
Abstract
The use of enzyme/prodrug system has gained attention because it could help improve the efficacy and safety of conventional cancer chemotherapies. In this approach, cancer cells are first transfected with a gene that can express an enzyme with ability to convert a nontoxic prodrug into its active cytotoxic form. As a result, the activated prodrug could kill the transfected cancer cells. Despite the significant progress of different suicide gene therapy protocols in preclinical studies and early clinical trials, none has reached the clinic due to several shortcomings. These include slow prodrug-drug conversion rate, low transfection/transduction efficiency of the vectors, and nonspecific toxicity/immunogenicity related to the delivery systems, plasmid DNA, enzymes, and/or prodrugs. This mini-review aims at providing an overview of the most widely used enzyme/prodrug systems with emphasis on reporting the results of the recent preclinical and clinical studies.
Similar content being viewed by others

Introduction
In recent years, cancer treatment methods are becoming more focused on targeted approaches in order to maximize treatment efficacy while reducing side effects. One of these targeted approaches is termed suicide gene therapy, also known as gene-directed enzyme prodrug therapy (GDEPT). The success of suicide gene therapy is dependent on its three major functional components: enzyme, prodrug, and gene delivery system (vector).
The function of the vector is to carry the gene that encodes an enzyme to the target cancer cells for expression. In general, gene delivery systems can be divided into two major groups including viral (e.g., adenovirus, lentivirus, etc.) and nonviral (e.g., synthetic polymers and lipids, bacteria-based and cell-based) (Table 1). Each vector type comes with a series of advantages and disadvantages. Since the focus of this mini-review is not on vectors but on enzyme/prodrug systems, we will not discuss them further here. For more information related to the use of vectors for cancer gene therapy, the readers are referred to excellent recent reviews by Forbes [27], Cattaneo et al. [11], and Mohit and Rafati [63].
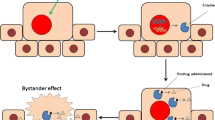
The function of the enzyme which is expressed by the transfected cancer cells is to convert the nontoxic/nonfunctional prodrug into its toxic (functional) form resulting in the death of the enzyme-producing cancer cells as well as neighboring cells through a phenomenon known as bystander effect (Fig. 1) [10, 26]. It is worth mentioning that the expression of the enzyme in transfected cells can be regulated by tumor-specific promoters. This regulatory element could enhance the enzyme/prodrug system’s safety by restricting the enzyme expression only to the tumor cells. By applying the cancer-specific promoters, the suicide gene could get selectively expressed in cancer cells, while sparing normal ones. The promoter of human telomerase reverse transcriptase (hTERT) is one of the most widely used promoters in the field and is the only transcriptional control element that has successfully entered into clinical trials [67]. However, the problem associated with the use of hTERT promoter is its low expression activity. In order to enhance promoter’s expression activity and also overcome the potential development of resistance by the tumor cells, several groups have tried to create more specific and efficacious ones. Recently, Rad 51, OPN, RAN, BRMS1, and MCM5 promoters were identified through screening of a large panel of normal and cancer cells, and interestingly, some of them showed significantly higher activity than hTERT promoter [14, 15, 32]. Another means of improving promoter activity is to artificially design a chimeric promoter. For example, a promoter with a stronger activity can be obtained by constructing the chimeric promoters with the fusion of two promoters [23] or various transcriptional regulatory elements [30, 89].

Schematic representation of the mechanism of action in suicide gene therapy. First, cancer cells are transfected with a gene that codes for an enzyme. The enzyme is able to convert the nontoxic prodrug into its cytotoxic form resulting in the death of the transfected cancer cells and the neighboring ones through a phenomenon known as bystander effect
In the following sections, we provide an overview of the eight most widely investigated enzyme/prodrug systems which are shown to be effective in inducing tumor cell death and inhibiting cancer progression. The purpose of this concise mini-review is to discuss the forte and weaknesses associated with the efficacy and safety of each system. Therefore, the coverage of the literature in this review is not encyclopedic; rather, selected examples have been chosen to highlight certain important points.
Enzyme/Prodrug Systems
Herpes Simplex Virus Thymidine Kinase (HSV-TK)/Ganciclovir
In this system, gancyclovir (GCV) undergoes a monophosphorylation step by herpes simplex virus thymidine kinase (HSV-TK) enzyme. After the first phosphorylation step, the triphosphate form of the GCV is produced with the help of endogenous kinase (Fig. 2a). In this form, the GCV-triphosphate can cause apoptosis by introducing chain termination and single-strand breaks into DNA [13]. One limitation of using GCV-triphosphate as anticancer agent is that it could only affect the dividing cells [4, 66, 87, 98]. The second drawback of this system is that the GCV-triphosphate transportation to surrounding cells is limited by the presence of gap junctional intercellular communications. Recently, Wu et al. showed that the bystander effect of the GCV-triphosphate can be enhanced by the expression of connexin32 (Cx32), which is responsible for producing gap junctions [112]. Furthermore, HSV-TK has a high affinity toward its natural substrate, thymidine, which leads to the need for using a high dose of GCV in order to win the competition [2]. This higher GCV dose could result in suppression of immune system and damage to bone marrow cells [50]. The enhanced activity of ATP-binding cassette (ABC) transporters in cancer-resistant cells is one of the main reasons of therapy resistance in cancer [55]. Therefore, ABCG2-mediated efflux of GCV in side population cells can limit the efficiency of this enzyme/prodrug system [35].

Chemical structures of the inactive prodrugs and their cytotoxic forms after activation by the corresponding enzyme
In the past decades, several attempts have been made in order to improve the efficiency of TK/GCV system. In one approach, modifications of enzyme active sites have resulted in generation of mutated forms of the enzyme, such as mutant 30 (L159I/I160L/F161A/A168Y/L169F), SR39 (L159I/I160F/F161L/A168F/L169M) [5, 50], A168H, and a mutated form of A168H (TK007) [78]. All these mutants have shown enhanced enzyme affinity toward GCV in comparison to wild type. In a different approach, an MDR-1-targeted small hairpin RNA (shRNA) was used to inhibit the ABC transporters which led to the accumulation and increase in drugs effect [73]. Other groups have also examined the use of other drugs such as acyclovir and valacyclovir instead of GCV and have observed more efficacy [93, 113].
