
Abstract
Age-related macular degeneration (AMD) is the primary cause of blindness in developed countries, and is the third leading cause worldwide. Emerging evidence suggests that beside environmental and genetic factors, epigenetic mechanisms, such as microRNA (miRNA) regulation of gene expression, are relevant to AMD providing an exciting new avenue for research and therapy. MiRNAs are short, non-coding RNAs thought to be imperative for coping with cellular stress. Numerous studies have analyzed miRNA dysregulation in AMD patients, although with varying outcomes. Four studies which profiled dysregulated circulating miRNAs in AMD yielded unique sets, and there is only minimal overlap in ocular miRNA profiling of AMD. Mouse models of AMD, including oxygen-induced retinopathy and laser-induced choroidal neovascularization, showed similarities to some extent with miRNA patterns in AMD. For example, miR-146a is an extensively researched miRNA thought to modulate inflammation, and was found to be upregulated in AMD mice and cellular systems, but also in human AMD retinae and vitreous humor. Similarly, mir-17, miR-125b and miR-155 were dysregulated in multiple AMD mouse models as well as in human AMD plasma or retinae. These miRNAs are thought to regulate angiogenesis, apoptosis, phagocytosis, and inflammation. A promising avenue of research is the modulation of such miRNAs, as the phenotype of AMD mice could be ameliorated with antagomirs or miRNA-mimic treatment. However, before meaningful strides can be made to develop miRNAs as a diagnostic or therapeutic tool, reproducible miRNA profiles need to be established for the various clinical outcomes of AMD.
Similar content being viewed by others
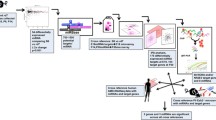


Several studies of microRNA, small non-protein coding RNA molecules, were conducted in blood and vitreous humor of AMD patients, but revealed few microRNAs consistently dysregulated in multiple studies. |
Similar profiling studies were reported in mouse models of distinct AMD features and demonstrated some similarities with the human AMD findings, including miR-146a, miR-17, miR-125b, and miR-155. |
Alteration of the levels of these miRNA in mice revealed an amelioration of the AMD-related damage, providing a promise for novel therapeutic approaches for this devastating blinding disease. |
1 Introduction
1.1 MicroRNAs: An Historical Perspective
MicroRNAs (miRNAs) are 19-24 nucleotide non-coding RNAs, which negatively regulate gene expression. Although miRNAs were not discovered until 1993, the idea that non-coding RNA molecules could interfere with gene expression, was first suggested in the late 1970s and early 1980s, when multiple groups found that exogenous oligonucleotides complementary to ribosomal RNA interfere with ribosome function [1–3].
Ironically, the motivation behind the project which ultimately lead to the discovery of miRNAs had only little to do with the field of noncoding RNAs or antisense gene regulation, but instead started with the study of neural development in microscopic worms [4, 5]. In the 1970s, Brenner [6] discovered a line of mutant lin-4(e912) Caenorhabditis elegans with remarkable developmental timing defects [7]. The “adult” larvae contained many cell types ordinarily only present during an early larval stage, and were devoid of many of the cell types and morphological structures typical of wild-type adults [4, 8].
In 1984, Ferguson et al. [9] discovered a C. elegans worm with nearly normal morphology in a culture of the lin-4 strain, and determined that a previously unknown gene, lin-14 was the responsible suppressor mutation. Animals with null alleles of lin-14 were shown to have developmental timing defects opposite to those of lin-4 [10]. These findings suggested an epistatic interaction between lin-4 and lin-14, and it was theorized that lin-4 encodes a protein which negatively regulates lin-14 [5]. In 1993, almost four years after the initial findings, two articles were published in the same issue of the Cell journal which elucidated the molecular relationship between lin-14 and lin-4. Lee et al. [4] showed that lin-4 is not a protein-encoding gene, but instead a 22nt non-coding RNA transcript and a 60nt hairpin structure. The second study reported that seven nucleotides in the 3′ untranslated region (UTR) of lin-14 are complementary to the lin-4 small RNAs, and suggested that lin-4 downregulates lin-14 translation through RNA-mediated gene expression [11].
1.2 MiRNAs and Ocular Pathogenesis
It is difficult to understate the importance of miRNAs, as they regulate proper cell growth, development, and differentiation [12]. Much of the early work on miRNAs focused on their roles in tissue and organ development [4, 13]. In this context, loss of miRNA function produced gross phenotypic defects, and their impact was unmistakable [13]. As technology advanced, researchers began uncovering the function of miRNAs in the mature tissue of higher eukaryotes. In recent years, the theory emerged that miRNA in mature organisms and tissues tend not to affect the primary function of cells, but are imperative for coping with various forms of cellular and organismal stress [13–16]. To date, miRNAs are thought to buffer against fluctuations in gene expression due to either stochastic modulation or environmental stress, in an effort to maintain cellular homeostasis [13, 17–19]. To facilitate their function, miRNAs are specifically incorporated into the RNA-induced silencing complex (RISC) and bind to transcripts in the cytosol. This binding either results in the degradation of the transcript by nucleases associated with the RIS complex or lead to reduced translation speed by influencing the ribosomal machinery.
As photoreceptors and retinal pigment epithelium (RPE) cells of the retina must maintain their function under exceptionally high rates of metabolism and protein synthesis, there is arguably no system under more stress [13]. Specifically, in pathological conditions of the eye, the intense stress overwhelms the cells’ coping mechanisms resulting in photoreceptor death ultimately causing blindness. This makes the retina a highly interesting organ to elucidate the pathological repercussions of miRNA dysregulation in mature tissue.
1.3 Age-Related Macular Degeneration (AMD)
AMD is a complex disease of the central retina and the primary cause of blindness in developed countries, as well as the third leading cause worldwide [20]. Estimates put the number of people suffering from the late stage of the disease worldwide at 11 million by 2020 [21]. In a healthy eye, light is transduced to a chemical signal by the highly specialized photoreceptors, which have a multifaceted support system. In AMD, the dysfunction of this support system causes progressive damage to the photoreceptors, and ultimately results in vision loss in one or both eyes [22].
