
Abstract
Purpose of Review
Marek’s disease virus (MDV) is a highly oncogenic alphaherpesvirus that causes various clinical symptoms including fatal lymphomas in chickens. The virus encodes several MDV-specific genes that play a major role in viral pathogenesis. This review will focus on the recent advances in our understanding of how these viral factors contribute to pathogenesis and tumor formation.
Recent Findings
Several viral factors involved in MDV pathogenesis have been identified including the major oncoprotein Meq, the viral chemokine vIL-8, MDV-encoded microRNAs, RLORF4, RLORF5a, pp14, pp38, a virus-encoded telomerase RNA (vTR), and viral telomeric repeats (TMRs). Our current knowledge of the role of these viral factors in MDV pathogenesis has immensely increased over the last few years; however, more work needs to be done to completely understand the mechanisms for most of them.
Summary
MDV pathogenesis and tumor formation is a complex process. Deciphering the mechanisms of viral factors involved in MDV pathogenesis and lymphomagenesis will not only improve our understanding of this neoplastic disease but will also provide new strategies for vaccine development against this deadly pathogen.
Similar content being viewed by others


Introduction
Marek’s disease virus (MDV, Gallid herpesvirus 2) is a lymphotropic alphaherpesvirus and the causative agent of Marek’s disease (MD) [1]. MD is characterized by generalized nerve inflammation, immunosuppression, and T cell lymphomas [1]. These MDV-induced lymphomas are considered to be the most clinically diagnosed cancer in the animal kingdom [2]. In addition, MDV serves as a natural virus-host animal model for herpesvirus-induced tumorigenesis and pathogenesis [3]. MD is one of the most important infectious diseases in chickens and causes dramatic losses in poultry industry worldwide of up to 1–2 billion US-dollars annually [4]. To date, vaccines are the only option to protect chickens from this deadly disease [5].
Upon infection of the host, MDV infects and replicates efficiently in B cells. However, recent data revealed that MDV can also productively infect a plethora of cells including macrophages, dendritic cells [6•], natural killer (NK) cells (Christine Jansen, unpublished data) and T cells [7••, 8, 9]. In addition to lytic replication, MDV establishes latency in T cells. Intriguingly, the virus integrates its genome into the telomeres of host cell chromosomes in latently infected cells [10, 11]. This ensures replication of the virus genome with the host chromosomes and maintenance in the host for life. This integration event is also crucial for transformation of mostly CD4+ T cells and a prerequisite for lymphoma formation [11].
MDV pathogenesis is characterized by four overlapping phases: (i) the early cytolytic phase with an initial amplification of the virus in the infected animal, (ii) the latent phase with latency establishment predominantly in CD4+ T cells, (iii) the late cytolytic phase, and (iv) the transformation phase with a rapid lymphoma development and dissemination of these tumors preferentially into visceral organs and skeletal muscles [3, 12].
MDV encodes more than 100 proteins that play a role in various processes of the virus lifecycle [13]. This brief overview highlights the current advances in our understanding of the key viral factors that contribute to MDV pathogenesis and lymphomagenesis.
The Major Oncogene meq
The major oncogene of MDV is meq (MDV Eco Q-encoded protein, MDV005 and MDV076) that encodes a 339 amino acid nuclear basic leucine zipper protein (bZIP). The Meq protein has a DNA-binding domain and can form homodimers as well as heterodimers with cellular bZIP proteins such as c-Jun and Fos [14, 15], allowing it to regulate cellular and viral gene expression [16, 17]. Two copies are present in the MDV genome, one in the internal and one in the terminal repeat long region (IRL and TRL) (Fig. 1) [15]. Meq is constitutively expressed in both lytic and latent stages of MDV infection as well as in lymphoblastoid cell lines derived from MDV tumors [9]. In infected chickens, the percentage of Meq expressing peripheral blood mononuclear cells (PBMCs) increases in the early latent phase, but decreases thereafter [18]. A recent publication indicated that also some infected lymphocytes do not express Meq [18]. meq itself is dispensable for MDV replication in vitro [19]; however, deletion of both copies of the meq gene or parts of it completely abrogates tumor formation [20,21,22,23].