TK/GCV system has also been used in combination with other therapeutic modalities in order to enhance its anticancer efficacy. For example, TK/valacyclovir in combination with surgery and accelerated radiation showed an increase in patient survival rates in malignant glioma patients [16]. This approach has also shown to not only increase the efficacy of the treatment but also decrease the toxicity in pancreatic adenocarcinoma [4]. Currently, there are a couple of clinical trials in progress for patients with high-risk acute leukemia (NCT00914628) and in patients with recurrent prostate cancer (NCT01913106) (www.clinicaltrials.gov).
Cytosine Deaminase (CD)/5-Fluorocytosine
In this system, 5-fluorocytosine (5-FC) that is a nontoxic prodrug is converted to its active form, 5-fluorouracil (5-FU), by CD activity (Fig. 2b) [37]. Although 5-FU has been used for cancer chemotherapy, at its therapeutic dose, it causes significant side effects such as mucositis, myelosuppression, dermatitis, and cardiac toxicity. Due to these side effects, its application as prodrug in the form of 5-FC and in combination with cytodeaminase enzyme has gained momentum in the past decades. Two forms of cytosine deaminase, bacteria (bCD) and yeast (yCD), have been investigated in the context of cancer therapy. After activation by CD, 5-FU can be further changed into potent pyrimidine antimetabolites by other cellular enzymes. 5-FU can inhibit thymidylate synthase which consequently leads to the cell cycle arrest and apoptosis. Since 5-FU traverse into the neighboring cells independent of gap junctions and even can pass the blood-brain barrier by diffusion, it induces a significant bystander effect [25, 45]. Because of its unique properties, CD/5-FC system has been used for the treatment of different types of cancer, such as colon carcinoma, glioma, and pancreatic cancer [24, 41, 42]. Having been combined with radiotherapy, CD/5-FC has shown quite a promising result due to the radio-sensitizing effect of 5-FU on the treated cells [65]. In comparison to HSV-TK/GCV, CD/5-FC has shown better results in renal and colorectal carcinoma probably due to its more potent bystander effect [92, 99].
Although use of CD/5-FC system has several strengths as mentioned above, there are some drawbacks that yet need to be addressed. The first drawback is due to the presence of normal flora in the gut which is able to convert 5-FC to 5-FU resulting in side effects. Several groups have worked to create bCD mutants in order to improve its kinetic showing promising results [24, 40, 41]. Yeast cytosine deaminase is another form of the enzyme which provides a few advantages over bCD including high affinity and better prodrug conversion kinetics. In addition, yCD can be fused with E. coli uracil phosphoribosyl transferase (UPRT) which is absent in mammalian cells. UPRT can directly convert 5-FU to 5-FdUMP [107] resulting in significant improvement of activity and enhanced cancer cell killing efficiency in prostate, ovarian, colon, and breast cancers [62, 81]. Some studies have also shown that yCD:UPRT/5-FC system may not get affected by acquired 5-FU resistance and could potentially kill both proliferating and nonproliferating cancer cells [52, 81].
While past clinical trials with CD/5-FC system alone have reported limited success [44], there are several new clinical trials in progress which could produce interesting outcomes. For instance, in one trial, genetically modified neural stem cells expressing bCD are used in combination with orally administered 5-FC for the treatment of recurrent gliomas (NCT01172964). In another phase I–II study, an investigational drug APS001F is currently being examined in combination with a recombinant bacterium (Bifidobacterium longum) which can produce CD (NCT01562626). Finally, there is a phase I study in progress for the treatment of grade III/IV gliomas by using retroviral replicating vector (Toca 511) in combination with 5-FC (NCT01470794). The results of these ongoing trials are not reported but can be followed at www.clinicaltrials.gov.
Cytochrome P450 (CYP450)/Oxazaphosphorine
Cytochrome P450 (CYP) enzymes are important factors in xenobiotics metabolism as they are responsible for deactivation of the toxic drugs in the liver. It has also been shown that these enzymes play an important role as an activating factor for oxazaphosphorine agents [72]. Oxazaphosphorine is a class of anticancer drugs that includes cyclophosphamide (CPA) and ifosfamide (IFA). Both of these two akylating agents are the substrates of P450 enzymes which metabolizes these compounds to yield a 4-hydroxy derivative. Then, the resulting metabolites continue their chemical reaction to produce phosphoramide or ifosphoramide mustard and acrolein (Fig. 2c). Ultimately, they form DNA cross-links and kill the cells. The resulted active reagents can be transported to the surrounding cells, independent of gap junctions, which gives them potent bystander effect [85]. Another advantage of this system in comparison to the other enzyme/prodrug systems is its less immunogenicity due to the presence of P450 enzymes in human body [70].
One major drawback of this system is the natural metabolism of these prodrugs by the liver P450 enzymes that generates active toxic metabolites in the whole body and eventually causes various side effects, including cardiotoxicity, bone marrow suppression, neurotoxicity, and nephrotoxicity. Some of these toxicities can be associated with the chloroacetaldehyde formation from CPA and IFA via N-dechloroethylation [12]. Therefore, the big challenge in this system is the inhibition of P450 enzyme activity in the liver so that the prodrug gets activated only in the tumor sites [29].
A few methods have been used to improve the cancer treatment efficacy of P450 GDEPT and to reduce its side effects. In one approach, combined or in parallel with CYP450/oxazaphosphorine treatment, anti-thyroid drugs, such as propylthiouracil and methimazole were used to inhibit hepatic P450 reductase activity and improve the prodrug activation in tumor cells [84]. In another approach, a CYP2B6/RED fusion protein was designed so that it can transfer electrons from NADPH to the heme moiety of the protein. This modification increases the efficiency of CYP2B6 substrate metabolism into active drugs [102].
In a phase I/II clinical trial, a recombinant retroviral vector that contains human CYP450 type 2B6 genes (CYP2B6), neomycin resistance marker gene and E. coli lacZ has been tested. In this study, gene-based delivery of CYP2B6 to the tumor site led to prodrug activation locally and produced higher concentrations of the active drug at the target site. The promising results of this study inspired the use of this approach in other cancer treatment studies [7]. However, regardless of the exciting results, there are still serious concerns about using this system for cancer therapy because of normal activity of CYP450 in human cells and difficulty in the delivery of the GDEPT elements to the targeted cancer cells without causing side effects on normal healthy cells [51, 82].