Although the pathogenesis of AMD is currently not completely understood, the complex etiology has been linked to cellular, biochemical, and molecular events and is influenced by multiple components involving both environmental factors and genetic predisposition [22–24]. Genome-wide association studies and large scale re-sequencing projects have identified a number of single nucleotide variants (SNVs) enriched in complement and complement-related genes that confer a strong risk for AMD [25–28]. Recent efforts have increased the number of independent genetic signals associated with AMD risk to 52 at 34 different loci [27], explaining around half of the genomic heritability of the disease. Six out of those 34 loci harbor one or more complement or complement-related genes. Together, the known genetic risk factors point to the involvement of oxidative stress, lipid metabolism, extracellular matrix biology, inflammation and dysregulation of the complement cascade and other immune responses in the pathogenesis of the disease.
Early stages of AMD are characterized by an abnormal RPE pigment distribution in the macula, and the formation of drusen-containing lipids and an array of proteins in the space between the basal lamina of the RPE and the inner collagenous layer of Bruch’s membrane [22, 29–31]. Early AMD is usually asymptomatic, but may cause a modest decline in visual acuity and function resulting in delayed dark adaptation [22].
Early AMD can progress to late-stage AMD, characterized by geographic atrophy (GA AMD) and choroidal or, less common, retinal neovascularization (both forms subsumed as NV AMD). GA AMD is defined by well-demarcated areas of RPE atrophy, which is followed by degeneration of the corresponding photoreceptors and choroidal capillaries [32]. Disease progression in GA AMD is usually slow, although it can result in significant visual deficits in reading, night vision, and dark adaptation [33, 34]. There is currently no approved or effective treatment to prevent the onset or progression of GA [24, 35].
The hallmark of NV AMD is the growth of new blood vessels generally sprouting from the choroid, penetrating the overlying Bruch’s membrane, and growing within the subretinal pigment epithelium space or the subretinal space [36, 37]. In NV AMD the growing neovascular membrane is initially capillary-like [36], and these fenestrated and friable vessels frequently leak serous fluid, lipid, and blood into the retina and surrounding spaces. The neovascularization is accompanied by the infiltration of macrophages and other inflammatory, matrix metalloproteinases producing cells, enabling the growing membrane to digest through tissue planes [38]. Macrophages release a range of other mediators, including pro-inflammatory and proangiogenic factors. At some point, the balance between growth factors and inflammatory mediators shifts toward an antiangiogenic, anti-proteolytic, and anti-migratory state, and the CNV lesion becomes fibrosed [36, 37]. This process is known as disciform scar formation, and is associated with irreversible visual loss [36]. Untreated patients with NV AMD experience a consistent, steady deterioration in visual acuity over the first few years following onset of neovascularization, with 75 % of patients reaching legal blindness within 3 years [39].
Choroidal neovascularization can be treated but not cured with inhibitors of vascular endothelial growth factor (VEGF) [40]. In the pathogenesis of NV AMD, increased VEGF production is a defensive response by the tissue to oxidative stress or hypoxia, triggering the proliferation of newly formed blood vessels and damaging the retinal tissues, especially the photoreceptors [41]. In the past several years, the inhibition of VEGF has become the gold standard in the treatment of NV AMD, but is far from an ideal therapy. Monthly injections of VEGF inhibitors are burdensome to patients and a major challenge to healthcare systems worldwide [42]. Additionally, AMD patients with NV exhibit a great deal of variability in response to anti-VEGF treatments, some patients even lose visual acuity during treatment [40, 43].
Despite advances in the prevention and treatment of the disease, the number of afflicted patients is expected to increase due to a rapidly growing elderly population [20]. In many cases, the damage is not contained to the photoreceptors and RPE cells, the mental health of these patients may also suffer. Depression affects 30 % of older adults with visual impairment, and those with AMD seem to be at particular risk with 39 % affected [44]. The mental deterioration is thought to stem from the frustration and hopelessness elderly patients can feel when the loss of functional capacity becomes apparent [44]. In a national opinion poll of US adults, when asked which disease is the worst that could happen to them, African Americans and Non-Hispanic Whites said that blindness was worse than cancer and HIV/AIDS [45]. This underscores the urgent need for an effective AMD treatment.
The purpose of this review is to inventory the work that has been done to profile miRNA dysregulation in AMD, and further summarize the miRNA work done in well-established mouse models of AMD (Fig. 1). The question arises whether miRNA-based approaches to treatment could be feasible to ameliorate AMD-related tissue damage in the retina and consequently to prevent vision impairment or even blindness.

Word-cloud representation of words found in abstracts related to the search term “miRNAs and AMD”. All words with an occurrence greater than 4 are plotted and their relative abundance is indicated by the size of the respective word. Figure was plotted with Tagxedo (http://www.tagxedo.com/app.html) using words from the abstracts of 21 publications related to miRNAs in AMD [51, 52, 56, 57, 71, 73–75, 77, 82, 89, 90, 99, 103–110]
2 MiRNA and AMD
2.1 Circulating miRNA Profiles of AMD Patients Show Inconsistencies
The function and mode of action of miRNAs in the cell are reasonably well understood and recent advances have yielded promising results for future miRNA-based therapies. MiRNAs have been detected in bodily fluids such as blood, saliva or urine [46]. Although the exact function and origin of those extracellular miRNA are still debated, these molecules seem to function as novel messengers allowing cells in the body to communicate with other cells [47]. A special class of extracellular miRNAs are called circulating miRNAs. They are found in plasma or serum and are theorized to be involved in cell to cell signaling [48]. Since circulating miRNAs can be easily obtained through a blood draw, they are promising targets to investigate as potential liquid biopsy markers. For example, if a marker profile could be linked to an increased risk of developing AMD, this could be used as a minimally invasive screening tool to monitor treatment success. Furthermore, dysregulated miRNAs could potentially be a treatment option themselves by using miRNA mimics or antagomirs to modulate miRNA levels in the cell [49, 50].
Four studies published between 2014 and 2016 analyzed the circulating miRNA expression profiles of AMD patients (Table 1; Fig. 2). Of note, there is little overlap in the findings between the four studies. Although some miRNAs were found in several studies, they do not necessarily agree on the orientation of the effect. For example, miR-424-5p was found to be dysregulated in two studies, one study found the miR-424-5p to be downregulated in patients with late-stage NV AMD (p corrected = 9.6 × 10−3) [51] and no significant differences between the expression of miR-424-5p in GA AMD patients and controls. The authors noted, however, that the miR-424-5p expression was significantly higher in GA AMD patients compared to NV AMD patients (p corrected <0.005). A second study, published almost one year later, showed the opposite results. They found that miR-424–5p was upregulated in NV and GA AMD patients versus controls (p < 0.0001) [52]. Both studies agreed on the finding that miR-424-5p expression is higher in GA than NV AMD. The studies used a similar approach, a discovery study to find potential candidates and a validation study relying on quantitative (q)RT-PCR to confirm the candidates’ association with AMD, and recruited similarly sized cohorts. Their approach differed in the method used for the discovery study [next-generation sequencing (NGS) vs. qRT-PCR], but this can hardly explain the different direction of dysregulation which each study confirmed by using qRT-PCR. A partial explanation for these discrepancies may lie in the patient inclusion criteria, as the first study included late-stage NV AMD patients, some of whom had received anti-VEGF therapy, while the second report used newly diagnosed patients, who had not received any therapy prior to their recruitment for the study. Further, some controls in the first study suffered from glaucoma, whereas in the second study, only samples from people without ocular abnormalities were included.