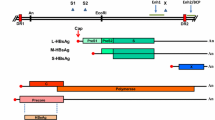
Overview of the MDV genome. Schematic representation of the MDV genome with a focus on the viral factors involved in pathogenesis and tumorigenesis. The two unique regions, unique long (UL) and short (US) are flanked by terminal (TRL and TRS) and internal (IRL and IRS) inverted repeat regions. The unique regions mainly harbor genes that are conserved among alphaherpesviruses and are involved in DNA replication and production of progeny virus. The repeat regions contain MDV-specific genes encoding for proteins or RNA that are important for pathogenesis, cellular tropism, tumorigenesis, and latency. The viral telomeric repeats (TMR) are crucial for integration of the virus genome into host telomeres and are highlighted with arrows. The position of the following genes is shown in the IRL and IRS: latency-associated transcripts (LAT), phosphoprotein 14 (pp14) and 38 (pp38), major oncogene meq, RLORF4 and 5a, viral chemokine vIL-8, viral telomerase RNA (vTR), miRNAs, and TMR
Several mechanistic pathways have been discovered for this viral oncogene. Meq represses p53-mediated transcriptional activity and apoptosis through its direct interaction with p53 [24]. It thereby plays a pivotal role in maintaining MDV latency in CD4+ T cells by blocking apoptosis [25]. Other Meq interaction partners that contribute to MDV pathogenesis include the heat shock protein 70 (hsp70) [26] and the retinoblastoma tumor suppressor protein (pRB) [4, 27], as well as Par-4 and SKP-2 [2]. Meq also was shown to trans-activate latent gene expression [28] and to suppress the promoters of the lytic MDV genes ICP4, pp38, and pp14 [29].
Repeat Long Open Reading Frame 4 and 5a
Repeat long open reading frame 4 (RLORF4) is located within the TRL/IRL regions of the MDV genome in the same orientation as meq (Fig. 1) [2, 30]. Comparative sequence analysis revealed that four out of six attenuated MDV-strains lack RLORF4, suggesting that RLORF4 plays a role in MDV pathogenesis [31]. To investigate the role of RLORF4 in MDV pathogenesis, Jarosinski and colleagues generated recombinant viruses that lack RLORF4 based on the very virulent MDV RB-1B strain [30]. Deletion of RLORF4 resulted in an increased virus replication and spread in vitro [30]; however, virus load was severely reduced in vivo. Furthermore, tumor development was severely impaired in the absence of RLORF4 compared to wild-type virus [30]. The exact mechanism that allows RLORF4 to contribute to MDV pathogenesis remains unknown. In addition, MDV encodes repeat long open reading frame 5a (RLORF5a) that is located upstream of RLORF4 (Fig. 1). Schat and colleagues initially demonstrated that deletion of RLORF5a in CVI988 did not alter virus replication [32]. This was confirmed by Jarosinski and colleagues for the very virulent RB-1B. In contrast to RLORF4, they observed that RLORF5a is dispensable for RB-1B pathogenesis and tumorigenesis [30].
Viral Interleukin-8
Viral interleukin-8 (vIL-8) (MDV003 and MDV078) is a secreted CXC chemokine that facilitates recruitment of target cells and plays a crucial role in MDV pathogenesis [33, 34]. It is the first CXC chemokine identified in an alphaherpesvirus and was initially named after interleukin-8 (cIL-8; cCXCL8), the first CXC chemokine identified in chickens [35, 36]. However, recent data demonstrated that vIL-8 is a functional orthologue to the chicken CXC ligand 13 (CXCL13) and binds to the cellular CXC receptor 5 (CXCR5) [37•]. It is expressed during lytic replication and has true late expression kinetics [36]. As mentioned above, vIL-8 plays an important role in pathogenesis as it allows recruitment of B cells, which serve as primary targets for lytic replication. In addition, it recruits CD4+ CD25+ T cells that could serve as a target for the establishment of latency and tumor formation [34]. Deletion of the vIL-8 gene or abrogation of vIL-8 expression decreases disease and tumor incidence in experimentally infected animals. In the case of a natural route of transmission, disease and tumor formation is completely abrogated [33, 34], underlining that the recruitment of target cells is crucial for MDV pathogenesis.
Splice Variants of meq, RLORF4/5a, and vIL-8
Multiple spliced transcripts have been identified in the region containing meq, RLORF4/5a, and vIL-8 [2, 38,39,40]. These splice variants include fusion proteins of Meq, RLORF4, and RLORF5a with exons II and III of vIL-8 [38]. The role of these splice variants in MDV pathogenesis remains poorly understood. Intriguingly, splice variants of meq and exons II and/or III of vIL-8 show differences regarding their localization and cellular dynamics compared to the full-length meq [41]. These splice forms are also expressed in MDV-induced tumor cells, suggesting that they might contribute to tumor formation. However, more work needs to be done to understand the contribution of these splice variants in MDV pathogenesis and tumorigenesis.