Nitroreductase/CB1954
This enzyme/prodrug system contains group of nitroreductase (NTR) enzymes that can activate prodrugs, such as CB1954 (5-(aziridin-1-yl)-2,4-dinitro-benzamide) (Fig. 2d). Two classes of enzymes and four types of prodrugs have been studied in this system. The two classes of enzymes are categorized based on their sensitivity to oxygen. Type I NTR can produce nitroso, hydroxylamine, and/or amine terminated products in the presence of molecular oxygen, while type II, on the other hand, can only produce these products when oxygen is absent [110]. The four classes of prodrugs include dinitroaziridinyl benzamides, dinitrobenzamide mustards, 4-nitrobenzyl carbamates, and nitroindolines. CB1954, which is the most widely used prodrug, is from dinitrobenzamide group [91]. The metabolites of this prodrug can freely diffuse to the surrounding cells which induce a strong bystander effect. They can damage DNA by their chelating property leading to p53 and cell cycle-independent apoptosis in both dividing and nondividing cells. This is an important advantage of the system over the ones that can only target the dividing cells [105]. While several studies have reported promising results, the low activation rate of CB1954 is one major limitation associated with this enzyme/prodrug system. At least two general approaches have been tested to improve the efficacy of this system. One is the engineering and application of alternative CB1954-activating nitroreductases, such as AzoR, NFsA, NemA, and flavin reductase I (FRase I) [79, 97]. The other approach is the synthesis of other nitroreductase prodrugs such as nitro-CBI-DEI and PR-104A [31, 80]. Both approaches have shown promising results in vitro and in vivo. Effective use of this enzyme/prodrug system has been demonstrated in a few clinical trials for the treatment of prostate and liver cancers [76]. However, the CB1954 low conversion rates and dose-dependent hepatotoxicity remained as major challenges for clinical application of this GDEPT method [46, 61]. For additional and more in depth information related to the nitroreductase/CB1954 system, readers are referred to an excellent review by Williams et al. [110].
Carboxypeptidase G2/Nitrogen Mustard
The enzyme carboxypeptidase G2 (CPG2) is derived from the Pseudomonas strain RS-16 and has no human analog. This enzyme cleaves glutamic acid from nitrogen mustard-based drugs, such as 4-[(2-chloroethyl)(2-mesyloxyethyl)amino]benzoyl-L-glutamic acid (CMDA) and bis-iodo phenol mustard (ZD2767P), into DNA alkylating agents, without any further modification (Fig. 2e). The resulted active drug can cause inter- and intra-strand DNA cross-linking, leading to apoptosis in both dividing and nondividing cells. It can also freely diffuse through cell membranes independent of gap junctions which offer potent bystander effect. Being independent from host enzymes for prodrug activation gives this system an outstanding benefit of a higher prodrug conversion rate, in comparison to the other enzyme/prodrug systems [22]. While different nitrogen mustard reagents have been developed and tested in the past two decades, 4-[(2-chloroethyl)(2-mesyloxyethyl)amino] benzoyl-l-glutamicacid (CMDA) is among the first successful ones. Several attempts have been made to increase the potency of CMDA. For example, ZD2767P is 300 times more potent than CMDA [28].
To increase the enzyme efficiency, a mutated form of the CPG2 gene was fused to vascular endothelial growth factor (VEGF). This modification resulted in binding of secreted CPG2 to the VEGF receptor, which kept CMDA around tumor mass and significantly reduced systemic side effects [96]. This enzyme prodrug system was used in attachment with tumor-specific antibodies in different clinical studies. However, some issues, like immunogenicity against mouse antibodies and CPG2 and also insufficient localization of enzyme\antibody conjugate, limited its success in cancer therapy [28, 56, 58].
Purine Nucleoside Phosphorylase/6-Methylpurine Deoxyriboside
In this system, prodrugs such as 6-methylpurine 2-deoxyriboside and fludarabine can be converted to 2-deoxyribose-1-phosphate by the E. coli purine nucleoside phosphorylase (PNP) (Fig. 2f). The advantage of using PNP/MEP is its high bystander activity based on gap junction-free transport of activated drugs, its effect on both proliferating and nonproliferating cells, and its unique mechanism of action that is independent of DNA synthesis. PNP catalyzes the cleavage in glycoside bond of (deoxy) adenosine-based substrates that produces either (MeP) or 2-fluoroadenine (F-Ade). Then, these substrates are converted to their triphosphate forms by cellular monophosphate and diphosphate kinases, which can inhibit both RNA and protein syntheses [74, 94]. Three studied prodrugs of this system are 6-methylpurine-2′-deoxyriboside (MeP-dR), 2-F-2′-deoxyadenosine (F-dAdo), and arabinofuranosyl-2-F-adenine monophosphate (F-araAMP). Among them, F-araAMP is clinically approved for chronic lymphocytic leukemia treatment [95]. These (deoxy) adenosine-based analogs are only the substrates of E. coli PNP and cannot be recognized by humane PNP. Thus, the prodrugs cannot be converted into toxic metabolites in normal tissues and consequently limits the off-target toxicities. However, the application of bacterial PNP can induce host immune response. A few groups have worked on the development of human PNP that can consume the (deoxy) adenosine-based prodrugs as its substrate [3, 75]. Among the recorded clinical trials, there is only one completed phase I clinical trial using this system. In this trial, the safety of E. coli PNP/fludarabine phosphate in patients with head and neck cancers has been studied. It was shown that localized production of fluoroadenine within tumor tissues, using fludarabine and E. coli PNP system, is successful in eradicating tumors without causing any significant toxicity [83].
Horseradish Peroxidase/Indole-3-Acetic Acid
This system consists of the horseradish peroxidase (HRP) enzyme and indole-3-acetic acid (IAA) as the prodrug. HRP enzyme oxidizes IAA at neutral pH to its cationic form that produces the carbon-centered skatolyl radical. This radical can be changed to peroxyl radical in the presence of oxygen which would turn into by-products, such as indole-3-carbinol, oxindole-3-carbinol, and 3-methylene-2-oxindole (Fig. 2g). These reactive by-products can induce DNA strand breaks, leading to apoptosis and cell death. This system also has a potent bystander effect because of the transportation of the activated drug via diffusion to the untransfected neighboring cells [30]. HRP/IAA system has been tested in several in vitro and in vivo models, including bladder, melanoma, and pancreatic cancer [36, 39, 47]. However, the major concern of using HRP/IAA system is the low expression level of the enzyme in transfected cells [19, 101]. The second drawback of this system is the origin of the enzyme which comes from plants, because it results in undefined mixture of isoenzyme with different glycosylation patterns which can cause side effect in body. To avoid this problem, different recombinant forms of HRP isoenzymes are produced, which have shown significant effects on breast and bladder carcinoma [6]. Although some promising results have been obtained from HRP/IAA system, there has been no clinical trial with this system by the time of writing this manuscript (www.clinicaltrials.gov).
