycosylation and thus “protection” of the RHTR/S cleavage site. The excess FGF23 proteolysis results in the ELISA detection of low iFGF23 but high cFGF23 in patient sera [38]. To test the molecular relationships between GALNT3 and FGF23, the GALNT3-KO mouse was bred with the autosomal dominant hypophosphatemic rickets (ADHR) knock-in mouse, which carries an orthologous human FGF23 stabilizing mutation (R176Q) [39]. Biochemical phenotyping of the GALNT3-KO/autosomal-dominant hypophosphatemic rickets (ADHR) mice demonstrated that the ADHR mutation completely rescued the serum phosphate and elevated cFGF23 (an indicator of “overcutting” and inactivation of FGF23) in the GALNT3-KO mice [40•], supporting the importance of the SPC motif in FGF23 stabilization and therefore activity. Additionally, recent results from the Mendelian disorder Raine’s syndrome (ARHR type 3), a recessive disorder that results from loss of function mutations in the family with sequence similarity 20, member C (FAM20c) pointed to FGF23 stabilizing events reciprocal to degradation in hfTC [41]. FAM20c is an “atypical” kinase localized to the trans-Golgi network (TGN) that phosphorylates proteins within multiple motifs [42, 43], including Serine-x-Aspartic acid (“S-x-E”) residues, which are found on secreted extracellular matrix proteins such as osteopontin (OPN) and DMP1 [43, 44]. Interestingly, in a patient with Raine’s syndrome, compound heterozygous mutations in FAM20c were associated with increased plasma iFGF23 [45•]. This patient was hypophosphatemic in childhood, had marked dental disease from infancy, as well as the formation of ectopic brain calcifications. Similar to Raine’s patients, Fam20c-KO mice also have increased iFGF23 and severe hypophosphatemia [46].
It was noted that the C-terminal portion of the FGF23 SPC protease site contains an S-x-E motif: 176RHT178R179/S 180 AE. Subsequently, mass spectrometry revealed that FAM20c phosphorylated FGF23 on S180 [47••], and in vitro studies demonstrated that this phosphorylation event prohibits GALNT3 O-glycosylation of T178, making FGF23 susceptible to intracellular protease cleavage [47••]. Wild-type FAM20c almost completely blocked FGF23 glycosylation, whereas coexpressing FAM20c cDNA carrying a Raine’s syndrome mutation only partially inhibited GALNT3 glycosylation of FGF23. This observation supports that patients with Raine syndrome and elevated FGF23 likely have reduced ability to phosphorylate FGF23 S180, enhancing GALNT3 glycosylation on T178 and thus leading to a greater proportion of iFGF23 and the hypophosphatemic phenotype [47••]. The SPC protease that catalyzes the cleavage of FGF23 was uncertain, but of the isoforms tested, PSCK1-3 (furin) was capable of cleaving native (unphosphorylated) or FGF23 phosphorylated at S180, consistent with the idea that furin could be another key target for modifying the balance of circulating concentrations of iFGF23 and cFGF23 (Fig. 1). Solving the crystal structure for FAM20c demonstrated that there are no classic kinase activation loop domains [48] indicating that the cellular secretion of FAM20c could be the primary mode to reduce activity, consistent with the ability to sense minute to minute changes in serum phosphate or respond to hormonal stimuli. Additionally, inactivating mutations in a related gene, FAM20a, cause amelogenesis imperfecta and gingival fibromatosis syndrome [49], and mouse deletion of FAM20a is associated with severe enamel anomalies [50]. FAM20a was determined to be a pseudokinase that forms a functional complex with FAM20c, giving FAM20c higher activity [51]. Thus, modification of interactions between FAM20c and −20a could also affect the bioactivity of circulating iFGF23 through enabling or preventing FGF23 phosphorylation and ultimately its degradation.