Neurovirulence Factor pp14
In addition to tumor formation, MDV can also cause various neurological symptoms. A viral protein associated with an increased neurovirulence in MDV-infected chickens is pp14 (MDV006 and MDV075), a 14 kilodalton (kDa) polypeptide (Fig. 1) [42,43,44]. pp14 is expressed with immediate early (IE) kinetics and is dispensable for virus replication and tumorigenesis [44]. Two splice variants of pp14 are expressed that differ in their N-terminal amino acid compositions and are expressed at different levels [45]. The pp14 transcript with a 5′ leader intronic internal ribosome entry site (IRES) is more abundant in MDV infected and transformed cells than its counterpart lacking this element. This is due to the ability of the 5′ IRES to mediate cap-independent translation initiation and may enable this mRNA to overcome translation inhibition [45,46,47]. In vivo, a pp14 deletion virus showed significantly less clinical MD signs compared to the wild-type virus. In addition, nerve lesions including cellular infiltration, proliferation of lymphoblastic cells, and edema in the nerve tissue were reduced in the absence of pp14 (wild type: 62.8%, pp14 deletion mutant 16.6%) [44]. However, the exact molecular mechanisms of MDV-mediated neurovirulence and how pp14 contributes to these symptoms remain elusive.
Phosphoprotein pp38
pp38 (MDV073) is a 38 kDa immediate early protein that is encoded in the junction of the UL and the IRL (Fig. 1). Deletion of the pp38 gene severely impaired tumor formation [48, 49], underlining its role in MDV pathogenesis. Besides the full-length protein, two splice variants of pp38 (Spl A and Spl B) were identified in vitro and in vivo [50]. The full-length pp38 is primarily expressed in early lytic replication, while the splice variants are present during the establishment of latency from 7 to 14 days post infection. This differential expression has been linked to an increased metabolic activity of infected cells that could contribute to the establishment of latency and/or to transformation [50, 51]. Like Meq, pp38 is also involved in the inhibition of apoptosis in MDV-infected and -transformed cells [51,52,53]. For pp38, it remains unclear if this is due to a direct block of apoptosis or the inhibition of a cytotoxic T cell response [52]. In contrast, there is evidence that full-length pp38 can induce apoptosis via the oxidative phosphorylation pathway [51, 54], a phenomenon that was not observed for the two splice variants Spl A and Spl B [51].
MDV MicroRNAs
A number of MDV-encoded microRNAs (miRNAs) have been discovered that are classified into three distinct miRNA clusters (Fig. 1). These clusters encode 14 precursor sequences and 26 mature miRNAs that are highly conserved between virus isolates [55]. They play an important role in MDV-induced pathogenesis and tumorigenesis [55]. The first cluster, termed the Meq-cluster, is located upstream of the meq oncogene and contains six pre-miRNAs (Fig. 1) [56, 57]. Deletion of this cluster severely impaired disease and tumor development, indicating that some miRNAs in this cluster play an important role in MDV-induced pathogenesis and tumorigenesis [58, 59]. The most highly expressed member of the Meq-cluster is mdv1-miR-M4-5p, a functional orthologue of the cellular gga-miR-155 [60]. gga-miR-155 is highly conserved from humans to chickens [61••] and is involved in virus-induced cancers such as Epstein-Barr virus-induced lymphomas in humans [62]. Similarly, miR-M4-5p of MDV plays a crucial role in MDV-lymphomagenesis as reviewed by Zhuang and colleagues [58, 59, 61••]. The mid-cluster is located downstream of meq and includes three pre-miRNAs (Fig. 1) [63]. This cluster is dispensable for MDV replication; however, deletion of one of the mid-cluster miRNAs, miR-M31, also leads to a decrease in MD incidence and tumorigenesis [64]. The last cluster is termed LAT-cluster as it is present within the latency-associated transcripts (LAT) and consists of five pre-miRNAs (Fig. 1) [56, 57]. The LAT-cluster encodes for at least one IE gene-specific miRNA, miR-M7-5p, that may contribute to the establishment and maintenance of latency [65].
Viral Telomerase RNA
Telomerase is a large ribonucleoprotein complex that is involved in the maintenance of telomeres at the end of eukaryotic chromosomes [66]. The telomerase complex contains two major components, the catalytic subunit telomerase reverse transcriptase (TERT) and a telomerase RNA (TR or TERC), which provides the template for the extension of the telomeres. Beyond that, the complex contains a number of species-specific telomerase-associated proteins that regulate telomerase activity and biogenesis [67, 68]. MDV encodes a viral TR (vTR) homologue that is crucial for efficient MDV-induced lymphoma formation [69]. vTR has an 88% sequence identity to the cellular TR in chickens (chTR) [70] and is the most abundant viral transcript detected in MDV-induced tumor cells [69]. Chbab and colleagues demonstrated that these high expression levels are crucial for MDV-induced tumor formation [71].
vTR is incorporated into the telomerase complex and enhances its activity when compared to chTR [72]. To investigate whether the tumor-promoting functions of vTR are dependent on its role in telomerase activity, Kaufer and colleagues generated recombinant viruses with a mutation in vTR that abrogated incorporation into the telomerase complex [73]. Intriguingly, lymphoma formation was not altered in the absence of the vTR-induced telomerase activity and only the onset of disease was slightly delayed [73]. Furthermore, tumor dissemination was also comparable to wild-type virus [73], suggesting that the tumor-promoting functions of vTR are independent of its role in the telomerase complex [73]. We recently demonstrated that another viral RNA, the Epstein-Barr virus-encoded RNA-2 (EBER-2), can complement the loss of vTR in MDV-induced tumor formation [74], suggesting conserved mechanism(s) between these viral RNAs. Further investigations are needed to decipher the mechanism of vTR in MDV-induced transformation.