Carboxylesterase (CE)/Irinotecan
Carboxylesterases are active endogenous enzymes in human body that play roles in hydrolysis of different ester and amide-containing chemicals [34]. Because of this property, they can convert irinotecan (CPT-11), a less cytotoxic agent, into its highly toxic form, SN-38 (Fig. 2h). SN-38 inhibits topoisomerase I and thus alters the DNA relaxation machinery [34, 77]. This enzyme/prodrug system has been used for the treatment of colorectal cancer [104], glioma [17], and various other tumor models [49, 57, 114]. While this enzyme has different isoforms, carboxylesterase-1 (CE1), which is mostly localized in the liver, is the most widely used one. Unfortunately, CE1 suffers from the low catalytic efficiency that limits the conversion rate of CPT-11 to SN-38. To overcome this deficiency, another isoform of this enzyme also known as CE2 has been studied. CE2 isoform is commonly expressed in intestine. CE2 has a higher catalytic efficiency than CE1 [38, 86] and has been used for the treatment of pancreatic and colon cancer [9, 71]. The rabbit analog of CE1 (rCE1) has also been engineered and used for conversion of CPT-11 to SN-38. While this isoform has shown a higher catalytic efficiency in comparison to human CE1 [20], it can cause immune response in human host. To avoid this problem, mutant CE1 (hCE1m6) was engineered using homology alignment of human and rabbit version of CE1 followed by replacing eight amino acids in human CE1. This change in sequence resulted in higher conversion efficiency [109].
Given that SN-38 has poor water solubility, its bystander activity may get reduced [103]. In an attempt to improve the bystander effect and/or overall anticancer efficacy, secretory form of CE2 has been genetically engineered [71]. This approach has shown promising success in several preclinical models [59, 90]. Currently, there are a few ongoing clinical trials using this enzyme/prodrug system. For instance, there is a phase I clinical trial in progress in which the patients with rrecurrent high-grade gliomas are undergoing treatment. Here, genetically modified neural stem cells which can express carboxylesterase are used in combination with irinotecan hydrochloride (NCT02192359).
Conclusion
Significant amount of preclinical work have been performed in the past two decades on gene-directed enzyme prodrug therapy of cancer. Based on the preclinical data, numerous clinical trials have been performed. While substantial progress has been made, no cancer suicide gene therapy protocol has been approved for clinical use. Important issues that have yet to be addressed include development of effective vectors for efficient and targeted gene transfer to the tumor cells, design of prodrugs that can be efficiently converted into the active drugs with potent bystander effect, and engineering enzymes with low immunogenicity and high affinity toward prodrugs. Overcoming these barriers may open new doors into effective eradication of many of yet incurable and stubborn cancers through GDEPT.
References
Aboody KS, Najbauer J, Metz MZ, D'Apuzzo M, Gutova M, Annala AJ et al. Neural stem cells mediated enzyme/prodrug therapy for glioma: preclinical studies. Sci Transl Med. 2013;5(184):184ra59–ra59.
Adachi M, Sampath J, Lan L-b, Sun D, Hargrove P, Flatley R, et al. Expression of MRP4 confers resistance to ganciclovir and compromises bystander cell killing. J Biol Chem. 2002;277(41):38998–9004.
Afshar S, Asai T, Morrison SL. Humanized ADEPT comprised of an engineered human purine nucleoside phosphorylase and a tumor targeting peptide for treatment of cancer. Mol Cancer Ther. 2009;8(1):185–93.
Aguilar LK, Shirley LA, Chung VM, Marsh CL, Walker J, Coyle W, et al. Gene-mediated cytotoxic immunotherapy as adjuvant to surgery or chemoradiation for pancreatic adenocarcinoma. Cancer Immunol Immunother. 2015;64(6):727–36.
Black M, Newcomb T, Wilson H, Loeb L. Creation of drug-specific herpes simplex virus type 1 thymidine kinase mutants for gene therapy. Proceed Natl Acad Sci USA. 1996;93(8):3525–9.
Bonifert G, Folkes L, Gmeiner C, Dachs G, Spadiut O. Recombinant horseradish peroxidase variants for targeted cancer treatment. Cancer Med. 2016;5(6):1194–203.
Braybrooke JP, Slade A, Deplanque G, Harrop R, Madhusudan S, Forster MD, et al. Phase I study of metXia-P450 gene therapy and oral cyclophosphamide for patients with advanced breast cancer or melanoma. Clin Cancer Res. 2005;11(4):1512–20.
Canine BF, Wang YH, Hatefi A. Biosynthesis and characterization of a novel genetically engineered polymer for targeted gene transfer to cancer cells. J Control Release. 2009;138(3):188–96.
Capello M, Lee M, Wang H, Babel I, Katz MH, Fleming JB, et al. Carboxylesterase 2 as a determinant of response to irinotecan and neoadjuvant FOLFIRINOX therapy in pancreatic ductal adenocarcinoma. J Natl Cancer Inst. 2015;107(8):132–41.
Caruso M, Panis Y, Gagandeep S, Houssin D, Salzmann J, Klatzmann D. Regression of established macroscopic liver metastases after in situ transduction of a suicide gene. Proceed Natl Acad Sci USA. 1993;90(15):7024–8.
Cattaneo R, Miest T, Shashkova EV, Barry MA. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol. 2008;6(7):529–40.
Chen CS, Jounaidi Y, Waxman DJ. Enantioselective metabolism and cytotoxicity of R-ifosfamide and S-ifosfamide by tumor cell-expressed cytochromes P450. Drug Metab Dispos. 2005;33(9):1261–7.
Chen H, Beardsley GP, Coen DM. Mechanism of ganciclovir-induced chain termination revealed by resistant viral polymerase mutants with reduced exonuclease activity. Proceed Natl Acad Sci USA. 2014;111(49):17462–7.
Chen X, Godbey W. The potential of the human osteopontin promoter and single-nucleotide polymorphisms for targeted cancer gene therapy. Curr Gene Ther. 2015;15(1):82–92.