Interaction between AMD related miRNAs and genes, pathways and tissues. We selected four miRNAs (plotted in blue) with the most evidence for an involvement in AMD and used Tarbase v7.0 [111] to extract genes (plotted in yellow) with validated interactions with those miRNAs. Genes that are regulated by at least three out of four miRNAs were used to search for significantly enriched pathways using WebGestalt [112]. Pathways containing at least six of these genes and being significantly enriched (Bonferroni corrected p value <0.01) are plotted in purple. The role of the miRNAs in AMD pathology was evaluated in different tissues and/or mouse models (plotted in green)
These inconsistencies are not unique to those two studies, in fact all four studies varied in the methods applied to quantify miRNAs, normalizing for qRT-PCR expression and profiling of patients included. Since the reported results differ substantially, the possibility that the different results could be due to methodological characteristics needs to be considered. Additionally, some studies had small sample sizes (13–33 patients, and as many controls), which draws into question the statistical power issue and consequently the validity of the conclusions. Conversely, two of the studies had large sample sizes (188–300 patients, and 147–200 controls). This, and the fact that AMD profiling studies of AMD mouse models revealed some overlapping results, appears reasonable to not entirely disregard the patient cmiRNA profiling studies, but instead to consider these results as preliminary, and to enjoy them with caution.
A more promising candidate may be miR-301. One study found the miR-301-3p arm to be downregulated in NV AMD [51], whereas a second study found the miR-301-5p arm to be upregulated in NV and GA AMD [52]. In miRNA biogenesis, the 5p and 3p arms of a mature miRNA are processed from a long precursor miRNA [53]. The 5p and 3p arms are separated, the guide strand is incorporated into the RNA-induced silencing complex, RISC, and subsequently becomes a functional gene inhibitor. The second strand, termed the complementary strand, usually becomes degraded. These two studies may indicate that miR-301-5p is a negative gene regulator in AMD, while miR-301-3p is subsequently degraded, and therefore downregulated in the patient.
In some cases the 5p and 3p arms can be directly correlated with another [54, 55]. Unfortunately, most publications do not specify which miRNA arm was studied, making it difficult to interpret their results. Furthermore, multiple members of the same miRNA families have been found in the various studies, i.e. let-7 and let-7c of the let-7 family, and miR-106a and b of the miR-17 family, among others. This is not as helpful as it might appear. MiRNAs are grouped into families based on similar sequence or their location in the genome, not based on their function. So, even though two members of the mir-188 family are indicated to be dysregulated in AMD, it still is difficult to draw conclusions from this.
2.2 MiRNA Dysregulation in AMD Ocular Tissue
To date, three studies profiled miRNA from samples of AMD retinae and RPE cells, and one study reported miRNA measurements from vitreous humor (VH) of AMD patients (Table 2; Fig. 2). MiR-146a was found to be upregulated in both forms of AMD, and in different tissue. One study showed the miRNA to be upregulated in the retina of GA AMD patients [56], whereas a second study found a 3.0-fold increase in the VH of NV AMD patients [57]. Both studies used similar sample sizes (12–13 patients and 9–13 controls), and methods to assess miRNA levels. The second study further discovered an upregulation of the miRNA in the plasma of NV AMD patients. Interestingly, a third group confirmed the upregulation of miR-146a in the serum of NV and GA AMD patients, although these findings were not validated using qRT-PCR [52]. Unfortunately, two of these studies did not specify whether they analyzed the 5p or 3p arm [52, 56].
MiR-146a is a well characterized small RNA and has been linked to progressive, age-related, inflammatory neurodegenerative disorders [58]. MiR-146a is theorized to modulate innate immune responses, inflammation, and the microglial activation state, although it is not yet clear whether the induction of miR-146a is protective or detrimental to the cell. It is under transcriptional control by nuclear factor-kappaB (NF-κB), and has been found to be upregulated by reactive oxygen species, pro-inflammatory cytokines, and amyloid peptides. Multiple studies have established that upregulation of miR-146a in stressed human neural cells such as astroglia and microglia, results in the downregulation of complement factor H (CFH) [59, 60]. CFH is a major repressor of the innate-immune and inflammatory response, and one of the key players in AMD pathogenesis [58, 61, 62]. It has been suggested that the upregulation of miR-146a has detrimental effects and promotes the pathogenesis of AMD [61]. There is evidence suggesting the opposite behavior as MiR-146a has been shown to interfere with interleukin-1-receptor-associated kinase-1 (IRAK1) and TNF receptor-associated factor 6 (TRAF6) [63, 64] as well as directly down-regulating interleukin-6 (IL-6) [57, 65]. IRAK1 and TRAF6 are known modulators of the NF-κB pathway, and their translational repression inhibits pro-inflammatory cytokine signaling [57, 66–68]. IL-6 acts as a pro-inflammatory cytokine, and is routinely used in the diagnosis of sepsis [69]. Since IL-6 is associated with progression of NV [64, 70] miR-146a may be a protective regulator of inflammation in NV AMD [57]. However, as the effect of miR-146a is not fully protective nor fully detrimental for AMD, the molecule is unlikely a good candidate for a therapeutic intervention.
A closely related small RNA to miR-146a, is miR-146b-5p, which is also theorized to be a negative regulator of inflammation, although it has not been characterized nearly as well as miR-146a [71]. MiR-146b is known to target IRAK1 and TRAF6 thereby inhibiting pro-inflammatory cytokine signaling [63, 65, 71, 72]. One study showed that the expression of miR-146a and miR-146b is highly induced by a combination of pro-inflammatory cytokines (IFN-γ, TNF-α, and IL-1β) in human RPE cells [71]. Conversely, miR-146b was shown to be downregulated in NV AMD plasma. Since both miRNAs bind similar targets, this could be a method of harnessing an immune reaction.