MDV Telomeric Repeats
MDV establishes latency in CD4+ T cells and integrates its genome into the telomeres of their chromosomes [10, 11]. Interestingly, the integrated virus genome is usually detected in multiple chromosomes of latently infected and tumor cells [11]. Integration is facilitated by telomeric repeat (TMR) arrays present within the a-like sequences at both ends of the virus genome and at the IRL-IRS junction [75, 76]. Each a-like sequence harbors two TMR arrays: short telomeric repeats (sTMR) with a fixed number of 6 repeats and multiple telomeric repeats (mTMR) with a variable number of repeats [11, 77, 78]. Deleting or mutating the mTMR severely impaired integration, pathogenesis, and tumor formation [11]. The sTMR have a dual function in the MDV life cycle. On the one hand, the sTMR play a key role in the integration of MDV, as mutation of the sequence reduces integration frequency and decreases MDV pathogenesis and tumor formation. One the other hand, the sTMR serve as essential spacers between the packaging signal pac-1 and the DR-1 cleavage sites, as its deletion completely abrogates MDV replication [77]. Truncation analyses revealed that the exact length of the sTMR is crucial for virus replication [77]. Intriguingly, several other herpesviruses harbor TMR arrays at the ends of their genomes [79], which also contribute to the integration of the respective virus into host telomeres [78, 80]. While our understanding of the role of the TMR arrays has tremendously increased over the last years, it remains completely unknown which viral and/or cellular proteins facilitate this integration into host telomeres.
Conclusions
Most of the genes encoded in the MDV genome have homologues in other alphaherpesviruses. They play important roles in DNA replication, particle formation, egress, and many other processes essential for the virus lifecycle. In addition, MDV encodes several virus-specific genes that are not primarily involved in replication but play a key role in viral pathogenesis. The best characterized gene by far is the major oncogene meq that is crucial for tumor formation as it regulates gene expression and blocks apoptosis [81]. The viral chemokine vIL-8 ensures that the target cells are recruited to the site of infection, a prerequisite for the success of this highly cell-associated pathogen. RLORF4 and the two phosphoproteins pp14 and pp38 also drive MDV pathogenesis; however, more work needs to be done to understand how these proteins contribute to this process. In addition to these proteins, MDV also encodes several RNAs that are crucial for MDV pathogenesis such as vTR and the MDV-encoded miRNAs. Furthermore, the MDV genome also harbors sequence elements such as the viral telomeric repeats that facilitate integration into host telomeres and maintenance of its genetic material with the host chromosomes. This complex set of proteins, RNAs, and sequence elements in the virus genome contributes to MDV pathogenesis and makes it such a successful pathogen.
Abbreviations
- MDV:
-
Marek’s disease virus
- MD:
-
Marek’s disease
- vTR:
-
Viral telomerase RNA
- vIL-8:
-
Viral interleukin-8
- RL:
-
Repeat long regions
- RLORF4:
-
Repeat long open reading frame 4
- TMRs:
-
Telomeric repeats
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Calnek BW. Pathogenesis of Marek’s disease virus infection. Curr Top Microbiol Immunol. 2001;255:25–55.
Parcells MS, Burnside J, Morgan RW. Marek’s disease virus-induced T-cell lymphomas. Cancer Associated Viruses; 2012:307–335.
Osterrieder N, Kamil JP, Schumacher D, Tischer BK, Trapp S. Marek’s disease virus: from miasma to model. Nat Rev Microbiol. 2006;4(4):283–94.
Davison F, Nair V. Marek’s disease: an evolving problem. London: Elsevier; 2004.
Boodhoo N, Gurung A, Sharif S, Behboudi S. Marek’s disease in chickens: a review with focus on immunology. Vet Res. 2016;47(1):119.
• Chakraborty P, et al. Marek’s disease virus infection of phagocytes: a de novo in vitro infection model. J Gen Virol. 2017;98(5):1080–8. Chakraborty et al. developed an in vitro model for the infection of macrophages and dendritic cells with MDV and showed that the virus can spread in those cultures.