Chen X, Scapa JE, Liu DX, Godbey WT. Cancer-specific promoters for expression-targeted gene therapy: ran, brms1 and mcm5. J Gene Med. 2016;18(7):89–101.
Chiocca EA, Aguilar LK, Bell SD, Kaur B, Hardcastle J, Cavaliere R, et al. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma. J Clin Oncol. 2011;29(27):3611–9.
Choi SA, Lee YE, Kwak PA, Lee JY, Kim SS, Lee SJ, et al. Clinically applicable human adipose tissue-derived mesenchymal stem cells delivering therapeutic genes to brainstem gliomas. Cancer Gene Ther. 2015;22(6):302–11.
Cortez MA, Godbey WT, Fang Y, Payne ME, Cafferty BJ, Kosakowska KA, et al. The synthesis of cyclic poly(ethylene imine) and exact linear analogues: an evaluation of gene delivery comparing polymer architectures. J Am Chem Soc. 2015;137(20):6541–9.
Dai M, Liu J, Chen DE, Rao Y, Tang ZJ, Ho WZ, et al. Tumor-targeted gene therapy using Adv-AFP-HRPC/IAA prodrug system suppresses growth of hepatoma xenografted in mice. Cancer Gene Ther. 2012;19(2):77–83.
Danks MK, Morton CL, Krull EJ, Cheshire PJ, Richmond LB, Naeve CW, et al. Comparison of activation of CPT-11 by rabbit and human carboxylesterases for use in enzyme/prodrug therapy. Am Assoc Cancer Res. 1999;5(4):917–24.
Dass CR, Choong PFM. Non-viral methods for gene transfer towards osteosarcoma therapy. J Drug Target. 2007;15(3):184–9.
Davies LC, Friedlos F, Hedley D, Martin J, Ogilvie LM, Scanlon IJ, et al. Novel fluorinated prodrugs for activation by carboxypeptidase G2 showing good in vivo antitumor activity in gene-directed enzyme prodrug therapy. J Med Chem. 2005;48(16):5321–8.
Davis JJ, Wang L, Dong F, Zhang L, Guo W, Teraishi F, et al. Oncolysis and suppression of tumor growth by a GFP-expressing oncolytic adenovirus controlled by an hTERT and CMV hybrid promoter. Cancer Gene Ther. 2006;13(7):720–3.
Deng LY, Wang JP, Gui ZF, Shen LZ. Antitumor activity of mutant bacterial cytosine deaminase gene for colon cancer. World J Gastroenterol. 2011;17(24):2958–64.
Fischer U, Steffens S, Frank S, Rainov NG, Schulze-Osthoff K, Kramm CM. Mechanisms of thymidine kinase/ganciclovir and cytosine deaminase/5-fluorocytosine suicide gene therapy-induced cell death in glioma cells. Oncogene. 2005;24(7):1231–43.
Floeth F, Shand N, Bojar H, Prisack H, Felsberg J, Neuen-Jacob E, et al. Local inflammation and devascularization ¾ in vivo mechanisms of the “bystander effect” in VPC-mediated HSV-Tk/GCV gene therapy for human malignant glioma. Cancer Gene Ther. 2001;8(11):843–51.
Forbes NS. Engineering the perfect (bacterial) cancer therapy. Nat Rev Cancer. 2010;10(11):785–94.
Francis R, Sharma S, Springer C, Green A, Hope-Stone L, Sena L, et al. A phase I trial of antibody directed enzyme prodrug therapy (ADEPT) in patients with advanced colorectal carcinoma or other CEA producing tumours. Br J Cancer. 2002;87(6):600–7.
Giraud B, Hebert G, Deroussent A, Veal GJ, Vassal G, Paci A. Oxazaphosphorines: new therapeutic strategies for an old class of drugs. Expert Opin Drug Metab Toxicol. 2010;6(8):919–38.
Greco O, Dachs GU, Tozer GM, Kanthou C. Mechanisms of cytotoxicity induced by horseradish peroxidase/indole-3-acetic acid gene therapy. J Cell Biochem. 2002;87(2):221–32.
Green LK, Syddall SP, Carlin KM, Bell GD, Guise CP, Mowday AM, et al. Pseudomonas aeruginosa NfsB and nitro-CBI-DEI—a promising enzyme/prodrug combination for gene directed enzyme prodrug therapy. Mol Cancer. 2013;12(1):1–6.
Hine CM, Seluanov A, Gorbunova V. Rad51 promoter-targeted gene therapy is effective for in vivo visualization and treatment of cancer. Mol Ther. 2012;20(2):347–55.
Hoffman RM, Zhao M. Methods for the development of tumor-targeting bacteria. Expert Opin Drug Dis. 2014;9(7):741–50.
Hosokawa M. Structure and catalytic properties of carboxylesterase isozymes involved in metabolic activation of prodrugs. Molecules. 2008;13(2):412–31.
Hu W, Liu W. Side populations of glioblastoma cells are less sensitive to HSV-TK/GCV suicide gene therapy system than the non-side population. In vitro cellular & developmental biology - animal. 2010;46(6):497–501.
Huang C, Liu L-Y, Song T-S, Ni L, Yang L, Hu X-Y, et al. Apoptosis of pancreatic cancer BXPC-3 cells induced by indole-3-acetic acid in combination with horseradish peroxidase. World J Gastroenterol WJG. 2005;11(29):4519–23.
Huber BE, Austin EA, Good SS, Knick VC, Tibbels S, Richards CA. In vivo antitumor activity of 5-fluorocytosine on human colorectal carcinoma cells genetically modified to express cytosine deaminase. Cancer Res. 1993;53(19):4619–26.
Humerickhouse R, Lohrbach K, Li L, Bosron WF, Dolan ME. Characterization of CPT-11 hydrolysis by human liver carboxylesterase isoforms hCE-1 and hCE-2. Cancer Res. 2000;60(5):1189–92.
Jeong YM, Oh MH, Kim SY, Li H, Yun HY, Baek KJ, et al. Indole-3-acetic acid/horseradish peroxidase induces apoptosis in TCCSUP human urinary bladder carcinoma cells. Die pharmazie - an Int J Pharm Sci. 2010;65(2):122–6.