2.3 Dysregulated miRNA in AMD Models Provide Clues to Understanding the Function of miRNA Dysregulated in AMD
Multiple studies have profiled miRNAs in RPE cells of mice and rats in experimental models meant to mimic characteristic pathological changes seen in AMD patients (Supplementary Table 1; Fig. 2). These studies have shown a combined total of 176 miRNAs to be dysregulated. Fourteen of these miRNAs were also dysregulated in the same direction in AMD patients, which makes them exciting contenders for further research (Table 3). Importantly, the miRNA with the highest number of studies supporting its involvement in AMD is miR-146a. Two studies performed in patients, and three studies performed in human RPE cells or in mouse retinae revealed a miR-146a upregulation, although one study indicated this miRNA species to be downregulated after oxygen-induced retinopathy (OIR) [56, 57, 71, 73–75]. Although technically not an AMD model, OIR is a well-established protocol used to study hyperoxia-induced retinal neovascularization and inflammation in mice, two well-known characteristics of AMD [76, 77]. One upregulated miRNA in an OIR model, and oxidative stress model in RPE cells, as well as in NV AMD plasma was miR-17, a regulator of angiogenesis [78]. MiR-17 targets several pro-angiogenic genes, and the inhibition of miR-17 along with miR-20 increased the number of perfused vessels in Matrigel plugs [78]. Besides its antiangiogenic activity, miR-17 was recently shown to be anti-apoptotic as well [79]. Pretreatment with miR-17 significantly prevented the norepinephrine-induced apoptosis of cardiomyocytes, by inhibiting apoptotic protease activating factor 1 (Apaf-1), a facilitator of apoptosome formation. miR-17 is part of the miR-17-92 cluster, which is a negative regulator of angiogenesis, and encodes miR-17, -18a, -19a/b, -20a, and miR-92a [50]. Of the other members of this cluster, only miR-92a was upregulated in RPE cells after oxidative stress, and in mouse retinae after OIR [80]. miR-92a was not dysregulated in AMD patients. MiR-92a is a well-studied miRNA which also regulates angiogenesis. First, Bonauer et al. [50] showed the miRNA was upregulated in a hind limb mouse ischemia model in response to ischemic injury. The authors then systemically injected mice with a miR-92 antagomir before inducing ischemia, and showed that these mice had improved functional recovery of ischemic limbs, due to improved neovascularization and perfusion [50]. Similarly, treating mice with the miR-92 antagomir after inducing an acute myocardial infarction, resulted in reduced infarct size and improved left ventricular systolic and diastolic function [50]. miR-92a was significantly reduced in patients with coronary artery disease, a major risk factor for acute myocardial infarction [81].
Another miRNA which may regulate neovascularization and apoptosis is miR-106b, which was downregulated in NV AMD plasma and VH, and in RPE cells after oxidative stress [57, 82]. MiR-106b-25 cluster knockout mice showed impaired neovascularization after hind limb ischemia in mice [83]. This effect was confirmed in a study which induced cerebral ischemia in mice which had received a miR-106b antagomir, and observed decreased infarct volume and neuronal injury [84]. The same miR-106b antagomir-protected cells derived from a pheochromocytoma of the rat adrenal medulla (PC12) against glutamate-induced apoptosis and oxidative damage, as evidenced by decreased lactate dehydrogenase release, and enhanced superoxide dismutase activity [84]. MiR-106b showed an age-dependent expression, and was downregulated in serum from patients aged 70 versus 22 years [85].
With neovascularization as the hallmark of NV AMD, it is not surprising that many of the miRNAs associated with the disease have also been linked to angiogenesis. MiR-125b was upregulated in patient retinae, in mouse retinae after OIR, and in ARPE-19 cells under oxidative stress [56, 75, 82]. Overexpression of miR-125b was negatively correlated with VEGF mRNA, and inhibited tumor-induced angiogenesis in ovarian cancer cells [86]. In brain endothelial cells (ECs), miR-125b overexpression resulted in a trend towards reduced tubule length [87]. This inverse correlation between miR-125b and in vitro tube formation by ECs was confirmed by Muramatsu et al. [88] who theorized that prolonged overexpression of miR-125b could result in blood vessel regression due to a transient proangiogenic stimulation of miR-125b induction in EC cells.
MiR-184 was downregulated in primary culture of human RPE cells isolated from eyes of AMD donors and in OIR mouse retinae [77, 89, 90]. One of the studies showed that miR-184 negatively regulates Wnt signaling in OIR retinae, a conserved intracellular signaling pathway also involved in regulating angiogenesis and inflammation [77]. This interaction was confirmed by other findings that showed corneal neovascularization induced by suture was ameliorated by topical administration of miR-184 in rats [91]. Inhibition of miR-184 significantly reduced phagocytosis efficiency in adult RPE cells [89]. Phagocytosis is crucial for the maintenance of photoreceptors, and an age-related decline in phagocytosis is thought to contribute to AMD [89, 92, 93]. Interestingly, a group studying ocular Chlamydia trachomatis infection demonstrated that miR-184 and miR-155 had a direct relationship with the degree of clinical inflammation, where miR-184 was downregulated, and miR-155 was upregulated as the severity increased [94]. This inverse relationship may also be true in AMD, since miR-184 was downregulated in AMD RPE cell culture, while miR-155 was upregulated in AMD retina and macula [56, 89].
MiR-424, which was dysregulated in opposite directions in two independent studies with AMD patients was upregulated in OIR mouse retinae [90]. This may indicate that the upregulation is a true effect, although the evidence of upregulation in OIR mouse retinae is far from conclusive. OIR mimics two key traits of AMD such as inflammation and angiogenesis. In AMD, however, the macula is primarily affected, and mice do not have a macula. Therefore, information gained from mouse models of AMD should be considered with caution.
2.4 Manipulation of miRNA in AMD Mouse Models Ameliorate Damage
Multiple studies have manipulated miRNA levels in vivo following a pathologic insult and tested the effect. MiRNA were either over-expressed using a pre-miR or miR-mimic, or under-expressed using an antisense-miR or a knockout mouse. Subsequently, neovascularization was quantified and many miRNAs were shown to ameliorate the inflicted damage (Table 4; Fig. 2). Unfortunately, most of the miRNAs tested were not homologous to those which were dysregulated in the AMD profiling studies (see above), but there are two that overlap, miR-155 and miR-184.
