
Abstract
The National Institute for Health and Care Excellence, as part of the institute’s single technology appraisal process, invited the manufacturer of ribociclib (Kisqali®, Novartis) to submit evidence regarding the clinical and cost effectiveness of the drug in combination with an aromatase inhibitor for the treatment of previously untreated, hormone receptor-positive, human epidermal growth factor receptor 2-negative, locally advanced or metastatic breast cancer. Kleijnen Systematic Reviews Ltd and Erasmus University Rotterdam were commissioned as the Evidence Review Group for this submission. The Evidence Review Group reviewed the evidence submitted by the manufacturer, corrected and validated the manufacturer’s decision analytic model, and conducted exploratory analyses to assess the robustness and validity of the presented clinical and cost-effectiveness results. This article describes the company submission, the Evidence Review Group assessment and National Institute for Health and Care Excellence subsequent decisions. The main clinical effectiveness evidence was obtained from the MONALEESA-2 trial, a randomised controlled trial comparing ribociclib plus letrozole with placebo plus letrozole. Progression-free survival was significantly longer in the ribociclib group (95% confidence interval, 19.3–not reached) vs. 14.7 months (95% confidence interval 13.0–16.5) in the placebo group. To assess the cost effectiveness of ribociclib in combination with an aromatase inhibitor, the company developed an individual patient-level model using a discrete-event simulation approach in Microsoft® Excel. In the model, simulated patients move through a series of three health states until death, i.e. first-line progression-free survival, second-line progression-free survival and progressive disease. The length of progression-free survival during the first line was informed by the MONALEESA-2 trial. The benefit in progression-free survival in the first line was transferred to a benefit in overall survival assuming full progression-free survival to overall survival surrogacy (because of the immaturity of overall survival data from the MONALEESA-2 trial). Patient-level data from the BOLERO-2 trial, evaluating the addition of everolimus to exemestane in the second-line treatment of postmenopausal HR-positive advanced breast cancer, were used to inform the length of progression-free survival during the second line. Costs included in the model were treatment costs (e.g. technology acquisition costs of first, second, third and/or later line treatments), drug administration costs, monitoring costs and health state costs (including terminal care). Additionally, the costs of adverse events associated with the first-line treatment were incorporated. The Evidence Review Group recalculated the incremental cost-effectiveness ratio using data from a different data cut-off date from the MONALEESA-2 trial and by changing some assumptions (e.g. progression-free survival to overall survival surrogacy approach and post-progression third and/or later line treatment-related costs). After two appraisal committee meetings and a revised base case submitted by the company (including a second enhanced patient access scheme discount), the committee concluded that taking into account the uncertainties in the calculation of the cost effectiveness, there were plausible cost-effectiveness estimates broadly in the range that could be considered as a cost-effective use of National Health Service resources. Therefore, ribociclib was recommended as a treatment option for the first-line treatment of hormone receptor-positive, human epidermal growth factor receptor 2-negative breast cancer, contingent on the company providing ribociclib with the discount agreed in the second enhanced patient access scheme.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Centrally assessed progression-free survival data might be more reliable than locally assessed progression-free survival data in the presence of differences in adverse-event rates. |
When the overall survival data from the trial are immature, different (e.g. partial or full) progression-free survival to overall survival surrogacy approaches should be explored in different scenario analyses, with different parameter realisations, which all should be taken into account in the decision-making process. |
When there are more than one simultaneous/recent appraisals in the same indication, this can be beneficial as the results of the other appraisal might be used for external validation purposes. |
The iterative nature of appraisal meetings may provide an opportunity to find a middle ground between the company assumptions and the Evidence Review Group point of view. The company may provide enhanced patient access scheme discounts to reduce the uncertainty surrounding the cost effectiveness of the drug in this process. |
1 Introduction
The National Institute for Health and Care Excellence (NICE) is an independent organisation providing national guidance on promoting good health and preventing and treating ill health. The single technology appraisal (STA) process is designed to provide recommendations on a single product, device or other technology with a single indication [1]. The process covers new technologies and enables NICE to produce guidance shortly after the technology is introduced into UK. The NICE Appraisal Committee (AC) obtains relevant evidence from several sources: the company submission (CS) [2], a report from the appointed independent Evidence Review Group (ERG) [3] and advice from consultees (i.e. patients, experts and other stakeholders). The CS includes a written report and a mathematical model describing the clinical and cost effectiveness of the technology under investigation. The ERG, an external organisation independent of NICE, reviews the CS and produces a summary report and provides a critique of the submitted evidence. After consideration of all the relevant evidence, the AC formulates preliminary guidance in the form of the Appraisal Consultation Document (ACD) as to whether or not to recommend the intervention. The stakeholders are invited to comment on this ACD and the submitted evidence. A subsequent ACD may be produced or a Final Appraisal Determination is issued. Once published, NICE technology guidance provides a legal obligation for National Health Service (NHS) providers to reimburse technologies that have been approved. This article presents a summary of the ERG report and the development of NICE guidance for the STA of ribociclib in combination with an aromatase inhibitor (AI), for treating previously untreated, hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2−), locally advanced or metastatic breast cancer. Full details of all the relevant appraisal documents can be found on the NICE website [4].
2 Decision Problem
Breast cancer is one of the most common cancers affecting women worldwide and a leading cause of cancer deaths [5]. Despite advances in the understanding of different forms of breast cancer and improvements in the treatment of patients, breast cancer remains a potentially life-threatening diagnosis, in large part because of breast cancer recurrence and the incurability of advanced disease; both remain major clinical challenges [6]. Advanced breast cancer is considered to include both locally advanced and stage IV (metastatic) cancer [7, 8], and the median survival of patients with advanced breast cancer is just 2–3 years [9].