Johnson AJ, Ardiani A, Sanchez-Bonilla M, Black ME. Comparative analysis of enzyme and pathway engineering strategies for 5FC-mediated suicide gene therapy applications. Cancer Gene Ther. 2011;18(8):533–42.
Kaliberov SA, Market JM, Gillespie GY, Krendelchtchikova V, Della Manna D, Sellers JC, et al. Mutation of Escherichia coli cytosine deaminase significantly enhances molecular chemotherapy of human glioma. Gene Ther. 2007;14(14):1111–9.
Kaliberova LN, Della Manna DL, Krendelchtchikova V, Black ME, Buchsbaum DJ, Kaliberov SA. Molecular chemotherapy of pancreatic cancer using novel mutant bacterial cytosine deaminase gene. Mol Cancer Ther. 2008;7(9):2845–54.
Kang Y, Zhang X, Jiang W, Wu C, Chen C, Zheng Y, et al. Tumor-directed gene therapy in mice using a composite nonviral gene delivery system consisting of the piggyBac transposon and polyethylenimine. BMC Cancer. 2009;9:126–33.
Karjoo Z, Chen X, Hatefi A. Progress and problems with the use of suicide genes for targeted cancer therapy. Adv Drug Deliv Rev. 2016;99(Pt A):113–28.
Kerr IG, Zimm S, Collins JM, O'Neill D, Poplack DG. Effect of intravenous dose and schedule on cerebrospinal fluid pharmacokinetics of 5-fluorouracil in the monkey. Cancer Res. 1984;44:4929–32.
Kestell P, Pruijn F, Siim B, Palmer B, Wilson W. Pharmacokinetics and metabolism of the nitrogen mustard bioreductive drug 5. Cancer Chemother Pharmacol. 2000;46(5):365–74.
Kim DS, Jeon SE, Park KC. Oxidation of indole-3-acetic acid by horseradish peroxidase induces apoptosis in G361 human melanoma cells. Cell Signal. 2004;16(1):81–8.
Kim KH, Dmitriev I, O'Malley JP, Wang M, Saddekni S, You Z, et al. A phase I clinical trial of Ad5.SSTR/TK.RGD, a novel infectivity-enhanced bicistronic adenovirus, in patients with recurrent gynecologic cancer. Clin Cancer Res. 2012;18(12):3440–51.
Kojima A, Hackett NR, Crystal RG. Reversal of CPT-11 resistance of lung cancer cells by adenovirus-mediated gene transfer of the human carboxylesterase cDNA. Cancer Res. 1998;58(19):4368–74.
Kokoris MS, Black ME. Characterization of herpes simplex virus type 1 thymidine kinase mutants engineered for improved ganciclovir or acyclovir activity. Protein Sci. 2002;11(9):2267–72.
Kumar S. Engineering cytochrome P450 biocatalysts for biotechnology, medicine, and bioremediation. Expert Opin Drug Metab Toxicol. 2011;6(2):115–31.
Leveille S, Samuel S, Goulet ML, Hiscott J. Enhancing VSV oncolytic activity with an improved cytosine deaminase suicide gene strategy. Cancer Gene Ther. 2011;18(6):435–43.
MacDiarmid JA, Brahmbhatt H. Minicells: versatile vectors for targeted drug or si/shRNA cancer therapy. Curr Opin Biotechnol. 2011;22(6):909–16.
Majhen D, Calderon H, Chandra N, Fajardo CA, Rajan A, Alemany R, et al. Adenovirus-based vaccines for fighting infectious diseases and cancer: progress in the field. Hum Gene Ther. 2014;25(4):301–17.
Malekshah OM, Lage H, Bahrami AR, Afshari JT, Behravan J. PXR and NF-kB correlate with the inducing effects of IL-1β and TNF-α on ABCG2 expression in breast cancer cell lines. Eur J Pharm Sci. 2012;47(2):474–80.
Martin J, Stribbling MS, Poon KG, Begent JRH, Napier M, Sharma KS, et al. Antibody-directed enzyme prodrug therapy: pharmacokinetics and plasma levels of prodrug and drug in a phase I clinical trial. Cancer Chemother Pharmacol. 1997;40(3):189–201.
Matzow T, Cowen RL, Williams KJ, Telfer BA, Flint PJ, Southgate TD, et al. Hypoxia-targeted over-expression of carboxylesterase as a means of increasing tumour sensitivity to irinotecan (CPT-11). J Gene Med. 2007;9(4):244–52.
Mayer A, Francis RJ, Sharma SK, Tolner B, Springer CJ, Martin J, et al. A phase I study of single administration of antibody-directed enzyme prodrug therapy with the recombinant anti-carcinoembryonic antigen antibody-enzyme fusion protein MFECP1 and a bis-iodo phenol mustard prodrug. Clin Cancer Res. 2006;12(21):6509–16.
Metz MZ, Gutova M, Lacey SF, Abramyants Y, Vo T, Gilchrist M, et al. Neural stem cell-mediated delivery of irinotecan-activating carboxylesterases to glioma: implications for clinical use. Stem Cells Transl Med. 2013;2(12):983–92.
Mingozzi F, High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122(1):23–36.
Mitchell D, Minchin R. E. coli nitroreductase/CB1954 gene-directed enzyme prodrug therapy: role of arylamine N-acetlytransferase 2. Cancer Gene Ther. 2008;15(11):758–64.
Miyagi T, Koshida K, Hori O, Konaka H, Katoh H, Kitagawa Y, et al. Gene therapy for prostate cancer using the cytosine deaminase/uracil phosphoribosyltransferase suicide system. J Gene Med. 2003;5(1):30–7.
Mohit E, Rafati S. Biological delivery approaches for gene therapy: strategies to potentiate efficacy and enhance specificity. Mol Immunol. 2013;56(4):599–611.
Münch RC, Janicki H, Volker I, Rasbach A, Hallek M, Buning H, et al. Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol Ther. 2013;21(1):109–18.
Nagy B, Mucsi I, Molnár J, Thurzo L. Combined effect of cisplatin and 5-fluorouracil with irradiation on tumor cells in vitro. Anticancer Res. 2002;22(1A):135–8.
Nasu Y, Saika T, Ebara S, Kusaka N, Kaku H, Abarzua F, et al. Suicide gene therapy with adenoviral delivery of HSV-tK gene for patients with local recurrence of prostate cancer after hormonal therapy. Mol Ther. 2007;15(4):834–40.