As mentioned above, strong data suggest that miR-184 is involved in angiogenesis. The studies showed that miR-184 is downregulated in OIR mouse retinae, and one of the groups also showed that intraocular injection with pre-miR-184 reduced retinal neovascularization in OIR mice [90]. The authors also tested the miRNA in the most widely accepted NV AMD model, the laser-induced choroidal neovascularization [95–98]. This model relies on laser injury to perforate Bruch’s membrane, resulting in subretinal blood vessel recruitment from the choroid, recapitulating the main characteristic of NV AMD [95]. Of note, in this context treatment with pre-miR-184 failed to reduce the area of choroidal NV.
A far more complex candidate is miR-155 as it regulates not only angiogenic but also inflammatory responses in the retina. MiR-155 was upregulated in GA AMD retina and macula, in OIR mouse retina, and in rat retinas after light-induced retinal degeneration [56, 75, 99]. Yan et al. [75] subjected miR-155 knockout mice to OIR, but found no protective effects against OIR provided by miR-155 deficiency. Instead, it induced rapid revascularization and prevented the development of aberrant neovascularization. Next, the authors demonstrated that intraocular injection of a miR-155 mimic in WT mice (postnatal day 3) delayed migration of the regularly and radially expanding superficial capillary plexus and hindered advancement toward the retinal edge. To elucidate the effect of miR-155 on inflammation, they first showed that miR-155 knockout mice had significantly reduced numbers of microglia. miR-155-deficient mice showed a decreased expression of mitochondrial translocator protein, a selective marker of microglia in their highly reactive state [100], but an increased expression of CD200R, which blocks pro-inflammatory activation in microglial cells [75, 101]. The OIR miR-155 knockout mice showed reduced translocator protein (TPSO) and inflammatory cytokines [75]. It is of note, however, that miR-155 has been shown to have a cell-type specific function. The growth of six breast cancer cell lines showed a wide range of sensitivities when transfected with a miR-155 mimic [102]. MiR-155 may have a similar cell-type specific function in the microglia, endothelial cells, RPE cells, or some of other cell types implicated in AMD pathogenesis.
3 Conclusions
Taken together, increasing research efforts on small RNAs suggests that dysregulated miRNAs may regulate key aspects of AMD pathology. While data on circulating miRNA in AMD are still scarce, findings on dysregulated ocular miRNA show more promise. For example, mir-146a and miR-155 are regulators of inflammation and microglial activation state in response to stress, and are both dysregulated in AMD retinae. Furthermore, angiogenesis and response to varying oxygen levels is likely modulated by miR125b, and miR-17, which may also regulate apoptosis. These key candidate miRNAs could be useful as novel therapeutic approaches in AMD, as exemplified by miR-155 which was shown to modulate damage in AMD mouse models in vivo. Still, studies in mouse models are not ideal in the long term, and must be complemented by comprehensive studies establishing miRNA profiles in AMD development and progression.
References
Carroll AP, Tooney PA, Cairns MJ. Context-specific microRNA function in developmental complexity. J Mol Cell Biol. 2013;5:73–84.
Jayaraman K, McParland K, Miller P, Ts’o PO. Selective inhibition of Escherichia coli protein synthesis and growth by nonionic oligonucleotides complementary to the 3′ end of 16S rRNA. Proc Natl Acad Sci USA. 1981;78:1537–41.
Eckhardt H, Lührmann R. Blocking of the initiation of protein biosynthesis by a pentanucleotide complementary to the 3′ end of Escherichia coli 16 S rRNA. J Biol Chem. 1979;254:11185–8.
Lee R, Feinbaum R, Ambros V. A short history of a short RNA. Cell. 2004;116:S89–92.
Orellana EA, Kasinski AL. Micrornas in cancer: a historical perspective on the path from discovery to therapy. Cancers (Basel). 2015;7:1388–405.
Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94.
Horvitz HR, Sulston JE. Isolation and genetic characterization of cell-lineage mutants of the nematode Caenorhabditis elegans. Genetics. 1980;96:435–54.
Chalfie M, Horvitz HR, Sulston JE. Mutations that lead to reiterations in the cell lineages of C. elegans. Cell. 1981;24:59–69.
Ferguson EL, Sternberg PW, Horvitz HR (1987) A genetic pathway for the specification of the vulval cell lineages of Caenorhabditis elegans. Nature 326:259–67.
Ambros V, Horvitz HR. Heterochronic mutants of the nematode Caenorhabditis elegans. Science. 1984;226:409–16.
Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin- 14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–62.
Grasso M, Piscopo P, Confaloni A, Denti MA. Circulating miRNAs as biomarkers for neurodegenerative disorders. Molecules. 2014;19:6891–910.
Sundermeier TR, Palczewski K. The physiological impact of microRNA gene regulation in the retina. Cell Mol Life Sci. 2012;69:2739–50.
Leung AKL, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40:205–15.
Emde A, Hornstein E. miRNAs at the interface of cellular stress and disease. EMBO J. 2014;33:1428–37.
Schober A, Nazari-Jahantigh M, Weber C. MicroRNA-mediated mechanisms of the cellular stress response in atherosclerosis. Nat Rev Cardiol. 2015;12:361–74.
Malhotra S, Kim T, Zager J, Bennett J, Ebright M, D’Angelica M, et al. Use of an oncolytic virus secreting GM-CSF as combined oncolytic and immunotherapy for treatment of colorectal and hepatic adenocarcinomas. Surgery. 2007;141:520–9.
Ebert MS, Sharp PA. Roles for MicroRNAs in conferring robustness to biological processes. Cell. 2012;149:505–24.
Vidigal JA, Ventura A. The biological functions of miRNAs: lessons from in vivo studies. Trends Cell Biol. 2015;25:137–47.
Pascolini D, Mariotti SP. Global estimates of visual impairment: 2010. Br J Ophthalmol. 2012;96:614–8.
Wong WL, Su X, Li X, Cheung CMG, Klein R, Cheng C-Y, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Heal. 2014;2:e106–16.
Fritsche LG, Fariss RN, Stambolian D, Abecasis GR, Curcio CA, Swaroop A. Age-related macular degeneration: genetics and biology coming together. Annu Rev Genom Hum Genet. 2014;15:151–71.
Grassmann F, Ach T, Brandl C, Heid IM, Weber BHF. What does genetics tell us about age-related macular degeneration? Annu Rev Vis Sci. 2015;1:73–96.
Grassmann F, Fauser S, Weber BHF. The genetics of age-related macular degeneration (AMD)—novel targets for designing treatment options? Eur J Pharm Biopharm. 2015;95:194–202.
Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–62.