Breast cancer is a heterogeneous disease and there are a number of recognisable histological and intrinsic subtypes distinguished by the expression of oestrogen receptors, progesterone receptors and epidermal growth factor receptors (e.g. HER2), and by distinct gene profiles that affect prognosis and outlook [10]. Around 75% of postmenopausal women with breast cancer have tumours that are HR+ [11], and the majority of HR+ patients are HER2−; therefore, HR+/HER2− is the most common form of breast cancer [11, 12].
The current NICE pathway of care and guidance regarding the therapeutic management of advanced breast cancer recommends endocrine therapy as the first-line treatment for the majority of patients [13]. According to the NICE pathway of care, chemotherapy should only be a first-line option in patients whose disease is imminently life threatening or requires early relief of symptoms because of significant visceral organ involvement. In such patients, endocrine therapy should then be offered on completion of chemotherapy [13].
Ribociclib (Kisqali®) is an oral cyclin-dependent kinase 4 and 6 inhibitor that aims to prevent the phosphorylation of retinoblastoma protein and restore its growth-inhibitory effects. Ribociclib in combination with an AI (ribociclib plus an AI) has a UK marketing authorisation for the following indication: “treatment of postmenopausal women with HR+, HER2− locally advanced or metastatic breast cancer as initial endocrine based therapy” [14].
The remit of this appraisal was specified by NICE’s final scope [15], which was to assess the clinical and cost effectiveness of ribociclib plus an AI within its licenced indication. The comparators listed in the scope were AIs, such as letrozole or anastrozole.
3 Independent Evidence Review Group Report
Kleijnen Systematic Reviews Ltd, in collaboration with Erasmus University Rotterdam, acted as the ERG, and reviewed the evidence on the clinical and cost effectiveness of ribociclib plus an AI for the treatment of postmenopausal women with HR+/HER2−, locally advanced or metastatic breast cancer as initial endocrine-based therapy. The review embodied three aims: (1) to assess whether the CS conformed to the methodological guidelines issued by NICE [1]; (2) to assess whether the company’s interpretation and analysis of the evidence were appropriate; and (3) to indicate the presence of other sources of evidence or alternative interpretations of the evidence that could inform NICE guidance.
The ERG critically reviewed the evidence in the CS, the response to clarification questions and evidence provided after the publication of the ACD. Furthermore, it conducted additional searches, explored the impact of assumptions on the incremental cost-effectiveness ratio (ICER), revised the economic model and explored additional scenario analyses.
3.1 Critique of the Decision Problem
In the CS, findings from the MONALEESA-2 trial [16] were used, in which ribociclib in combination with letrozole was compared with letrozole alone. Other AIs such as anastrozole were not considered. However, the ERG believed that the company provided sufficient justification for the generalisability of letrozole to other AIs. In addition, the ERG noted that the MONALEESA-2 trial included very few patients from England and Wales, and that the trial population differed from the UK population particularly in terms of endocrine sensitivity and the proportion of patients with de-novo advanced breast cancer. Thus, the ERG concluded that the MONALEESA-2 trial might not be entirely representative of the population in the NICE scope.
3.2 Summary of the Clinical Evidence
One phase III trial, MONALEESA-2 [16], with 668 patients was presented as the main source of evidence. The MONALEESA-2 study included postmenopausal women with HR+/HER2− recurrent or metastatic breast cancer who had not received previous systemic therapy for advanced disease.
The trial was conducted at 223 trial centres in 29 countries including very few patients from England and Wales. Patients were randomised 1:1 to receive ribociclib (600 mg once daily, days 1–21 of a 28-day cycle) plus letrozole (2.5 mg once daily, continuous treatment) or placebo plus letrozole (2.5 mg once daily, continuous treatment). Dose reductions for ribociclib (from 600 to 400 to 200 mg per day) were permitted to manage adverse events (AEs); no dose reductions were permitted for letrozole. Treatment was continued until disease progression, unacceptable toxicity, death, or discontinuation of ribociclib or letrozole for any other reason.
The primary outcome was progression-free survival (PFS) as per RECIST version 1.1 criteria, based on local radiological assessment; assessments were also carried out by a blinded independent review committee (BIRC). The key secondary endpoint was overall survival (OS) [defined as the time from the date of randomisation to the date of death as a result of any cause].
A total of 334 patients were randomised to ribociclib and 334 to placebo in the intention-to-treat population. At the time of the data cut-off (29 January, 2016), a total of 349 patients (52.2%) were still receiving treatment (ribociclib, n = 195; placebo, n = 154). The rates of discontinuation were 41.6% in the ribociclib group compared with 53.9% in the placebo group. The most frequent reason for discontinuation was disease progression in both groups (ribociclib, 26.0%; placebo, 43.7%). Discontinuations because of AEs were 7.5% in the ribociclib group and 2.1% in the placebo group. The median duration of follow-up from randomisation to the data cut-off was 15.3 months.
In the clinical effectiveness section, the CS presented data from the first interim analysis only (cut-off January 2016) and focused on results based on local assessments. Progression-free survival was significantly longer in the ribociclib group (95% confidence interval 19.3–not reached) vs. 14.7 months (95% confidence interval 13.0–16.5) in the placebo group. The addition of ribociclib to letrozole reduced the risk of death or progression by 44% (hazard ratio = 0.56; 95% confidence interval 0.43–0.72).
Although there were some differences in effectiveness, subgroup analyses showed that results for PFS favoured ribociclib for all subgroups including both those with newly diagnosed disease and those with existing disease and those who had received prior therapy and patients who had not. Although the occurrence of any AEs was overall similar in ribociclib and placebo groups, a greater number of AEs and severe AEs were attributable to ribociclib. The most common event was neutropenia. Gastrointestinal events such as nausea, vomiting and diarrhoea occurred more frequently in the ribociclib group. The submission did not meet the criteria for end of life as the life expectancy for patients with newly diagnosed HR+/HER2− advanced breast cancer is greater than 24 months.