•• Schermuly J, et al. In vitro model for lytic replication, latency, and transformation of an oncogenic alphaherpesvirus. Proc Natl Acad Sci U S A. 2015;112(23):7279–84. Schermuly et al. established an in vitro infection system for B and T cells, the main target cells of MDV during infection in the host. This system allows the analysis of viral and cellular factors that contribute to virus replication, establishment of latency, and transformation in those lymphocytes.
Calnek BW, Schat KA, Ross LJN, Shek WR, Chen CLH. Further characterization of Marek’s disease virus-infected lymphocytes. I. In vivo infection. Int J Cancer. 1984;33(3):389–98.
Arumugaswami V, Kumar PM, Konjufca V, Dienglewicz RL, Reddy SM, Parcells MS. Latency of Marek’s disease virus (MDV) in a reticuloendotheliosis virus-transformed T-cell line. I: uptake and structure of the latent MDV genome. Avian Dis. 2009;53(2):149–55.
Delecluse HJ, Hammerschmidt W. Status of Marek’s disease virus in established lymphoma cell lines: herpesvirus integration is common. J Virol. 1993;67(1):82–92.
Kaufer BB, Jarosinski KW, Osterrieder N. Herpesvirus telomeric repeats facilitate genomic integration into host telomeres and mobilization of viral DNA during reactivation. J Exp Med. 2011;208(3):605–15.
Addinger HK, Calnek BW. Pathogenesis of Marek’s disease: early distribution of virus and viral antigens in infected chickens. J Natl Cancer Inst. 1973;50(5):1287–98.
Tulman ER, Afonso CL, Lu Z, Zsak L, Rock DL, Kutish GF. The genome of a very virulent Marek’s disease virus. J Virol. 2000;74(17):7980–8.
Qian Z, Brunovskis P, Rauscher F 3rd, Lee L, Kung HJ. Transactivation activity of Meq, a Marek’s disease herpesvirus bZIP protein persistently expressed in latently infected transformed T cells. J Virol. 1995;69(7):4037–44.
Jones D, Lee L, Liu JL, Kung HJ, Tillotson JK. Marek disease virus encodes a basic-leucine zipper gene resembling the Fos/Jun oncogenes that is highly expressed in lymphoblastoid tumors. Proc Natl Acad Sci U S A. 1992;89(9):4042–6.
Suchodolski PF, Izumiya Y, Lupiani B, Ajithdoss DK, Lee LF, Kung HJ, et al. Both homo and heterodimers of Marek’s disease virus encoded Meq protein contribute to transformation of lymphocytes in chickens. Virology. 2010;399(2):312–21.
Subramaniam S, Preeyanon L, Cheng HH. Transcriptional profiling of mEq-dependent genes in Marek’s disease resistant and susceptible inbred chicken lines. PLoS One. 2013;8(10):e78171.
Tai SHS, Hearn C, Umthong S, Agafitei O, Cheng HH, Dunn JR, et al. Expression of Marek’s disease virus oncoprotein Meq during infection in the natural host. Virology. 2017;503:103–13.
Kung HJ, et al. Meq: an MDV-specific bZIP transactivator with transforming properties. Curr Top Microbiol Immunol. 2001;255:245–60.
Lee LF, Lupiani B, Silva RF, Kung HJ, Reddy SM. Recombinant Marek’s disease virus (MDV) lacking the Meq oncogene confers protection against challenge with a very virulent plus strain of MDV. Vaccine. 2008;26(15):1887–92.
Silva RF, Dunn JR, Cheng HH, Niikura M. A MEQ-deleted Marek’s disease virus cloned as a bacterial artificial chromosome is a highly efficacious vaccine. Avian Dis. 2010;54(2):862–9.
Zhang Y, Liu C, Yan F, Liu A, Cheng Y, Li Z, et al. Recombinant Gallid herpesvirus 2 with interrupted meq genes confers safe and efficacious protection against virulent field strains. Vaccine. 2017;35(36):4695–701.
Lupiani B, Lee LF, Cui X, Gimeno I, Anderson A, Morgan RW, et al. Marek’s disease virus-encoded Meq gene is involved in transformation of lymphocytes but is dispensable for replication. Proc Natl Acad Sci U S A. 2004;101(32):11815–20.
Deng X, Li X, Shen Y, Qiu Y, Shi Z, Shao D, et al. The Meq oncoprotein of Marek’s disease virus interacts with p53 and inhibits its transcriptional and apoptotic activities. Virol J. 2010;7:348.
Levy AM, Gilad O, Xia L, Izumiya Y, Choi J, Tsalenko A, et al. Marek’s disease virus Meq transforms chicken cells via the v-Jun transcriptional cascade: a converging transforming pathway for avian oncoviruses. Proc Natl Acad Sci U S A. 2005;102(41):14831–6.