Klein RJ, Zeiss C, Chew EY, Tsai J, Sackler RS, Haynes C, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–9.
Fritsche LG, Igl W, Bailey JNC, Grassmann F, Sengupta S, Bragg-Gresham JL, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet. 2016;48:134–43.
Grassmann F, Cantsilieris S, Schulz-Kuhnt A-S, White SJ, Richardson AJ, Hewitt AW, et al. Multiallelic copy number variation in the complement component 4A (C4A) gene is associated with late-stage age-related macular degeneration (AMD). J Neuroinflamm. 2016;13:81.
Curcio CA, Johnson M, Huang JD, Rudolf M. Aging, age-related macular degeneration, and the response-to-retention of apolipoprotein B-containing lipoproteins. Prog Retin Eye Res. 2009;28:393–422.
Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99:14682–7.
Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000;14:835–46.
McLeod DS, Grebe R, Bhutto I, Merges C, Baba T, Lutty GA. Relationship between RPE and choriocapillaris in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50:4982–91.
Owsley C, McGwin G, Jackson GR, Kallies K, Clark M. Cone- and rod-mediated dark adaptation impairment in age-related maculopathy. Ophthalmology. 2007;114:1728–35.
Bhutto I, Lutty G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol Aspects Med. 2012;33:295–317.
Holz FG, Strauss EC, Schmitz-Valckenberg S, Van Lookeren Campagne M. Geographic atrophy: clinical features and potential therapeutic approaches. Ophthalmology. 2014;121:1079–91.
Grossniklaus HE, Green WR. Choroidal neovascularization. Am J Ophthalmol. 2004;137:496–503.
Keane PA, de Salvo G, Sim DA, Goverdhan S, Agrawal R, Tufail A. Strategies for improving early detection and diagnosis of neovascular age-related macular degeneration. Clin Ophthalmol. 2015;9:353–66.
Balasubramanian SA, Krishna Kumar K, Baird PN. The role of proteases and inflammatory molecules in triggering neovascular age-related macular degeneration: basic science to clinical relevance. Transl Res. 2014;164:179–92.
Wong T, Chakravarthy U, Klein R, Mitchell P, Zlateva G, Buggage R, et al. The Natural History and Prognosis of Neovascular Age-Related Macular Degeneration. Ophthalmology. 2008;115(116–126):e1.
Rosenfeld PJ, Shapiro H, Tuomi L, Webster M, Elledge J, Blodi B. Characteristics of patients losing vision after 2 years of monthly dosing in the phase III ranibizumab clinical trials. Ophthalmology. 2011;118:523–30.
Mavija M, Alimanovic E, Jaksic V, Kasumovic S, Cekic S, Stamenkovic M. Therapeutic modalities of exudative age-related macular degeneration. Med Arch. 2014;68:204.
Day S, Acquah K, Lee PP, Mruthyunjaya P, Sloan FA. Medicare costs for neovascular age-related macular degeneration, 1994-2007. Am J Ophthalmol. 2011;152:1014–20.
McKibbin M, Ali M, Bansal S, Baxter PD, West K, Williams G, et al. CFH, VEGF and HTRA1 promoter genotype may influence the response to intravitreal ranibizumab therapy for neovascular age-related macular degeneration. Br J Ophthalmol. 2012;96:208–12.
Cimarolli VR, Casten RJ, Rovner BW, Heyl V, Sörensen S, Horowitz A. Anxiety and depression in patients with advanced macular degeneration: current perspectives. Clin Ophthalmol. 2016;10:55–63.
Scott AW, Bressler NM, Ffolkes S, Wittenborn JS, Jorkasky J. Public attitudes about eye and vision health. JAMA Ophthalmol. 2016;56:1672.
Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18:997–1006.
Turchinovich A, Weiz L, Burwinkel B. Extracellular miRNAs: the mystery of their origin and function. Trends Biochem Sci. 2012;37:460–5.
Zhang Y, Liu D, Chen X, Li J, Li L, Bian Z, et al. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol Cell. 2010;39:133–44.
Zhao H, Wang J, Gao L, Wang R, Liu X, Gao Z, et al. MiRNA-424 Protects against permanent focal cerebral ischemia injury in mice involving suppressing microglia activation. Stroke. 2013;44:1706–13.
Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–3.
Grassmann F, Schoenberger PG, Brandl C, Schick T, Hasler D, Meister G, et al. A circulating MicroRNA profile is associated with late-stage neovascular age-related macular degeneration. PLoS ONE. 2014;9:e107461.
Szemraj M, Bielecka-Kowalska A, Oszajca K, Krajewska M, Goś R, Jurowski P, et al. Serum MicroRNAs as potential biomarkers of AMD. Med Sci Monit. 2015;21:2734–42.
Naidoo A, Naidoo K, Yende-Zuma N, Gengiah TN, Padayatchi N, Gray AL, et al. Changes to antiretroviral drug regimens during integrated TB–HIV treatment: results of the SAPiT trial. Antivir Ther. 2014;19:161–9.
Choo KB, Soon YL, Nguyen PNN, Hiew MSY, Huang C-J. MicroRNA-5p and -3p co-expression and cross-targeting in colon cancer cells. J Biomed Sci. 2014;21:95.
Mitra R, Lin C-C, Eischen CM, Bandyopadhyay S, Zhao Z. Concordant dysregulation of miR-5p and miR-3p arms of the same precursor microRNA may be a mechanism in inducing cell proliferation and tumorigenesis: a lung cancer study. RNA. 2015;21:1055–65.
Bhattacharjee S, Zhao Y, Dua P, Rogaev EI, Lukiw WJ. microRNA-34a-mediated down-regulation of the microglial-enriched triggering receptor and phagocytosis-sensor TREM2 in age-related macular degeneration. PLoS One. 2016;11:e0150211.
Ménard C, Rezende FA, Miloudi K, Wilson A, Tétreault N, Hardy P, et al. MicroRNA signatures in vitreous humour and plasma of patients with exudative AMD. Oncotarget. 2016;7:19171–84.
Alexandrov PN, Dua P, Lukiw WJ. Up-regulation of miRNA-146a in progressive, age-related inflammatory neurodegenerative disorders of the human CNS. Front Neurol. 2014;5:1–5.
Pogue AI, Li YY, Cui J-G, Zhao Y, Kruck TPA, Percy ME, et al. Characterization of an NF-kappaB-regulated, miRNA-146a-mediated down-regulation of complement factor H (CFH) in metal-sulfate-stressed human brain cells. J Inorg Biochem. 2009;103:1591–5.