Responses to Systemic Iron Metabolism
Emerging data from clinical and translational studies demonstrated that FGF23 is controlled in bone by pathways not previously associated with phosphate metabolism. In this regard, early clinical studies reported that patients with anemia and treated with specific iron preparations (such as iron maltose but not iron dextran) had associated elevations in FGF23, as well as hallmark manifestations of excess FGF23 including hypophosphatemia with inappropriately normal or reduced 1,25D [52, 53]. Maintaining proper iron handling is important for bone growth due to deleterious direct and indirect effects of excess iron and anemia. Iron metabolism in bone is important for proper collagen synthesis through modifications controlling protein hydroxylation [54], as well as renal 1,25D production as an integral portion of the CYP family of metabolic enzymes [55], and iron overload leads to oxidative stress in bone and other tissues [56]. Additional clues to FGF23 regulation by factors outside of the characterized feedback loops known to exist for phosphate metabolism came from the close examination of ADHR patients [39]. ADHR is a disorder of FGF23 excess due to FGF23 gain of function mutations occurring within the FGF23 SPC proteolytic cleavage site (176RHTR179/S180AE) [39], and substitute the charged arginines (R) at positions 176 or 179 with glutamine (Q) or tryptophan (W) [39, 57, 58]. Upon modification of the SPC recognition motif by the ADHR mutations, an FGF23 species more resistant to intracellular furin-like cleavage and inactivation is produced.
The late-onset ADHR disease phenotypes arise in physiological situations of iron deficiency, such as puberty and during pregnancy [59]. In patients with ADHR, increases in iFGF23 and cFGF23 occur in parallel with disease onset as determined using the intact and C-terminal ELISAs. It was discovered that iFGF23 and cFGF23 were inversely correlated with serum iron concentrations in ADHR, but only cFGF23 was elevated coincident with low iron in control subjects [60]. This may indicate that normal individuals “sense” the amount of FGF23 required for proper phosphate homeostasis and proactively cleave iFGF23 to maintain appropriate circulating concentrations. Additionally, the inverse nature between serum iron and iFGF23 was evident for 1,25D and serum phosphate in ADHR patients during longitudinal analyses [60], as would be expected if low serum iron increased iFGF23 as the “driver” of the biochemical manifestations in ADHR. Interestingly, when tested in XLH patients, serum cFGF23 and iFGF23 were higher in XLH patients, but similar to normal individuals, the ratio of cFGF23 to iron was inversely proportional and there was no significant relationships between serum iron and iFGF23 [61]. These studies support that the pathogenesis of elevated iFGF23 in XLH is different from ADHR and it would be expected therefore that iron repletion therapy would be likely to impact patients with ADHR versus those with XLH (see below).
Due to the fact that ADHR is unique among the FGF23-related disorders of phosphate handling in that patients display early or late onset, the ADHR knock-in mice were placed on an iron-deficient diet to mechanistically test the role of anemia in FGF23 regulation [62]. After receiving low iron diets, adult ADHR mice increased bone FGF23 mRNA, and similar to ADHR patients, had elevated serum iFGF23 and cFGF23 with significant hypophosphatemia. In contrast, WT mice also increased FGF23 mRNA but had normal iFGF23 with elevated cFGF23. Taken together, these results suggested that anemia stimulated FGF23 transcription, but ADHR mice could not compensate by cleaving FGF23 due to the proteolytic-resistant R176Q-FGF23. However, WT mice could inactivate FGF23 through proteolysis, evident by the elevated cFGF23 and normal iFGF23, and accompanying normal serum phosphate [62], in parallel to the above clinical ADHR results. To examine FGF23 in pregnancy and during neonatal life, in a translational study, pregnant WT and ADHR breeder mice were placed on low iron diets in the final week of pregnancy (days 14–21) to mimic iron deficiency anemia in last trimester and maintained on this diet to nurse pups until weaning (3 weeks of age) [63•]. Both WT and ADHR pups were hypophosphatemic with markedly increased iFGF23, with the ADHR mice being more severely affected due to the stabilizing R176Q FGF23 mutation. Additionally, hypoxia was tested under cell culture conditions and FGF23 mRNA was significantly increased coordinate with stabilization of the iron-sensing transcription factor hypoxia-inducible factor (HIF)-1α [63•]. Hypophosphatemia is also associated with states of inflammation such as sepsis. Recent evidence supports that agents associated with inflammation such as TNF and IL-1β stimulate FGF23 production in cultured osteocyte cell lines and human bone samples in an NF-κB-dependent manner [64]. Further research is needed in this interesting area, as these studies suggest that iron and inflammation could potentially affect circulating FGF23 in vivo; however, in a study examining children, FGF23 was associated with iron status and not with inflammation markers [65].