Zhao Y, Kurian D, Xu H, Petherbridge L, Smith LP, Hunt L, et al. Interaction of Marek’s disease virus oncoprotein Meq with heat-shock protein 70 in lymphoid tumour cells. J Gen Virol. 2009;90(Pt 9):2201–8.
Brown AC, Baigent SJ, Smith LP, Chattoo JP, Petherbridge LJ, Hawes P, et al. Interaction of MEQ protein and C-terminal-binding protein is critical for induction of lymphomas by Marek’s disease virus. Proc Natl Acad Sci U S A. 2006;103(6):1687–92.
Parcells MS, et al. Marek’s disease virus reactivation from latency: changes in gene expression at the origin of replication. Poult Sci. 2003;82(6):893–8.
Levy AM, Izumiya Y, Brunovskis P, Xia L, Parcells MS, Reddy SM, et al. Characterization of the chromosomal binding sites and dimerization partners of the viral oncoprotein Meq in Marek’s disease virus-transformed T cells. J Virol. 2003;77(23):12841–51.
Jarosinski KW, Osterrieder N, Nair VK, Schat KA. Attenuation of Marek’s disease virus by deletion of open reading frame RLORF4 but not RLORF5a. J Virol. 2005;79:11647–59.
Jarosinski KW, O’Connell PH, Schat KA. Impact of deletions within the bam HI-L fragment of attenuated Marek’s disease virus on vIL-8 expression and the newly identified transcript of open reading frame LORF4. Virus Genes. 2003;26(3):255–69.
Schat KA, et al. Open reading frame L1 of Marek’s disease herpesvirus is not essential for in vitro and in vivo virus replication and establishment of latency. J Gen Virol. 1998;79(Pt 4):841–9.
Cui XP, et al. Marek’s disease virus-encoded vIL-8 gene is involved in early cytolytic infection but dispensable for establishment of latency. J Virol. 2004;78(9):4753–60.
Engel AT, Selvaraj RK, Kamil JP, Osterrieder N, Kaufer BB. Marek’s disease viral interleukin-8 promotes lymphoma formation through targeted recruitment of B cells and CD4+ CD25+ T cells. J Virol. 2012;86(16):8536–45.
Kaiser P, Hughes S, Bumstead N. The chicken 9E3/CEF4 CXC chemokine is the avian orthologue of IL8 and maps to chicken chromosome 4 syntenic with genes flanking the mammalian chemokine cluster. Immunogenetics. 1999;49(7–8):673–84.
Parcells MS, Lin SF, Dienglewicz RL, Majerciak V, Robinson DR, Chen HC, et al. Marek’s disease virus (MDV) encodes an interleukin-8 homolog (vIL-8): characterization of the vIL-8 protein and a vIL-8 deletion mutant MDV. J Virol. 2001;75(11):5159–73.
• Haertle, S., et al., Identification of the receptor and cellular ortholog of the Marek’s disease virus (MDV) CXC chemokine. Front Microbiol. 2017;8. Härtle et al. show that vIL-8 is an orthologue of the cellular CXCL13 chemokine, which recruits B cells and a certain subset of T cells. In addition, they identified CXCR5 as the cellular receptor of vIL-8. The study thereby elucidates the origin and function of this viral chemokine.
Jarosinski KW, Schat KA. Multiple alternative splicing to exons II and III of viral interleukin-8 (vIL-8) in the Marek’s disease virus genome: the importance of vIL-8 exon I. Virus Genes. 2007;34(1):9–22.
Kumar P, et al. Spliced gene products of the Meq oncoprotein of Marek’s disease virus (MDV) are expressed during latency, bind to the MDV genome, are potent transcriptional repressors, and induce cellular proliferation. J Virol. 2010.
Okada T, Takagi M, Murata S, Onuma M, Ohashi K. Identification and characterization of a novel spliced form of the meq transcript in lymphoblastoid cell lines derived from Marek’s disease tumours. J Gen Virol. 2007;88(Pt 8):2111–20.
Anobile JM, Arumugaswami V, Downs D, Czymmek K, Parcells M, Schmidt CJ. Nuclear localization and dynamic properties of the Marek’s disease virus oncogene products Meq and Meq/vIL8. J Virol. 2006;80(3):1160–6.
Hong Y, Coussens PM. Identification of an immediate-early gene in the Marek’s disease virus long internal repeat region which encodes a unique 14-kilodalton polypeptide. J Virol. 1994;68(6):3593–603.
Hong Y, Frame M, Coussens PM. A 14-kDa immediate-early phosphoprotein is specifically expressed in cells infected with oncogenic Marek’s disease virus strains and their attenuated derivatives. 1995.