Yallapu MM, Jaggi M, Chauhan SC. Curcumin nanoformulations: a future nanomedicine for cancer. Drug Discov Today. 2012;17:71–80.
Lukiw WJ, Surjyadipta B, Dua P, Alexandrov PN. Common micro RNAs (miRNAs) target complement factor H (CFH) regulation in Alzheimer’s disease (AD) and in agerelated macular degeneration (AMD). Int J Biochem Mol Biol. 2012;3:105–16.
Hill JM, Pogue AI, Lukiw WJ. Pathogenic microRNAs common to brain and retinal degeneration; recent observations in Alzheimer’s disease and age-related macular degeneration. Front Neurol. 2015;6:232.
Taganov KD, Boldin MP, Chang K-J, Baltimore D. NF- B-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses interferon-γ. Proc Natl Acad Sci USA. 2006;103:12481–6.
Iyer A, Zurolo E, Prabowo A, Fluiter K, Spliet WGM, van Rijen PC, et al. MicroRNA-146a: a key regulator of astrocyte-mediated inflammatory response. PLoS One. 2012;7:e44789.
Bhaumik D, Scott GK, Schokrpur S, Patil CK, Orjalo AV, Rodier F, et al. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging (Albany NY). 2009;1:402–11.
Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci USA. 2011;108:5003–8.
Zernecke A, Bidzhekov K, Noels H, Shagdarsuren E, Gan L, Denecke B, et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci Signal. 2009;2:ra81.
Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. 2011;13:423–33.
Kitanovski L, Jazbec J, Hojker S, Derganc M. Diagnostic accuracy of lipopolysaccharide-binding protein for predicting bacteremia/clinical sepsis in children with febrile neutropenia: comparison with interleukin-6, procalcitonin, and C-reactive protein. Supp Care Cancer. 2014;22:269–77.
Lavalette S, Raoul W, Houssier M, Camelo S, Levy O, Calippe B, et al. Interleukin-1 inhibition prevents choroidal neovascularization and does not exacerbate photoreceptor degeneration. Am J Pathol. 2011;178:2416–23.
Kutty RK, Nagineni CN, Samuel W, Vijayasarathy C, Jaworski C, Duncan T, et al. Differential regulation of microRNA-146a and microRNA-146b-5p in human retinal pigment epithelial cells by interleukin-1β, tumor necrosis factor-α, and interferon-γ. Mol Vis. 2013;19:737–50.
Perry MM, Moschos SA, Williams AE, Shepherd NJ, Larner-Svensson HM, Lindsay MA. Rapid changes in MicroRNA-146a Expression negatively regulate the IL-1-induced inflammatory response in human lung alveolar epithelial cells. J Immunol. 2008;180:5689–98.
Garcia TY, Gutierrez M, Reynolds J, Lamba DA. Modeling the dynamic AMD-associated chronic oxidative stress changes in human ESC and ipsc-derived RPE cells. Investig Ophthalmol Vis Sci. 2015;56:7480–8.
Chung SH, Gillies M, Sugiyama Y, Zhu L, Lee S, Shen W. Profiling of MicroRNAs involved in retinal degeneration caused by selective Müller cell ablation. PLoS One. 2015;10:e0118949.
Yan L, Lee S, Lazzaro DR, Aranda J, Grant MB, Chaqour B. Single and compound knock-outs of microRNA (miRNA)-155 and its angiogenic gene target CCN7 in mice alter vascular and neovascular growth in the retina via resident microglia. J Biol Chem. 2015;290:23264–81.
Connor KM, Krah NM, Dennison RJ, Aderman CM, Chen J, Guerin KI, et al. Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat Protoc. 2009;4:1565–73.
Weder N, Zhang H, Jensen K, Yang BZ, Simen A, Jackowski A, et al. Child abuse, depression, and methylation in genes involved with stress, neural plasticity, and brain circuitry. J Am Acad Child Adolesc Psychiatry. 2014;53(417–424):e5.
Doebele C, Bonauer A, Fischer A, Scholz A, Reiss Y, Urbich C, et al. Members of the microRNA-17-92 cluster exhibit a cell-intrinsic antiangiogenic function in endothelial cells. Blood. 2010;115:4944–50.
Song S, Seo H-H, Lee S-Y, Lee CY, Lee J, Yoo K-J, et al. MicroRNA-17-mediated down-regulation of apoptotic protease activating factor 1 attenuates apoptosome formation and subsequent apoptosis of cardiomyocytes. Biochem Biophys Res Commun. 2015;465:299–304.
Anand S, Cheresh DA. Emerging role of micro-RNAs in the regulation of angiogenesis. Genes Cancer. 2011;2:1134–8.
Fichtlscherer S, De Rosa S, Fox H, Schwietz T, Fischer A, Liebetrau C, et al. Circulating microRNAs in patients with coronary artery disease. Circ Res. 2010;107:677–84.
Howell JC, Chun E, Farrell AN, Hur EY, Caroti CM, Iuvone PM, et al. Global microRNA expression profiling: curcumin (diferuloylmethane) alters oxidative stress-responsive microRNAs in human ARPE-19 cells. Mol Vis. 2013;19:544–60.
Semo J, Sharir R, Afek A, Avivi C, Barshack I, Maysel-Auslender S, et al. The 106b-25 microRNA cluster is essential for neovascularization after hindlimb ischaemia in mice. Eur Heart J. 2014;35:3212–23.
Li P, Shen M, Gao F, Wu J, Zhang J, Teng F, et al. An Antagomir to MicroRNA-106b-5p Ameliorates Cerebral Ischemia and Reperfusion Injury in Rats Via Inhibiting Apoptosis and Oxidative Stress. Mol Neurobiol. 2016. doi:10.1007/s12035-016-9842-1.
Zhang H, Yang H, Zhang C, Jing Y, Wang C, Liu C, et al. Investigation of MicroRNA expression in human serum during the aging process. J Gerontol A Biol Sci Med Sci. 2014;70:1–8.
He J, Jing Y, Li W, Qian X, Xu Q, Li F-S, et al. Roles and mechanism of miR-199a and miR-125b in tumor angiogenesis. PLoS One. 2013;8:e56647.
Smits M, Wurdinger T, van het Hof B, Drexhage JA, Geerts D, Wesseling P, et al. Myc-associated zinc finger protein (MAZ) is regulated by miR-125b and mediates VEGF-induced angiogenesis in glioblastoma. FASEB J. 2012;26:2639–47.