Nemunaitis J, Tong AW, Nemunaitis M, Senzer N, Phadke AP, Bedell C, et al. A phase I study of telomerase-specific replication competent oncolytic adenovirus (telomelysin) for various solid tumors. Mol Ther. 2010;18(2):429–34.
Nouri FS, Banerjee D, Hatefi A. Practical issues with the use of stem cells for cancer gene therapy. Stem cell review. 2015;11(5):688–98.
Nouri FS, Wang X, Dorrani M, Karjoo Z, Hatefi A. A ecombinant biopolymeric platform for reliable evaluation of the activity of pH-responsive amphiphile fusogenic peptides. Biomacromolecules. 2013;14(6):2033–40.
On K, Leigh G, Dilair B, Sharifah I, Mark U, Hayley S, et al. Direct retroviral delivery of human cytochrome P450 2B6 for gene-directed enzyme prodrug therapy of cancer. Cancer Gene Ther. 2001;8(7):473–82.
Oosterhoff D, Pinedo HM, van der Meulen IH, de Graaf M, Sone T, Kruyt FA, et al. Secreted and tumour targeted human carboxylesterase for activation of irinotecan. Br J Cancer. 2002;87(6):659–64.
Ortiz de Montellano PR. Cytochrome P450-activated prodrugs. Future medicinal chemistry. 2013;5(2):213–28.
Park SY, Lee W, Lee J, Kim IS. Combination gene therapy using multidrug resistance (MDR1) gene shRNA and herpes simplex virus-thymidine kinase. Cancer Lett. 2008;261(2):205–14.
Parker WB, Allan PW, Shaddix SC, Rose LM, Speegle HF, Gillespie GY, et al. Metabolism and metabolic actions of 6-methylpurine and 2-fluoroadenine in human cells. Biochem Pharmacol. 1998;55(10):1673–81.
Parker WB, Allan PW, Waud WR, Hong JS, Sorscher EJ. Effect of expression of adenine phosphoribosyltransferase on the in vivo anti-tumor activity of prodrugs activated by E. coli purine nucleoside phosphorylase. Cancer Gene Ther. 2011;18(6):390–8.
Patel P, Young JG, Mautner V, Ashdown D, Bonney S, Pineda RG, et al. A phase I/II clinical trial in localized prostate cancer of an adenovirus expressing nitroreductase with CB1984. Mol Ther. 2009;17(7):1292–9.
Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6(10):789–802.
Preuß E, Muik A, Weber K, Otte J, von Laer D, Fehse B. Cancer suicide gene therapy with TK.007: superior killing efficiency and bystander effect. J Mol Med. 2011;89(11):1113–24.
Prosser G, Copp J, Syddall S, Williams E, Smaill J, Wilson W, et al. Discovery and evaluation of Escherichia coli nitroreductases that activate the anti-cancer prodrug CB1954. Biochem Pharmacol. 2010;79(5):678–87.
Prosser GA, Copp JN, Mowday AM, Guise CP, Syddall SP, Williams EM, et al. Creation and screening of a multi-family bacterial oxidoreductase library to discover novel nitroreductases that efficiently activate the bioreductive prodrugs CB1954 and PR-104 A. Biochem Pharmacol. 2013;85(8):1091–103.
Richard C, Duivenvoorden W, Bourbeau D, Massie B, Roa W, Yau J, et al. Sensitivity of 5-fluorouracil-resistant cancer cells to adenovirus suicide gene therapy. Cancer Gene Ther. 2006;14(1):57–65.
Rodríguez-Antona C, Donato M, Boobis A, Edwards R, Watts P, Castell JV, et al. Cytochrome P450 expression in human hepatocytes and hepatoma cell lines: molecular mechanisms that determine lower expression in cultured cells. Xenobiotica. 2002;32(6):505–20.
Rosenthal E, Chung T, Parker W, Allan P, Clemons L, Lowman D, et al. Phase I dose-escalating trial of Escherichia coli purine nucleoside phosphorylase and fludarabine gene therapy for advanced solid tumors. Ann Onc. 2015;26:1481–7.
Ross AD, Varghese G, Oporto B, Carmichael FJ, Israel Y. Effect of propylthiouracil treatment on NADPH-cytochrome P450 reductase levels, oxygen consumption and hydroxyl radical formation in liver microsomes from rats fed ethanol or acetone chronically. Biochem Pharmacol. 1995;49(7):979–89.
Roy P, Waxman DJ. Activation of oxazaphosphorines by cytochrome P450: application to gene-directed enzyme prodrug therapy for cancer. Toxicol in Vitro. 2006;20(2):176–86.
Sanghani SP, Quinney SK, Fredenburg TB, Davis WI, Murry DJ, Bosron WF. Hydrolysis of irinotecan and its oxidative metabolites, 7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino] carbonyloxycamptothecin and 7-ethyl-10-[4-(1-piperidino)-1-amino]-carbonyloxycamptothecin, by human carboxylesterases CES1A1, CES2, and a newly expressed carboxylesterase isoenzyme, CES3. Drug Metab Dispos. 2004;32(5):505–11.
Sangro B, Mazzolini G, Ruiz M, Ruiz J, Quiroga J, Herrero I, et al. A phase I clinical trial of thymidine kinase-based gene therapy in advanced hepatocellular carcinoma. Cancer Gene Ther. 2010;17(12):837–43.
Santiago O, Jorge L, Schaffer DV. Adeno-associated virus (AAV) vectors in cancer gene therapy. J Control Release. 2016:In press.
Schlabach MR, Hu JK, Li M, Elledge SJ. Synthetic design of strong promoters. Proceed Natl Acad Sci USA. 2010;107(6):2538–43.
Seo G-M, Rachakatla RS, Balivada S, Pyle M, Shrestha TB, Basel MT, et al. A self-contained enzyme activating prodrug cytotherapy for preclinical melanoma. Mol Biol Rep. 2012;39(1):157–65.
Sharma K, Sengupta K, Chakrapani H. Nitroreductase-activated nitric oxide (NO) prodrugs. Bioorg Med Cemistry Lett. 2013;23(21):5964–7.
Shirakawa T, Gardner TA, Ko S-C, Bander N, Woo S, Gotoh A, et al. Cytotoxicity of adenoviral-mediated cytosine deaminase plus 5-fluorocytosine gene therapy is superior to thymidine kinase plus acyclovir in a human renal cell carcinoma model. J Urol. 1999;162(3 Pt 1):949–54.