3.3 Critique of the Clinical Evidence
The company searched a good range of databases and conference proceedings and carried out searches in accordance with NICE guidance. However, no literature searches were conducted to identify AE data, indirect and mixed treatment comparisons or non-randomised and non-controlled evidence.
The clinical effectiveness evidence in the submission was based on one trial, the MONALEESA-2 study. The ERG was not aware of any other evidence relevant to the decision problem. Overall, the MONALEESA-2 trial was a good-quality randomised controlled trial. However, increased rates of AEs, such as neutropenia (74% in the ribociclib group vs. 5% in the letrozole group), could have unblinded physicians and/or patients. Therefore, results based on independent reviews were deemed to be more reliable.
The main concern regarding the methodology of the MONALEESA-2 trial was that the initial results presented in the CS were based on data from the January 2016 cut-off for PFS. At this point, the OS data were immature as the required number of deaths had not been reached, with 43 deaths (23 in the ribociclib group and 20 in the placebo group) at the time of the data cut-off. The ERG identified two more recent interim analyses from the MONALEESA-2 trial after the January 2016 cut-off (June 2016 and January 2017), and requested these data as part of the clarification process. The ERG considered OS results from the most recent data cut-off (January 2017) the most informative. Additionally, the ERG considered the BIRC results more plausible for PFS, partly because the NICE committee preferred these data in a recent technology appraisal (TA495: palbociclib in the first line treatment of HR+/HER2− advanced breast cancer) [17], and partly because the increased AEs in the ribociclib arm could have unblinded physicians and/or patients; therefore, making results based on independent reviews more reliable. Unfortunately, BIRC results were not available at the latest (January 2017) data cut-off; therefore, the ERG considered the BIRC PFS results from the June 2016 cut-off to be the most appropriate.
The PFS results were more favourable for ribociclib in the company preferred results compared with the ERG preferred results. Using the latest data cut-off, OS Kaplan–Meier (KM) curves were still rather immature and there appeared to be insufficient evidence from the MONALEESA-2 trial to argue that ribociclib improves OS.
3.4 Summary of the Cost-Effectiveness Evidence
The company developed an individual patient-level model, following a discrete-event simulation approach, to assess the cost effectiveness of ribociclib plus an AI for previously untreated advanced or metastatic HR+/HER2− breast cancer. Letrozole was considered as representative of the available AI therapies, both in terms of clinical effectiveness and costs. In the model, simulated patients move through a series of health states; these include first-line PFS (PFS1), second-line PFS (PFS2), progressed disease (later lines) and death. A lifetime horizon was chosen and costs and effects were discounted with an annual rate of 3.5%.
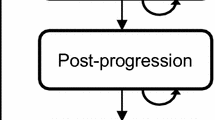
All patients start in the PFS1 state, in which they receive either ribociclib in combination with letrozole or letrozole alone. Patients stay in this state until they progress and move to the PFS2 state, or until they die. Second-line PFS represents the time between disease progression in first- and second-line treatment cessation (as a proxy for disease progression). In the PFS2 state, patients receive one of the following treatments: everolimus in combination with exemestane, exemestane (representing a single-agent endocrine therapy) or capecitabine (representing chemotherapy). Patients stay in this state until they progress and move to the progressed disease state, or until they die. The progressed disease state represents the time from second-line therapy cessation (as a proxy for progression) until death, and in this state, patients receive subsequent treatments and/or supportive/palliative care. The death state is an absorbing state. The model structure is illustrated in Fig. 1.

Model structure. PFS 1 first-line progression-free survival, PFS 2 second-line progression-free survival
For the PFS1 state, the relevant clinical model inputs for the ribociclib plus an AI and AI monotherapy arms were time to treatment discontinuation (TTD), PFS and the proportion of death among the PFS events. These inputs were derived from the analysis of data from the June 2016 cut-off of the MONALEESA-2 trial.
For the extrapolation of the PFS of the ribociclib plus an AI and AI arms from the MONALEESA-2 trial, an exponential distribution was considered to be the most appropriate, considering the plausibility of the long-term extrapolation to the external KM data from other trials in which letrozole monotherapy was a comparator (PALOMA-2 [18], LEA [19] and ALLIANCE trials [20]). The survival extrapolation of the TTD in the first line was conducted independently and based on clinical expert opinion and model predictions. The company selected the exponential distribution for the base case and alternative distributions were explored in scenario analyses, taking into account a time constraint, which assured that the TTD was never greater than PFS.
After the first-line treatment, it was assumed that the distribution of treatments received in the PFS2 state (exemestane monotherapy, everolimus plus exemestane or chemotherapy) were different after ribociclib plus an AI and AI monotherapy arms. These estimates were based on clinical opinion and the impact of assuming different treatment distributions was explored in scenario analyses.
In the second line, TTD data from the BOLERO-2 trial [21] (comparing everolimus plus exemestane with exemestane in second-line HR+/HER2− breast cancer) were used as a proxy for PFS. For the patients who received everolimus plus exemestane, a Weibull distribution was deemed to be the most appropriate by the company, based on its visual fit to the TTD KM curve from the trial. For the TTD of exemestane monotherapy and chemotherapy, hazard ratios from the BOLERO-2 trial and a study by Li et al. [22] were applied to the fitted Weibull curve, respectively.
The probability of death before treatment discontinuation in the second line was derived from the analysis of patient-level data from the BOLERO-2 trial for patients receiving everolimus plus exemestane combination therapy or exemestane monotherapy in the PFS2 state. No death before treatment discontinuation was assumed for patients receiving chemotherapy in the PFS2 state.