Emerging Treatments
Iron Therapy for ADHR
The findings that ADHR patients [60] and ADHR mice [62, 63•] had elevated iFGF23 during iron deficiency, coupled with the fact that iron supplementation in ADHR mice reversed the increased iFGF23 and hypophosphatemia [63•], and FGF23 is associated with iron levels in children [65] provided the basis for the hypothesis that if anemic, ADHR patients may reduce FGF23 and benefit from iron repletion. In a case study, a child with ADHR (confirmed FGF23 R179Q mutation) and a family history of waxing and waning of disease was provided high dose iron supplementation. This patient was withdrawn from phosphate and alfacalcidol (1,25D analog) due to the fact that her father’s symptoms spontaneously resolved; however, her hypophosphatemia did not improve [66•]. The patient then received increasing doses of iron supplements (iron (II) sulfate), which increased serum iron and in parallel, improved serum phosphate and TmP/GFR and was associated with elevated serum 1,25D. Over a period of 6 months, serum iron was stabilized and the rickets medication was reduced, then withdrawn, and iron supplementation ceased [66•]. After all medication withdrawal, iFGF23 was normalized in the patient. Although, it cannot be completely ruled out that patient’s improvement could be due to factors outside of iron homeostasis given that she was followed for almost 9 years and considering the documented waxing and waning of ADHR. Although more patients should be observed, the steadily improved patient biochemical profile and pronounced effects on 1,25D while receiving iron supplementation are consistent with the potential use of iron as a novel stand alone, or combination therapy, for clinical management of ADHR.
FGF23 Neutralizing Antibody
The current therapy for FGF23-mediated hypophosphatemic disorders consists of high-dose oral calcitriol (1,25D analog) and phosphate, which usually leads to healing of the rickets and osteomalacia. However, the treatment can have complications including diarrhea from phosphate, hyperparathyroidism, and nephrocalcinosis. It was reported that the hypophosphatemic rickets phenotype of the Hyp mouse model of XLH could be almost completely reversed using FGF23 neutralizing antibodies [67]. With the development of humanized monoclonal antibodies specifically targeting iFGF23 (“KRN23”), a single dose early phase trial to treat XLH patients was undertaken [68••]. Injection of the antibody subcutaneously resulted in increased TMP/GFR, as well as elevated serum phosphate and 1,25D concentrations. Hyperphosphatemia was not a complication although plasma 1,25D was elevated at higher doses over a time course of up to 50 days. An escalation and extended time course study of KRN23 was then undertaken in a subset of patients with a dosing regimen of up to 1 year [69]. During the extended phase of the trial, monthly KRN23 delivery increased serum phosphate and TmP/GFR in all subjects. Although 1,25D concentrations transiently increased, average serum and urinary calcium remained within the normal ranges [69]. Thus, long-term treatment blocking the activity of FGF23 may be a novel therapeutic approach for XLH and other FGF23-related diseases, including tumor-induced osteomalacia (TIO), an acquired hypophosphatemic disease where patients have FGF23-producing mesenchymal tumors that are often difficult to localize [70].
Conclusions
In conclusion, recent work has shown that the interplay of systemic factors such as ions, including calcium, phosphate, and iron, as well as hormones including PTH and 1,25D, in combination with local factors such as FGFs, play essential roles in bone FGF23 production, intracellular processing, and function. The downstream actions on bone caused by modifying circulating FGF23 impacts important features of bone homeostasis including mineralization. Arising from this collective work are several new therapeutic paradigms targeting FGF23 and its effectors for treating diseases of altered mineral metabolism.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology. 2005;146(12):5358–64.
Shimada T, Hasegawa H, Yamazaki Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19(3):429–35.