Shirakawa T, Terao S, Hinata N, Tanaka K, Takenaka A, Hara I, et al. Long-term outcome of phase I/II clinical trial of Ad-OC-TK/VAL gene therapy for hormone-refractory metastatic prostate cancer. Hum Gene Ther. 2007;18(12):1225–32.
Silamkoti AV, Allan PW, Hassan AE, Fowler AT, Sorscher EJ, Parker WB, Secrist JA 3rd. Synthesis and biological activity of 2-fluoro adenine and 6-methyl purine nucleoside analogs as prodrugs for suicide gene therapy of cancer. Nucleosides Nucleotides Nucleic Acids. 2005;24(5–7):881–5.
Sorscher EJ, Hong JS, Allan PW, Waud WR, Parker WB. In vivo antitumor activity of intratumoral fludarabine phosphate in refractory tumors expressing E. coli purine nucleoside phosphorylase. Cancer Chemother Pharmacol. 2012;70(2):321–9.
Spooner R, Friedlos F, Maycroft K, Stribbling S, Roussel J, Brueggen J, et al. A novel vascular endothelial growth factor-directed therapy that selectively activates cytotoxic prodrugs. Br J Cancer. 2003;88(10):1622–30.
Swe P, Copp J, Green L, Guise C, Mowday A, Smaill J, et al. Targeted mutagenesis of the vibrio fischeri flavin reductase FRase I to improve activation of the anticancer prodrug CB1954. Biochem Pharmacol. 2012;84(6):775–83.
Traversari C, Marktel S, Magnani Z, Mangia P, Russo V, Ciceri F, et al. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood. 2007;109(11):4708–15.
Trinh QT, Austin EA, Murray DM, Knick VC. E. HB. Enzyme/prodrug gene therapy: comparison of cytosine deaminase/5-fluorocytosine versus thymidine kinase/ganciclovir enzyme/prodrug systems in a human colorectal carcinoma cell line. Cancer Res. 1995;55(21):4808–12.
Tsuji S, Chen X, Hancock B, Hernandez V, Visentin B, Reil K et al. Preclinical evaluation of VAX-IP, a novel bacterial minicell-based biopharmaceutical for nonmuscle invasive bladder cancer. Mol Ther Oncol. 2016;3:In press.
Tupper J, Stratford MR, Hill S, Tozer GM, Dachs GU. In vivo characterization of horseradish peroxidase with indole-3-acetic acid and 5-bromoindole-3-acetic acid for gene therapy of cancer. Cancer Gene Ther. 2010;17(6):420–8.
Tychopoulos M, Corcos L, Genne P, Beaune P, de Waziers I. A virus-directed enzyme prodrug therapy (VDEPT) strategy for lung cancer using a CYP2B6//NADPH-cytochrome P450 reductase fusion protein. Cancer Gene Ther. 2005;12(5):497–508.
Vangara KK, Ali HI, Lu D, Liu JL, Kolluru S, Palakurthi S. SN-38-cyclodextrin complexation and its influence on the solubility, stability, and in vitro anticancer activity against avarian cancer. AAPS Pharm Sci Tech. 2014;15(2):472–82.
Vanhoefer U, Harstrick A, Achterrath W, Cao S, Seeber S, Rustum YM. Irinotecan in the treatment of colorectal cancer: clinical overview. J Clin Oncol. 2001;19(5):1501–18.
Vass SO, Jarrom D, Wilson WR, Hyde EI, Searle PF. E. coli NfsA: an alternative nitroreductase for prodrug activation gene therapy in combination with CB1954. Br J Cancer. 2009;100(12):1903–11.
Voges J, Reszka R, Gossmann A, Dittmar C, Richter R, Garlip G, et al. Imaging-guided convection-enhanced delivery and gene therapy of glioblastoma. Ann Neurol. 2003;54(4):479–87.
Wang F, Zamora G, Sun C-H, Trinidad A, Chun C, Kwon YJ, et al. Increased sensitivity of glioma cells to 5-fluorocytosine following photo-chemical internalization enhanced nonviral transfection of the cytosine deaminase suicide gene. J Neuro-Oncol. 2014;118(1):29–37.
Wang Y, Canine BF, Hatefi A. HSV-TK/GCV cancer suicide gene therapy by a designed recombinant multifunctional vector. Nanomedicine. 2011;7(2):193–200.
Wierdl M, Tsurkan L, Hyatt JL, Edwards CC, Hatfield MJ, Morton CL, et al. An improved human carboxylesterase for enzyme/prodrug therapy with CPT-11. Cancer Gene Ther. 2008;15(3):183–92.
Williams EM, Little RF, Mowday AM, Rich MH, Chan-Hyams JVE, Copp JN, et al. Nitroreductase gene-directed enzyme prodrug therapy: insights and advances toward clinical utility. Biochem J. 2015;471(2):131–53.
Wold WSM, Toth K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Current gene therapy. 2013;13(6):421–33.
Wu L, Zhou W-B, Shen F, Liu W, Wu H-W, Zhou S-J, et al. Connexin32 mediated antitumor effects of suicide gene therapy against hepatocellular carcinoma: in vitro and in vivo anticancer activity. Mol Med Rep. 2016;13(4):3213–9.
Xiong T, Li Y, Ni F, Zhang F. Monitoring of bystander effect of herpes simplex virus thymidine kinase/acyclovir system using fluorescence resonance energy transfer technique. J Biomed Nanotechnol. 2012;8(1):74–9.
Yano H, Kayukawa S, Iida S, Nakagawa C, Sanda T, Kusumoto S, et al. Overexpression of carboxylesterase-2 results in enhanced efficacy of topoisomerase I inhibitor, irinotecan (CPT-11), for refractory multiple myeloma. Blood. 2006;108(11):5107.
Yi Y, Noh MJ, Lee KH. Current advances in retroviral gene therapy. Curr Gene Ther. 2011;11(3):218–28.
Acknowledgments
This work was supported by a grant from the National Institutes of Health/ National Cancer Institute (R01CA175318) to Arash Hatefi.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Drug Delivery
Rights and permissions
About this article

Cite this article
Malekshah, O.M., Chen, X., Nomani, A. et al. Enzyme/Prodrug Systems for Cancer Gene Therapy. Curr Pharmacol Rep 2, 299–308 (2016). https://doi.org/10.1007/s40495-016-0073-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40495-016-0073-y