The company used the pooled post-treatment discontinuation survival data as a proxy for the post-progression survival in the BOLERO-2 trial, based on the observed similarity of the KM curves shown in the CS. A range of parametric survival models were fitted to the pooled data and the Weibull distribution was chosen to model the post-discontinuation survival after everolimus plus exemestane combination therapy or exemestane monotherapy in the PFS2 state, based on the statistical and visual fit. For the post-discontinuation survival under chemotherapy, the hazard ratios reported in Li et al. [22] were used. Alternative distributions for the modelling of the post-discontinuation survival were explored in the scenario analyses in the CS.
In the model, OS is modelled indirectly, and is a function of the time spent in each of the alive health states (PFS1, PFS2 and progressed disease). In the model, in the base case, a full PFS to OS surrogacy approach was followed. In this approach, it is assumed that a gain in PFS would lead to an equal gain in OS, for the patients who did not die upon progression as depicted in Fig. 2.

Source: company submission, Fig. 20, p. 102
Illustration of the full progression-free survival (PFS) to overall survival (OS) surrogacy approach. PD progressive disease, PFS1 first-line progression-free survival, PFS2 second-line progression-free survival.
In addition to the full PFS to OS surrogacy approach, a range of threshold-based PFS to OS surrogacy scenarios (from 4 to 24 months) were conducted. In these threshold-based scenarios, a gain in PFS was translated into an equivalent gain in OS only if a pre-defined threshold is exceeded. The threshold was defined either in terms of the absolute PFS under ribociclib with letrozole or in terms of the PFS gain of ribociclib in combination with letrozole compared to letrozole monotherapy. A schematic of the patient flow based on an absolute PFS-based threshold scenario is given in Fig. 3.

Source: company submission, Fig. 21, p. 103
Illustration of the patient flow in an absolute, progression-free survival (PFS)-based threshold scenario. OS overall survival, PD progressive disease, PFS1 first-line progression free survival, PFS2 second-line progression-free survival.
Utility values of patients (on and off treatment) in the PFS1 state were derived from the statistical analysis of EQ-5D-5L data collected in the MONALEESA-2 trial. Utility values for PFS2 were taken from Lloyd et al. [23] adjusted for age and treatment response (the latter based on the BOLERO-2 study). For patients treated with second-line chemotherapy, a utility decrement was applied, in line with the findings of Peasgood et al. [24]. Utility values for the progressed disease state were taken from TA495 [17], which were derived from Lloyd et al. [23], as well, adjusted for age.
Treatment costs (e.g. technology acquisition costs of first, second, third and later line treatments), drug administration costs, monitoring costs and health state costs (including terminal care) were included in the model. Additionally, the costs of AEs associated with the first-line treatment were incorporated. All costs used in the model calculations were based on their 2016 values.
The prices of generic treatments (e.g. letrozole) were taken from the electronic market information tool (eMIT) [25]. Non-generic drug prices were taken from the British National Formulary [26]. For ribociclib, the company proposed a patient access scheme (PAS) discount in its submission and for everolimus, another PAS price discount was already available for the NHS.
The licenced dose for ribociclib is 600 mg once daily for 21 days of a 28-day cycle, which consists of three 200-mg tablets. Ribociclib 200 mg is available in packs of 63, 42 and 21 tablets, with a pricing structure such that each 200-mg tablet has the same price regardless of the pack size. A proportion of patients in MONALEESA-2 had their dose reduced to 400 mg and 200 mg daily. The company assumed that patients who reduce their dose do not waste tablets as they can simply take fewer tablets daily, and therefore a pack lasts longer. The company used individual patient data to calculate the total number of days patients received 600 mg, 400 mg or 200 mg at each cycle until the ninth cycle, and cycle 10 data were used for cycle 10 and onwards owing to decreasing patient numbers.
The drug dosage and treatment duration data for everolimus and/or exemestane in the second line were derived from the BOLERO-2 study. Capecitabine was assumed as the representative chemotherapy administered in the second line and its dosage and duration data were taken from the NICE clinical guideline for breast cancer [27].
For the progression health state, drug acquisition costs were estimated to be £461.54 per week (i.e. £2000 per month), based on clinical expert opinion. Monitoring costs for ribociclib were incorporated in line with the anticipated licence of ribociclib. Ribociclib, letrozole, everolimus and exemestane are all administered orally, and the drug administration costs for chemotherapy were based on NHS reference costs and previous NICE technology appraisals. The costs associated with the management of the disease were estimated based on the package of resource use for advanced breast cancer recommended in the NICE clinical guideline 81, previous NICE technology appraisals in HR+/HER2− advanced breast cancer (TA421 [28] and TA495 [17]) and validation through clinical experts. Costs of grade 3 or 4 AEs were based on NHS reference costs.
Without the PAS, incremental quality-adjusted life-years (QALYs) were 0.96 and ICER values, which were confidential, were above the accepted willingness-to-pay threshold of £30,000/QALY. With the PAS, incremental costs reduced to such an extent that the resulting ICER was around £30,000/QALY. Quality-adjusted life-years were predominantly gained within the PFS1 state, and the increase in costs is mainly caused by the increase in first-line treatment costs. The probabilistic sensitivity analysis results were in line with the deterministic base-case results. Within the deterministic sensitivity analyses, the company varied some of the input parameters to its upper and lower limits. This analysis showed that the ICER was most sensitive to the discount rates.