Shimada T, Mizutani S, Muto T, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98(11):6500–5.
Farrow EG, Summers LJ, Schiavi SC, McCormick JA, Ellison DH, White KE. Altered renal FGF23-mediated activity involving MAPK and Wnt: effects of the Hyp mutation. J Endocrinol. 2010;207(1):67–75.
Urakawa I, Yamazaki Y, Shimada T, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444(7120):770–4.
Matsumura Y, Aizawa H, Shiraki-Iida T, Nagai R, Kuro-o M, Nabeshima Y. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem Biophys Res Commun. 1998;242(3):626–30.
Bloch L, Sineshchekova O, Reichenbach D, et al. Klotho is a substrate for alpha-, beta- and gamma-secretase. FEBS Lett. 2009;583(19):3221–4.
Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, Shi M, Eliseenkova AV, Razzaque MS, Moe OW, et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci U S A. 2009.
Kurosu H, Ogawa Y, Miyoshi M, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281(10):6120–3.
Li H, Martin A, David V, Quarles LD. Compound deletion of Fgfr3 and Fgfr4 partially rescues the Hyp mouse phenotype. Am J Physiol Endocrinol Metab. 2011;300(3):E508–17.
Gattineni J, Alphonse P, Zhang Q, Mathews N, Bates CM, Baum M. Regulation of renal phosphate transport by FGF23 is mediated by FGFR1 and FGFR4. Am J Physiol Renal Physiol. 2014;306(3):F351–8. In this in vivo animal study, FGFRs were conditionally or globally deleted from mice, and the results were consistent with FGFR1 and FGFR4 playing significant roles in mediating renal FGF23-dependent phosphate metabolism.
Bourgeois S, Capuano P, Stange G, et al. The phosphate transporter NaPi-IIa determines the rapid renal adaptation to dietary phosphate intake in mouse irrespective of persistently high FGF23 levels. Pflugers Arch. 2013;465(11):1557–72.
Xiao Z, Huang J, Cao L, Liang Y, Han X, Quarles LD. Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PLoS One. 2014;9(8):e104154. This study examined the effects on conditional deletion of FGFR1 from mouse osteocytes on FGF23. FGF23 is decreased in animals with deleted receptor and that FGFR1 deletion rescued the elevated FGF23 and biochemical phenotype of the Hyp mouse model of XLH, suggesting this receptor plays a role in normal FGF23 production and potentially during genetic hypophosphatemia.
Wu AL, Feng B, Chen MZ, et al. Antibody-mediated activation of FGFR1 induces FGF23 production and hypophosphatemia. PLoS ONE. 2013;8(2):e57322.
Xiao L, Esliger A, Hurley MM. Nuclear fibroblast growth factor 2 (FGF2) isoforms inhibit bone marrow stromal cell mineralization through FGF23/FGFR/MAPK in vitro. J Bone Miner Res. 2013;28(1):35–45.
Homer-Bouthiette C, Doetschman T, Xiao L, Hurley MM. Knockout of nuclear high molecular weight FGF2 isoforms in mice modulates bone and phosphate homeostasis. J Biol Chem. 2014;289(52):36303–14. With ubiquitous knock out of HMW-FGF2, mice showed increased BMD, and primary bone cultures from these mice had reduced bone Fgf23, Sost mRNA, and serum sclerostin, supporting that FGF2 isoforms may be upstream of genes involved in mineral ion handling.
Han X, Xiao Z, Quarles LD. Membrane and integrative nuclear fibroblastic growth factor receptor (FGFR) regulation of FGF-23. J Biol Chem. 2015;290(16):10447–59.
Brownstein CA, Adler F, Nelson-Williams C, et al. A translocation causing increased alpha-klotho level results in hypophosphatemic rickets and hyperparathyroidism. Proc Natl Acad Sci U S A. 2008;105(9):3455–60.
Smith RC, O'Bryan LM, Farrow EG, et al. Circulating alphaKlotho influences phosphate handling by controlling FGF23 production. J Clin Invest. 2012;122(12):4710–5. This publication was the first to show that a soluble form of alpha-Klotho, cKL, could increase FGF23 production in bone, and overexpression of this factor produced a similar hypophosphatemic osteomalacia observed in a patient with cKL overexpression due to a KLOTHO gene translocation.