Muramatsu F, Kidoya H, Naito H, Sakimoto S, Takakura N. microRNA-125b inhibits tube formation of blood vessels through translational suppression of VE-cadherin. Oncogene. 2013;32(4):414–21.
Murad N, Kokkinaki M, Gunawardena N, Gunawan MS, Hathout Y, Janczura KJ, et al. MiR-184 regulates ezrin, LAMP-1 expression, affects phagocytosis in human retinal pigment epithelium and is downregulated in age-related macular degeneration. FEBS J. 2014;281:5251–64.
Shen J, Yang X, Xie B, Chen Y, Swaim M, Hackett SF, et al. MicroRNAs regulate ocular neovascularization. Mol Ther. 2008;16:1208–16.
Zong R, Zhou T, Lin Z, Bao X, Xiu Y, Chen Y, et al. Down-regulation of MicroRNA-184 is associated with corneal neovascularization. Invest Ophthalmol Vis Sci. 2016;57:1398–407.
Li W. Phagocyte dysfunction, tissue aging and degeneration. Ageing Res Rev. 2013;12:1005–12.
Sun K, Cai H, Tezel TH, Paik D, Gaillard ER, Del Priore LV. Bruch’s membrane aging decreases phagocytosis of outer segments by retinal pigment epithelium. Mol Vis. 2007;13:2310–9.
Derrick T, Last AR, Burr SE, Roberts CH, Nabicassa M, Cassama E, et al. Inverse relationship between microRNA-155 and -184 expression with increasing conjunctival inflammation during ocular Chlamydia trachomatis infection. BMC Infect Dis. 2016;16:60.
Lambert V, Lecomte J, Hansen S, Blacher S, Gonzalez M-LA, Struman I, et al. Laser-induced choroidal neovascularization model to study age-related macular degeneration in mice. Nat Protoc. 2013;8:2197–211.
Jin Kim S, Ju Lee H, Yun J-H, Hwa Ko J, Choi DY, Youn OhJ. Intravitreal TSG-6 suppresses laser-induced choroidal neovascularization by inhibiting CCR2+ monocyte recruitment. Sci Rep. 2015;5:11872.
Huang H, Parlier R, Shen J-K, Lutty GA, Vinores SA. VEGF receptor blockade markedly reduces retinal microglia/macrophage infiltration into laser-induced CNV. PLoS One. 2013;8:e71808.
Nakajima T, Hirata M, Shearer TR, Azuma M. Mechanism for laser-induced neovascularization in rat choroid: accumulation of integrin α chain-positive cells and their ligands. Mol Vis. 2014;20:864–71.
Saxena K, Rutar MV, Provis JM, Natoli RC. Identification of miRNAs in a model of retinal degenerations. Investig Ophthalmol Vis Sci. 2015;56:1820–9.
Liu GJ, Middleton RJ, Hatty CR, Kam WWY, Chan R, Pham T, et al. The 18 kDa translocator protein, microglia and neuroinflammation. Brain Pathol. 2014;24:631–53.
Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KHG, Lynch MA. CD200 ligand-receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. J Neurosci. 2007;27:8309–13.
Goldgraben MA, Russell R, Rueda OM, Caldas C, Git A. Double-stranded microRNA mimics can induce length- and passenger strand–dependent effects in a cell type–specific manner. RNA. 2016;22:193–203.
Ertekin S, Yıldırım O, Dinç E, Ayaz L, Fidancı SB, Tamer L. Evaluation of circulating miRNAs in wet age-related macular degeneration. Mol Vis. 2014;20:1057–66.
Lin H, Qian J, Castillo AC, Long B, Keyes KT, Chen G, et al. Effect of miR-23 on oxidant-induced injury in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2011;52:6308–14.
Han S, Kong YC, Sun B, Han QH, Chen Y, Wang YC. microRNA-218 inhibits oxygen-induced retinal neovascularization via reducing the expression of roundabout 1. Chin Med J. 2016;129:709–15.
Westenskow PD, Kurihara T, Aguilar E, Scheppke EL, Moreno SK, Wittgrove C, et al. Ras pathway inhibition prevents neovascularization by repressing endothelial cell sprouting. J Clin Invest. 2013;123:4900–8.
Zhou Q, Gallagher R, Ufret-Vincenty R, Li X, Olson EN, Wang S. Regulation of angiogenesis and choroidal neovascularization by members of microRNA-23 ~ 27 ~ 24 clusters. Proc Natl Acad Sci USA. 2011;108:8287–92.
Liu C-H, Sun Y, Li J, Gong Y, Tian KT, Evans LP, et al. Endothelial microRNA-150 is an intrinsic suppressor of pathologic ocular neovascularization. Proc Natl Acad Sci USA. 2015;112:12163–8.
Zhou Q, Anderson C, Zhang H, Li X, Inglis F, Jayagopal A, et al. Repression of choroidal neovascularization through actin cytoskeleton pathways by MicroRNA-24. Mol Ther. 2014;22:378–89.
Haque R, Chun E, Howell JC, Sengupta T, Chen D, Kim H. MicroRNA-30b-mediated regulation of catalase expression in human ARPE-19 cells. PLoS One. 2012;7:e42542.
Vlachos IS, Paraskevopoulou MD, Karagkouni D, Georgakilas G, Vergoulis T, Kanellos I, et al. DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucl Acids Res. 2015;43:D153–9.
Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucl Acids Res. 2013;41:W77–83.
Kutty RK, Nagineni CN, Samuel W, Vijayasarathy C, Hooks JJ, Redmond TM. Inflammatory cytokines regulate microRNA-155 expression in human retinal pigment epithelial cells by activating JAK/STAT pathway. Biochem Biophys Res Commun. 2010;402:390–5.
Bai Y, Bai X, Wang Z, Zhang X, Ruan C, Miao J. MicroRNA-126 inhibits ischemia-induced retinal neovascularization via regulating angiogenic growth factors. Exp Mol Pathol. 2011;91:471–7.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors Patricia Berber, Felix Grassmann, Christina Kiel, and Bernhard HF Weber declare no conflict of interest.
Funding
The work and open access publication was funded in part by the institutional budget for Research and Teaching from the Freestate of Bavaria to BHFW.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Berber, P., Grassmann, F., Kiel, C. et al. An Eye on Age-Related Macular Degeneration: The Role of MicroRNAs in Disease Pathology. Mol Diagn Ther 21, 31–43 (2017). https://doi.org/10.1007/s40291-016-0234-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-016-0234-z