Tahiri-Alaoui A, Smith LP, Kgosana L, Petherbridge LJ, Nair V. Identification of a neurovirulence factor from Marek’s disease virus. Avian Dis. 2013;57(2 Suppl):387–94.
Tahiri-Alaoui A, Matsuda D, Xu H, Panagiotis P, Burman L, Lambeth LS, et al. The 5′ leader of the mRNA encoding the marek’s disease virus serotype 1 pp14 protein contains an intronic internal ribosome entry site with allosteric properties. J Virol. 2009;83(24):12769–78.
Tahiri-Alaoui A, Smith LP, Baigent S, Kgosana L, Petherbridge LJ, Lambeth LS, et al. Identification of an intercistronic internal ribosome entry site in a Marek’s disease virus immediate-early gene. J Virol. 2009;83(11):5846–53.
Tahiri-Alaoui A, Zhao Y, Sadigh Y, Popplestone J, Kgosana L, Smith LP, et al. Poly(A) binding protein 1 enhances cap-independent translation initiation of neurovirulence factor from avian herpesvirus. PLoS One. 2014;9(12):e114466.
Reddy SM, Lupiani B, Gimeno IM, Silva RF, Lee LF, Witter RL. Rescue of a pathogenic Marek’s disease virus with overlapping cosmid DNAs: use of a pp38 mutant to validate the technology for the study of gene function. Proc Natl Acad Sci U S A. 2002;99(10):7054–9.
Cui ZZ, Lee LF, Liu JL, Kung HJ. Structural analysis and transcriptional mapping of the Marek’s disease virus gene encoding pp38, an antigen associated with transformed cells. J Virol. 1991;65(12):6509–15.
Li X, Jarosinski KW, Schat KA. Expression of Marek’s disease virus phosphorylated polypeptide pp38 produces splice variants and enhances metabolic activity. Vet Microbiol. 2006;117(2–4):154–68.
Schat KA, Piepenbrink MS, Buckles EL, Schukken YH, Jarosinski KW. Importance of differential expression of Marek’s disease virus gene pp38 for the pathogenesis of Marek’s disease. Avian Dis. 2013;57(2 Suppl):503–8.
Gimeno IM, Witter RL, Hunt HD, Reddy SM, Lee LF, Silva RF. The pp38 gene of Marek’s disease virus (MDV) is necessary for cytolytic infection of B cells and maintenance of the transformed state but not for cytolytic infection of the feather follicle epithelium and horizontal spread of MDV. J Virol. 2005;79(7):4545–9.
Ross N. T-cell transformation by Marek’s disease virus. Trends Microbiol. 1999;7(1):22–9.
Piepenbrink MS, Li X, O’Connell PH, Schat KA. Marek’s disease virus phosphorylated polypeptide pp38 alters transcription rates of mitochondrial electron transport and oxidative phosphorylation genes. Virus Genes. 2009;39(1):102–12.
Yao Y, Nair V. Role of virus-encoded microRNAs in avian viral diseases. Viruses. 2014;6(3):1379–94.
Yao Y, Zhao Y, Xu H, Smith LP, Lawrie CH, Watson M, et al. MicroRNA profile of Marek’s disease virus-transformed T-cell line MSB-1: predominance of virus-encoded microRNAs. J Virol. 2008;82(8):4007–15.
Burnside J, Bernberg E, Anderson A, Lu C, Meyers BC, Green PJ, et al. Marek’s disease virus encodes MicroRNAs that map to meq and the latency-associated transcript. J Virol. 2006;80(17):8778–86.
Yu ZH, Teng M, Sun AJ, Yu LL, Hu B, Qu LH, et al. Virus-encoded miR-155 ortholog is an important potential regulator but not essential for the development of lymphomas induced by very virulent Marek’s disease virus. Virology. 2014;448:55–64.
Zhao Y, Xu H, Yao Y, Smith LP, Kgosana L, Green J, et al. Critical role of the virus-encoded microRNA-155 ortholog in the induction of Marek’s disease lymphomas. PLoS Pathog. 2011;7(2):e1001305.
Zhao Y, Yao Y, Xu H, Lambeth L, Smith LP, Kgosana L, et al. A functional MicroRNA-155 ortholog encoded by the oncogenic Marek’s disease virus. J Virol. 2009;83(1):489–92.
•• Zhuang G, et al. A tiny RNA that packs a big punch: the critical role of a viral miR-155 ortholog in lymphomagenesis in Marek’s disease. Front Microbiol. 2017;8:1169. Zhuang et al. provide an extensive overview of the role of MDV miR-155 orthologue (miR-M4-5p) in virus-induced lymphomagenesis.