Bacic D, Lehir M, Biber J, Kaissling B, Murer H, Wagner CA. The renal Na+/phosphate cotransporter NaPi-IIa is internalized via the receptor-mediated endocytic route in response to parathyroid hormone. Kidney Int. 2006;69(3):495–503.
Liu S, Tang W, Zhou J, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol. 2006;17(5):1305–15.
Krajisnik T, Bjorklund P, Marsell R, et al. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol. 2007;195(1):125–31.
Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117(12):4003–8.
Rhee Y, Bivi N, Farrow E, et al. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone. 2011;49(4):636–43.
Olauson H, Lindberg K, Amin R, et al. Parathyroid-specific deletion of Klotho unravels a novel calcineurin-dependent FGF23 signaling pathway that regulates PTH secretion. PLoS Genet. 2013;9(12):e1003975. Conditional deletion of alpha-Klotho from mouse parathyroid gland produced mice that were normal for calcium metabolism and PTH production. Investigation of alternative signaling pathways implicated NFAT signaling as controlling FGF23-mediated PTH secretion.
Farrow EG, Davis SI, Summers LJ, White KE. Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J Am Soc Nephrol. 2009;20(5):955–60.
Meir T, Durlacher K, Pan Z, et al. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. 2014;86(6):1106–15.
David V, Dai B, Martin A, Huang J, Han X, Quarles LD. Calcium regulates FGF-23 expression in bone. Endocrinology. 2013;154(12):4469–82. Using conditional deletions of target genes in parathyroid in combination with diet and in vitro promoter studies, these studies supported a role for calcium-mediated regulation of FGF23 production in bone.
Gravesen E, Mace ML, Hofman-Bang J, Olgaard K, Lewin E. Circulating FGF23 levels in response to acute changes in plasma Ca(2+). Calcif Tissue Int. 2014;95(1):46–53.
Quinn SJ, Thomsen AR, Pang JL, et al. Interactions between calcium and phosphorus in the regulation of the production of fibroblast growth factor 23 in vivo. Am J Physiol Endocrinol Metab. 2013;304(3):E310–20.
Rodriguez-Ortiz ME, Lopez I, Munoz-Castaneda JR, et al. Calcium deficiency reduces circulating levels of FGF23. J Am Soc Nephrol. 2012;23(7):1190–7.
Antoniucci DM, Yamashita T, Portale AA. Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. J Clin Endocrinol Metab. 2006;91(8):3144–9.
Burnett SM, Gunawardene SC, Bringhurst FR, Juppner H, Lee H, Finkelstein JS. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res. 2006;21(8):1187–96.
Jonsson KB, Zahradnik R, Larsson T, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348(17):1656–63.
Yamazaki Y, Okazaki R, Shibata M, et al. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab. 2002;87(11):4957–60.
Bergwitz C, Banerjee S, Abu-Zahra H, et al. Defective O-glycosylation due to a novel homozygous S129P mutation is associated with lack of fibroblast growth factor 23 secretion and tumoral calcinosis. J Clin Endocrinol Metab. 2009;94(11):4267–74.
Kato K, Jeanneau C, Tarp MA, et al. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem. 2006;281(27):18370–7.
Frishberg Y, Ito N, Rinat C, et al. Hyperostosis-hyperphosphatemia syndrome: a congenital disorder of O-glycosylation associated with augmented processing of fibroblast growth factor 23. J Bone Miner Res. 2007;22(2):235–42.
ADHR-Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26(3):345–8.
Ichikawa S, Gray AK, Padgett LR, et al. Genetic rescue of glycosylation-deficient Fgf23 in the Galnt3 knockout mouse. Endocrinology. 2014;155(10):3891–8. To determine the role of glycosylation in iFGF23 stability, the GALNT3-KO mouse was bred to the ADHR knock-in mouse. The ADHR point mutation (R176Q) stabilized FGF23 and completely rescued the GALNT3-KO mouse phenotypes of elevated cFGF23 and hyperphosphatemia demonstrating that glycosylation is required for FGF23 secretion but not in vivo bioactivity.