Furthermore, the company performed several scenario analyses. A time horizon of 5 or 10 years (instead of 40 years), the use of a Weibull or Gompertz parametric function for first-line PFS (instead of an exponential function) and the use of different threshold-based PFS to OS surrogacy approaches (instead of a full PFS to OS surrogacy approach) had the largest impacts on the ICER both with and without PAS. The use of lower post-progression treatment costs for the progressed disease health state (i.e. £1000, £425, or £0 per month instead of £2000 per month) increased the ICER.
3.5 Critique of the Cost-Effectiveness Evidence
The cost-effectiveness searches in the CS were well documented, reproducible and were carried out in line with the NICE guide to the methods of technology appraisal. The model structure followed in this appraisal (patient-level simulation with four health states; PFS1, PFS2, progressive disease and death) was different from most of the other oncology economic models in NICE technology appraisals, which have used a partitioned survival approach, extrapolating PFS and OS from clinical trial data [29, 30]. Because of the immaturity of the OS data, the OS in the CS was modelled indirectly and is a sum of the time spent in each of the alive health states. In the base case, it was assumed that any gain in PFS was 100% translated into OS gain. The ERG considered this assumption to be speculative, as there are studies indicating that the duration of PFS gain would translate into an OS gain that is shorter, especially in HER2− patients, which can be also seen in the PALOMA-1 trial [31] (comparing palbociclib plus letrozole vs. letrozole) where a “gain in median OS/gain in median PFS” ratio close to 38.5% was observed. The ERG considered the observed ratio of 38.5% to be more plausible than the completely arbitrary 100% or threshold-based PFS to OS surrogacy scenarios that the company assumed.
In addition to the PFS to OS surrogacy approach, using PFS and OS data from a different study (BOLERO-2) to inform the model on time spent in PFS2 and PD states, without any adjustments, assumes that the BOLERO-2 trial was conducted subsequent to the MONALEESA-2 trial, just after the patients progressed after the first-line therapy. Instead, the ERG would have preferred an approach where the OS and PFS parametric functions used from the BOLERO-2 trial were adjusted based on the patient characteristics at disease progression from the first-line treatment (e.g. age, previous treatment, Eastern Cooperative Oncology Group disease status, time since diagnosis at the time of first-line treatment progression). The use of such adjusted and matched OS and PFS survival functions from BOLERO-2 might have an impact on the cost-effectiveness results.
In the company base case, time-to-event related model inputs for the PFS1 state (i.e. PFS and TTD) were based on the June 2016 cut-off of the MONALEESA-2 trial. The ERG considered that data from the latest cut-off (January 2017) should have been used in the model. The company provided January 2017 data only for PFS that is based on the local assessment. Time to treatment discontinuation and PFS based on a central assessment from the January 2017 cut-off were not provided.
For the extrapolation of the PFS in the first line, the ERG disagreed with the company that the exponential distribution provided the best fit to the external data from other clinical trials. According to the ERG, the Weibull distribution, both for PFS and TTD, would be at least as plausible as the exponential distribution.
For the estimation of drug acquisition costs in the progression health state, the company used expert opinion. However, very little information was provided on the details of what was suggested by the experts to arrive at these costs. Thus, the ERG was not able to assess the validity of this cost estimate (approximately £2000 per month), and this cost appeared to be high, in comparison to later line drug acquisition costs in other breast cancer technology appraisals.
Other issues identified by the ERG include: not including wastage in treatment costs (because dispensed packages cannot be used for other patients once a patient discontinues treatment), unnecessary assumptions made while fitting a Weibull distribution for the modelling of the post-treatment discontinuation survival after chemotherapy, and differential proportions of treatments received in the second line after ribociclib plus letrozole and letrozole.
3.6 Additional Exploratory Analyses Conducted by the Evidence Review Group
Table 1 shows the adjustments made by the ERG to the company base case. The ERG preferred base case resulted in incremental QALYs of 0.53 and ICERs above accepted thresholds, even with the PAS price discount for ribociclib. The most influential adjustments/corrections made by the ERG were: (1) changing the full PFS to OS surrogacy approach to a partial PFS to OS surrogacy approach; (2) using model inputs derived from the most recent PFS dataset of the MONALEESA-2 trial (data cut-off January 2017) and; (3) using an inflation-adjusted third-line treatment-related cost estimate from a published NICE appraisal (TA239, fulvestrant) [32].
From the probabilistic sensitivity analysis results, the probability that ribociclib plus letrozole therapy is cost effective compared to letrozole monotherapy is considerably lower than 50% at a £30,000/QALY threshold (with the PAS price). The ERG conducted some additional scenario analyses on their preferred base case to assess structural uncertainty.
In one of the scenarios, the ERG explored the impact of using a Weibull distribution instead of an exponential distribution to generate the time to event for PFS in the first line. The ERG considered the Weibull distribution to be at least as plausible as the exponential distribution. Selecting the Weibull distribution instead of the exponential distribution increased the ICER substantially. Scenarios with a more modest impact on the ICER included different trial data-based letrozole acquisition costs from cycle 11 onwards, and assuming different second-line treatment proportions.
3.7 Conclusions of the Evidence Review Group Report
The ERG considered the searches conducted in the CS to be satisfactory and MONALEESA-2 [16] to be a good-quality randomised trial. However, the ERG would have preferred data from the latest cut-off and centrally assessed PFS instead of locally assessed PFS.
The economic model described in the CS was considered by the ERG to meet the NICE reference case to a reasonable extent and was mostly in line with the decision problem specified in the scope. After correcting the errors and implementing the changes described in the previous section, the ERG base-case analysis yielded an ICER that is above accepted NICE thresholds. In addition, owing to the high uncertainty surrounding the ICER because of several structural assumptions in the model (e.g. regarding PFS/OS surrogacy, the choice of the parametric distribution to extrapolate first-line PFS/TTD and third-line treatment acquisition costs), the ERG deemed that the uncertainty around the cost-effectiveness estimates of ribociclib was substantial.