Linnstaedt SD, Gottwein E, Skalsky RL, Luftig MA, Cullen BR. Virally induced cellular microRNA miR-155 plays a key role in B-cell immortalization by Epstein-Barr virus. J Virol. 2010;84(22):11670–8.
Luo J, Teng M, Fan JM, Wang FY, Zhou L, Deng RG, et al. Marek’s disease virus-encoded microRNAs: genomics, expression and function. Sci China Life Sci. 2010;53(10):1174–80.
Teng M, Yu ZH, Zhao P, Zhuang GQ, Wu ZX, Dang L, et al. Putative roles as oncogene or tumour suppressor of the mid-clustered microRNAs in Gallid alphaherpesvirus 2 (GaHV2) induced Marek’s disease lymphomagenesis. J Gen Virol. 2017;98(5):1097–112.
Strassheim S, Stik G, Rasschaert D, Laurent S. mdv1-miR-M7-5p, located in the newly identified first intron of the latency-associated transcript of Marek’s disease virus, targets the immediate-early genes ICP4 and ICP27. J Gen Virol. 2012;93(Pt 8):1731–42.
Blackburn EH. Telomere states and cell fates. Nature. 2000;408(6808):53–6.
Collins K. Physiological assembly and activity of human telomerase complexes. Mech Ageing Dev. 2008;129(1–2):91–8.
Autexier C, Lue NF. The structure and function of telomerase reverse transcriptase. Annu Rev Biochem. 2006;75:493–517.
Trapp S, Parcells MS, Kamil JP, Schumacher D, Tischer BK, Kumar PM, et al. A virus-encoded telomerase RNA promotes malignant T cell lymphomagenesis. J Exp Med. 2006;203(5):1307–17.
Fragnet L, Blasco MA, Klapper W, Rasschaert D. The RNA subunit of telomerase is encoded by Marek’s disease virus. J Virol. 2003;77(10):5985–96.
Chbab N, Egerer A, Veiga I, Jarosinski KW, Osterrieder N. Viral control of vTR expression is critical for efficient formation and dissemination of lymphoma induced by Marek’s disease virus (MDV). Vet Res. 2010;41(5):56.
Fragnet L, Kut E, Rasschaert D. Comparative functional study of the viral telomerase RNA based on natural mutations. J Biol Chem. 2005;280(25):23502–15.
Kaufer BB, Trapp S, Jarosinski KW, Osterrieder N. Herpesvirus telomerase RNA(vTR)-dependent lymphoma formation does not require interaction of vTR with telomerase reverse transcriptase (TERT). PLoS Pathog. 2010;6(8):e1001073.
Kheimar A, Kaufer BB. Epstein-Barr virus-encoded RNAs (EBERs) complement the loss of herpesvirus telomerase RNA (vTR) in virus-induced tumor formation. Sci Rep. 2018;8(1):209.
Kishi M, et al. A repeat sequence, GGGTTA, is shared by DNA of human herpesvirus 6 and Marek’s disease virus. J Virol. 1988;62(12):4824–7.
Volkening JD, Spatz SJ. Identification and characterization of the genomic termini and cleavage/packaging signals of gallid herpesvirus type 2. Avian Dis. 2013;57(2 Suppl):401–8.
Greco A, Fester N, Engel AT, Kaufer BB. Role of the short telomeric repeat region in Marek’s disease virus replication, genomic integration, and lymphomagenesis. J Virol. 2014;88(24):14138–47.
Osterrieder N, Wallaschek N, Kaufer BB. Herpesvirus genome integration into telomeric repeats of host cell chromosomes. Annu Rev Virol. 2014;1(1):215–35.
Kheimar A, Previdelli R, Wight D, Kaufer B. Telomeres and telomerase: role in Marek’s disease virus pathogenesis, integration and tumorigenesis. Viruses. 2017;9(7):173.
Wallaschek N, Sanyal A, Pirzer F, Gravel A, Mori Y, Flamand L, et al. The telomeric repeats of human herpesvirus 6A (HHV-6A) are required for efficient virus integration. PLoS Pathog. 2016;12(5):e1005666.
Jarosinski KW, Tischer BK, Trapp S, Osterrieder N. Marek’s disease virus: lytic replication, oncogenesis and control. Expert Rev Vaccines. 2006;5(6):761–72.
Acknowledgements
BBK and LDB were supported by the DFG grant KA 3492/3-1 awarded to BBK. AK and FAZA were supported by the Egyptian Ministry of High Education (MOHE).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Virology
Rights and permissions
About this article

Cite this article
Bertzbach, L.D., Kheimar, A., Ali, F.A.Z. et al. Viral Factors Involved in Marek’s Disease Virus (MDV) Pathogenesis. Curr Clin Micro Rpt 5, 238–244 (2018). https://doi.org/10.1007/s40588-018-0104-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40588-018-0104-z