Ababneh FK, AlSwaid A, Youssef T, Al Azzawi M, Crosby A, AlBalwi MA. Hereditary deletion of the entire FAM20C gene in a patient with Raine syndrome. Am J Med Genet A. 2013;161A(12):3155–60.
Tagliabracci VS, Wiley SE, Guo X, et al. A single kinase generates the majority of the secreted phosphoproteome. Cell. 2015;161(7):1619–32.
Tagliabracci VS, Engel JL, Wen J, et al. Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science. 2012;336(6085):1150–3.
Ishikawa HO, Xu A, Ogura E, Manning G, Irvine KD. The Raine syndrome protein FAM20C is a Golgi kinase that phosphorylates bio-mineralization proteins. PLoS ONE. 2012;7(8):e42988.
Rafaelsen SH, Raeder H, Fagerheim AK, et al. Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J Bone Miner Res. 2013;28(6):1378–85. This clinical genetic study demonstrated that mutations in FAM20c are associated with elevated FGF23 in Raine’s syndrome, linking the FAM20c kinase to phosphate homeostasis.
Wang X, Wang S, Li C, et al. Inactivation of a novel FGF23 regulator, FAM20C, leads to hypophosphatemic rickets in mice. PLoS Genet. 2012;8(5):e1002708.
Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzales DJ, Appaiah HN, Koller A, Nizet V, White KE, and Dixon JE. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. In press. Proc Natl Acad Sci U S A. 2014. The novel kinase FAM20c was shown to phosphorylate FGF23 and this action prevented GALNT3-mediated O -glycosylation of FGF23, leading to enhanced degradation of the mature hormone. This paper likely explains the manifestations of Raine's syndrome where patients have loss of function FAM20c mutations and increased FGF23 stability.
Xiao J, Tagliabracci VS, Wen J, Kim SA, Dixon JE. Crystal structure of the Golgi casein kinase. Proc Natl Acad Sci U S A. 2013;110(26):10574–9.
O'Sullivan J, Bitu CC, Daly SB, et al. Whole-Exome sequencing identifies FAM20A mutations as a cause of amelogenesis imperfecta and gingival hyperplasia syndrome. Am J Hum Genet. 2011;88(5):616–20.
Vogel P, Hansen GM, Read RW, et al. Amelogenesis imperfecta and other biomineralization defects in Fam20a and Fam20c null mice. Vet Pathol. 2012;49(6):998–1017.
Cui J, Xiao J, Tagliabracci VS, Wen J, Rahdar M, and Dixon JE. A secretory kinase complex regulates extracellular protein phosphorylation. eLife. 2015;4(e06120).
Prats M, Font R, Garcia C, Cabre C, Jariod M, Vea AM. Effect of ferric carboxymaltose on serum phosphate and C-terminal FGF23 levels in non-dialysis chronic kidney disease patients: post-hoc analysis of a prospective study. BMC Nephrol. 2013;14(1):167.
Schouten BJ, Hunt PJ, Livesey JH, Frampton CM, Soule SG. FGF23 elevation and hypophosphatemia after intravenous iron polymaltose: a prospective study. J Clin Endocrinol Metab. 2009;94(7):2332–7.
Homan EP, Lietman C, Grafe I, et al. Differential effects of collagen prolyl 3-hydroxylation on skeletal tissues. PLoS Genet. 2014;10(1):e1004121.
Sugimoto H, Shinkyo R, Hayashi K, et al. Crystal structure of CYP105A1 (P450SU-1) in complex with 1alpha,25-dihydroxyvitamin D3. Biochemistry. 2008;47(13):4017–27.
Toxqui L, Vaquero MP. Chronic iron deficiency as an emerging risk factor for osteoporosis: a hypothesis. Nutrients. 2015;7(4):2324–44.
White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001;60(6):2079–86.
Shimada T, Muto T, Urakawa I, et al. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology. 2002;143(8):3179–82.