3.7.1 Key Methodological Issues
The CS model type was different from most of the models submitted in previous related breast cancer appraisals, and a discrete-event simulation approach was followed. The ERG considered the modelling approach plausible; however, it found some of the assumptions (e.g. full PFS to OS surrogacy assumption, using unadjusted/unmatched data from the BOLERO-2 trial upon progression from the first-line treatment and selection of exponential distribution for modelling first-line PFS and TTD) as debateable. Furthermore, the ERG was doubtful on the choice of some of the input parameters for the cost-effectiveness model, such as the treatment costs in the progressed disease state (corresponding to third and further lines of therapy).
In the original CS, a detailed explanation of the underlying Visual Basic codes and functions operationalising the discrete-event simulation in the cost-effectiveness analysis calculations was not provided. Understanding and verification of the logic of the discrete-event simulation were possible only after the details and an explanation of the non-transparent codes were provided, upon the request of the ERG, in the company’s response to the clarification letter. Because of this delay of provision of the coding details, the ERG could not conduct a full-scale technical verification, but rather some black-box testing to check the plausibility of the results.
The recent appraisal of palbociclib [17], another cyclin-dependent kinase 4 and 6 inhibitor licenced for the same indication (first-line treatment of HR+/HER2− breast cancer), provided an opportunity to compare the evidence between two appraisals. In both of them, the comparator was the same, letrozole monotherapy; therefore, it was expected that the cost-effectiveness results, specifically pertaining to the control arm in both appraisals would be similar. However, the results differed substantially between TA495 (palbociclib) and TA496 (ribociclib). The ERG noticed the differences in modelling approaches (discrete-event simulation for TA496 vs. partitioned survival modelling for TA495) and some of the non-confidential input choices (e.g. PFS/OS parametric modelling based on MONALEESA-2 and BOLERO-2 for TA496 [4] vs. PALOMA-2 and PALOMA-1 for TA495 [17], and different utility inputs). However, the exact underlying reason of the gap between the results of these two appraisals could not be exactly pinpointed, as there was no access to the full details of the palbociclib submission.
4 National Institute for Health and Care Excellence Guidance
4.1 Company Changes to the Model and the First Appraisal Committee Meeting
The company, before the first ACM, updated its base case according to the ERG critique. In the new company base case, an enhanced PAS discount was proposed, and change #1, #2, #3 and #5 from Table 1 were implemented in the new company base case.
The company did not incorporate the fourth (considering the treatment costs in the progressed disease state) and the sixth (partial PFS to OS surrogacy reflecting OS/PFS gain of PALOMA-1 trial [31]) change of the ERG preferred base case in Table 1. Instead, the impact of these changes in the updated company base case was explored in the scenario analyses in the presence of the enhanced PAS discount. In the updated company base case and de-novo scenario analyses, incremental QALYs gained for ribociclib were between 0.53 and 0.89. The ICER values in the base-case and the scenario analyses with the new enhanced PAS were around the acceptable thresholds. The probabilistic sensitivity analysis results were in line with the deterministic base-case results.
The committee considered the company’s decision problem and noted that it was in line with the NICE scope. Additionally, the committee concluded that the MONALEESA-2 trial [16] formed suitable evidence on the clinical efficacy of ribociclib, in terms of PFS. However, the OS benefit of ribociclib was deemed uncertain, owing to the immaturity of the data. The increased incidence of AEs associated with ribociclib was acknowledged by the committee but it was deemed as acceptable.
The committee deferred the decision in this meeting because (1) the approach the company followed in the economic model was different from most of the other models used in oncology technology appraisals; (2) the gap between the cost-effectiveness results from the model used in this submission and the model used in the palbociclib appraisal was substantial; and (3) some of the ribociclib model results were deemed to be counterintuitive (e.g. a decrease in survival gain resulted in ribociclib becoming more cost effective).
Therefore, the Decision Support Unit (DSU) was asked to support the committee to assess the validity of the modelling and the PFS to OS surrogacy approach, to compare the evidence between the ribociclib and palbociclib appraisals and pinpoint the main source of uncertainty, and to appraise the quality of the evidence to support these assumptions.
4.2 Decision Support Unit Analyses and the Second Appraisal Committee Meeting
In the second ACM, the main results of the DSU analysis were discussed [33]. The DSU conducted a literature review on the PFS to OS surrogacy approach and conducted some additional validation efforts on the CS model and provided a detailed comparison between the evidence from both ribociclib and palbociclib appraisals. It agreed that the model structure of the company was different from most of the other appraisals in oncology, but reasonably acceptable for decision making. Additionally, the DSU added that the BOLERO-2 trial [21] data, which were used in modelling of the patients receiving second-line treatment, seemed to be representative of patients progressing after the first-line treatment in MONALEESA-2.
It was unclear to the DSU if a full PFS to OS surrogacy is a valid approach. While the findings from the literature suggested that a PFS gain would likely result in an OS gain, there is no clear relationship. The DSU noted that the update of the PALOMA-1 study indicates a ratio smaller than 38%, but provided results for both full and partial PFS to OS surrogacy approaches suggested by the ERG.
The DSU conducted several black-box tests and compared the CS ribociclib model with the CS palbociclib appraisal. It was suggested that the CS model had no additional programming errors other than those the ERG had identified, and that the difference between the cost-effectiveness results of these two models was because of differences in modelling assumptions and input choices and not the result of programming errors.
Further, the DSU scrutinised the impact of the third and later line treatment costs on the ICER under full and partial PFS to OS surrogacy assumptions. The DSU confirmed that the counterintuitive behaviour observed in some parameter realisations (e.g. ribociclib becomes more cost effective when there is less survival gain) is the result of the costs and effects assigned in the progressed state and how cost effective third and later line treatment is in comparison to first-line treatment.