Econs MJ, McEnery PT. Autosomal dominant hypophosphatemic rickets/osteomalacia: clinical characterization of a novel renal phosphate-wasting disorder. J Clin Endocrinol Metab. 1997;82(2):674–81.
Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, and Econs MJ. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. J Clin Endocrinol Metab. 2011.
Imel EA, Gray AK, Padgett LR, Econs MJ. Iron and fibroblast growth factor 23 in X-linked hypophosphatemia. Bone. 2014;60:87–92.
Farrow EG, Yu X, Summers LJ, et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc Natl Acad Sci U S A. 2011;108(46):E1146–55.
Clinkenbeard EL, Farrow EG, Summers LJ, et al. Neonatal iron deficiency causes abnormal phosphate metabolism by elevating FGF23 in normal and ADHR mice. J Bone Miner Res. 2014;29(2):361–9. This translational work tested FGF23 production in young ADHR and wild-type (WT) mice following challenge with a low iron diet. ADHR mice had similar increases in FGF23 mRNA, but more severe hypophosphatemic osteomalacia than WT mice most likely due to stabilization of FGF23. Additional studies demonstrated that hypoxia in the setting of normal iron also increased FGF23.
Ito N, Wijenayaka AR, Prideaux M, et al. Regulation of FGF23 expression in IDG-SW3 osteocytes and human bone by pro-inflammatory stimuli. Mol Cel Endocrinol. 2015;399:208–18.
Braithwaite V, Prentice AM, Doherty C, Prentice A. FGF23 is correlated with iron status but not with inflammation and decreases after iron supplementation: a supplementation study. Int J Pediatr Endocrinol. 2012;2012(1):27.
Kapelari K, Kohle J, Kotzot D, and Hogler W. Iron supplementation associated with loss of phenotype in autosomal dominant hypophosphatemic rickets. J Clin Endocrinol Metab. 2015:20152391. This case report described a therapeutic course of iron treatment in a child with iron deficiency ADHR. Iron (II) sulfate reduced FGF23 and increased 1,25D and the patient was eventually removed from clinical management of phosphate and 1,25D analog supplementation.
Aono Y, Shimada T, Yamazaki Y, et al. The neutralization of FGF-23 ameliorates hypophosphatemia and rickets in Hyp mice. J Bone Miner Metab. 2003;18:S16.
Carpenter TO, Imel EA, Ruppe MD, et al. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Invest. 2014;124(4):1587–97. A clinical trial of FGF23 neutralizing antibody as a novel treatment for XLH demonstrated improvement in patient hypophosphatemia and TmP/GFP with minimal reported side effects.
Imel EA, Zhang X, Ruppe MD, et al. Prolonged correction of serum phosphorus in adults with x-linked hypophosphatemia using monthly doses of KRN23. J Clin Endocrinol Metab. 2015;100(7):2565–73.
Ryan EA, Reiss E. Oncogenous osteomalacia. Review of the world literature of 42 cases and report of two new cases. Am J Med. 1984;77(3):501–12.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants R01-DK063934 and R01-DK95784 (KEW) and F32-AR065389 (ELC) and a Showalter Scholar Award funded in part through the Ralph W. and Grace M. Showalter Research Trust Fund (KEW).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Erica L. Clinkenbeard declares no conflict of interest.
Kenneth E. White received royalties from Kyowa Hakko Kirin Co., Ltd. for licensing of the FGF23 gene and the anti-FGF23 monoclonal antibody trials, as well as research funding from Eli Lilly.
Human and Animal Rights and Informed Consent
With regard to the authors’ research cited in this paper, all institutional and national guidelines for the care and use of laboratory animals were followed. Additionally, all procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Additional information
This article is part of the Topical Collection on Molecular Biology of Skeletal Development
Rights and permissions
About this article

Cite this article
Clinkenbeard, E.L., White, K.E. Systemic Control of Bone Homeostasis by FGF23 Signaling. Curr Mol Bio Rep 2, 62–71 (2016). https://doi.org/10.1007/s40610-016-0035-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40610-016-0035-5