Furthermore, the DSU noted the wide gap between modelled PFS and TTD, and added that this wide gap was not in line with the reported treatment discontinuation proportions in the trial. According to the DSU, the Weibull distribution would provide a more plausible fit for PFS and TTD, as it would lead to a smaller difference between them. Using Weibull distribution for PFS and/or TTD would increase the ICER.
Additionally, the DSU added that the utility input used for the PFS1 state from the MONALEESA-2 trial, which was based on EQ-5D-5L, should be mapped into EQ-5D-3L values, based on the NICE position statement published a few weeks before the second ACM [34]. Additionally, an alternative utility input for the PFS2 state was suggested, which was derived from Mitra et al. [35]. This value was based on EQ-5D scores and was in line with the NICE reference case, unlike the original CS values that were based on a vignette valuation. Using these utilities (NICE reference case) would increase the ICER.
The DSU concluded that the treatment cost in third and later lines has a big impact on the ICER, especially in the partial PFS to OS surrogacy approach. It deemed that both estimates from the company (£2000) and the ERG (£1140) probably overestimated the actual treatment costs but considered the ERG’s estimate to be more plausible. In line with the ERG’s analyses, the ICER increased with lower third/later line treatment costs in the partial PFS to OS surrogacy approach. Furthermore, the DSU emphasised that the company’s assumptions on dose reductions have a great impact on the ICER, and if all patients received a full licenced dose, the ICER would increase substantially.
The committee accepted that the model is appropriate for decision making and acknowledged the uncertainties and the differences in terms of ICERs between different scenarios presented by the ERG and by the DSU. However, it was decided that the majority of the plausible scenarios resulted in ICER figures that were higher than the range normally considered to be a cost-effective use of NHS resources.
4.3 Final Guidance
The company revised its base case (post first ACD) according to the findings of the DSU analysis. The company’s revisions in the new base case are summarised in Table 2.
The resulting ICER of the revised base case was below the threshold of £30,000/QALY. The company provided some exploratory scenario analyses to reflect the impact of some of the conflicting input choices (e.g. utility and third and later line treatment costs) and assumptions (e.g. PFS to OS surrogacy and PFS/TTD extrapolation) on the ICER. The ERG verified the results of the revised base case of the company.
The company considered some of the alternative inputs suggested by the DSU as implausible. First, the company suggested that a ribociclib dose reduction was based on the trial and would be observed in real clinical practice. Second, the use of the new utility inputs from Mitra et al. [35] was considered to be inconsistent with the previous breast cancer appraisals, where the utility values from Lloyd et al. [23] were used. In addition, the company maintained its position on the PFS/TTD extrapolation in the first line, and argued that the gap between the TTD and PFS was observed from the Kaplan–Meier curves from the MONALEESA-2 trial, as well. Finally the company disagreed with the third and later line treatment cost estimate of the ERG (£1140) and DSU (lower than £1140) because the source for the cost estimate (i.e. TA239, appraisal of fulvestrant in breast cancer) dated back to 2009 and the treatment costs would be higher owing to the newer treatments available in the market.
The committee concluded that a degree of partial PFS to OS surrogacy is probably more likely than full surrogacy; however, the magnitude of the relationship was highly uncertain and both the full and partial PFS to OS surrogacy assumption results would be considered in its decision making. The committee initially preferred the Weibull curve for PFS and TTD extrapolation because it reduced the difference between TTD and PFS in the first line to the expected levels. However, the Cancer Drug Fund clinical lead noted that extrapolation using the exponential curve appeared to provide clinically reasonable results for PFS, and that there was some justification in using the Weibull curve to model the TTD. The committee then agreed that there are a number of ways to extrapolate PFS and TTD in the model. It therefore concluded that there was uncertainty about which extrapolations are the most appropriate.
The committee deemed the utility values in the revised company base case as appropriate for decision making, but noted that the 3L mapped utilities from the MONALEESA-2 trial in the first line may undervalue the quality of life of patients in the progression-free health state. The committee also found the company’s approach of using trial-based ribociclib dose reduction data appropriate.
Finally, the committee noted different treatment cost estimates for the third and later line treatment costs from the company, the ERG, the DSU and the Cancer Drug Fund lead. It also noted that in the ongoing appraisal of palbociclib, an average cost of subsequent therapies was calculated as £1200 per month. Therefore, the committee concluded that it would consider costs in the region of £1140–£1200 in its decision making.
Overall, the committee agreed that the cost-effectiveness estimates are subject to high uncertainty given the assumptions about PFS to OS surrogacy, PFS and TTD extrapolations, and the cost of subsequent (i.e. third and later line) treatments. Taking into account the uncertainties in the calculation of the cost-effectiveness estimates, it was persuaded that there were plausible cost-effectiveness estimates broadly in the range that could be considered a cost-effective use of NHS resources. Therefore, ribociclib was recommended as a first-line treatment option for patients with HR+/HER2− breast cancer.
5 Conclusions
This STA illustrated that using data from different data cut-offs and using locally assessed or centrally assessed PFS data can have a substantial impact on both clinical and cost-effectiveness results. The ERG preferred all clinical inputs (i.e. PFS, TTD, death probabilities and AEs) to be based on the latest (the most informative) data cut-off and PFS based on a central assessment, as locally assessed PFS could be biased because the blinding might have been flawed as a result of different incidences of AEs.
The modelling approach followed in this appraisal, discrete-event simulation, was different from most of the other modelling approaches in oncology. This modelling approach, as in all other approaches, has its own advantages and disadvantages. New modelling approaches can be acceptable; however, it is essential that the underlying coding is transparent and the explanation of the programming is provided in the initial submission so that the critique can be completed in a timely manner.
Another interesting aspect of this STA was how the uncertainty surrounding the OS was tackled when the relevant data were lacking. As the OS data from the trial were immature, different (e.g. partial or full) PFS to OS surrogacy approaches were followed in different scenario analyses, which were all taken into account in the decision-making process. It was also observed that under some parameter realisations, the third-line treatment cost estimate became the most impactful parameter, hence characterisation of the uncertainty surrounding that parameter was essential.
This STA further uncovered the complexity of decision making if two simultaneous appraisals provide some conflicting results, which might lead to additional doubts. However, these differences do not mean that the modelling approach/input choices from one of the appraisals are automatically wrong. In the presence of such conflicting cases, it might be important to have a full understanding of the underlying causes of the differences. In these situations, an external member who can access data from both appraisals can prove useful. The committee may therefore (but also for other reasons), on occasion benefit from external points of view and external support (e.g. DSU) in addition to those of the ERG.
Last, this STA showed that the iterative nature of appraisal meetings may provide an opportunity to find a middle ground between the company assumptions and the ERG point of view. The company may provide enhanced PAS discounts to reduce the uncertainty surrounding the cost effectiveness of a drug in this process.
References
National Institute for Health and Care Excellence. Single technology appraisal: user guide for company evidence submission template. https://www.nice.org.uk/process/pmg24/resources/single-technology-appraisal-user-guide-for-company-evidence-submission-template-pdf-72286715419333. Accessed 30 Mar 2017.
Novartis Pharmaceuticals UK Limited. Ribociclib in combination with an aromatase inhibitor for previously untreated advanced or metastatic hormone receptor-positive, HER2-negative breast cancer [ID1026]. Company evidence submission. Single technology appraisal (STA). Frimley: Novartis Pharmaceuticals UK Limited; 2017.
Riemsma R, Büyükkaramikli N, de Groot S, Fayter D, Armstrong N, Wei C-Y, et al. Ribociclib in combination with an aromatase inhibitor for previously untreated advanced or metastatic hormone receptor-positive, HER2-negative breast cancer. https://www.nice.org.uk/guidance/ta496/documents/committee-papers. Accessed 24 Jul 2018.
National Institute for Health and Care Excellence. Ribociclib with an aromatase inhibitor for previously untreated, hormone receptor-positive, HER2-negative, locally advanced or metastatic breast cancer. NICE technology appraisal guidance 496. https://www.nice.org.uk/guidance/ta496. Accessed 25 May 2018.
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86.
Tripathy D, The BMC. Medicine breast cancer collection: an illustration of contemporary research and clinical care. BMC Med. 2015;13:223.
Cancer Research UK. Breast cancer: stages, types and grades: TNM staging. http://www.cancerresearchuk.org/about-cancer/type/breast-cancer/treatment/tnm-breast-cancer-staging. Accessed 6 Apr 2017.
Cancer Research UK. Breast cancer: stages, types and grades: number stages of breast cancer. http://www.cancerresearchuk.org/about-cancer/type/breast-cancer/treatment/number-stages-of-breast-cancer. Accessed 6 Apr 2017.
Cardoso F, Costa A, Senkus E, Aapro M, André F, Barrios CH, et al. 3rd ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 3). Ann Oncol. 2017;28(1):16–33.
Hurvitz SA, Pietras RJ. Rational management of endocrine resistance in breast cancer: a comprehensive review of estrogen receptor biology, treatment options, and future directions. Cancer. 2008;113(9):2385–97.
Howlader N, Altekruse SF, Li CI, Chen VW, Clarke CA, Ries LA, et al. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J Natl Cancer Inst. 2014;106(5):dju055.
Vidula N, Rugo HS. Cyclin-dependent kinase 4/6 inhibitors for the treatment of breast cancer: a review of preclinical and clinical data. Clin Breast Cancer. 2016;16(1):8–17.
National Institute for Health and Care Excellence. NICE pathways: managing advanced breast cancer. Last updated March 2018. http://pathways.nice.org.uk/pathways/advanced-breast-cancer. Accessed 25 May 2018.
European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP). Kisqali: ribociclib. Summary of opinion (initial authorisation). EMA/CHMP/383155/2017. http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion_-_Initial_authorisation/human/004213/WC500229893.pdf. Accessed 25 May 2018.
National Institute for Health and Care Excellence. Ribociclib in combination with an aromatase inhibitor for previously untreated advanced or metastatic hormone receptor-positive, HER2-negative breast cancer: final scope. London: National Institute for Health and Care Excellence; 2017.
Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375(18):1738–48.
National Institute for Health and Care Excellence. Palbociclib with an aromatase inhibitor for previously untreated, hormone receptor-positive, HER2-negative, locally advanced or metastatic breast cancer. NICE technology appraisal guidance 495. https://www.nice.org.uk/guidance/ta495. Accessed 25 May 2018.
Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. 2016;375(20):1925–36.
Martín M, Loibl S, von Minckwitz G, Morales S, Martinez N, Guerrero A, et al. Phase III trial evaluating the addition of bevacizumab to endocrine therapy as first-line treatment for advanced breast cancer: the letrozole/fulvestrant and avastin (LEA) study. J Clin Oncol. 2015;33(9):1045–52.
Dickler MN, Barry WT, Cirrincione CT, Ellis MJ, Moynahan ME, Innocenti F, et al. Phase III trial evaluating letrozole as first-line endocrine therapy with or without bevacizumab for the treatment of postmenopausal women with hormone receptor-positive advanced-stage breast cancer: CALGB 40503 (Alliance). J Clin Oncol. 2016;34(22):2602–9.
Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366(6):520–9.
Li N, Hao Y, Xie J, Lin PL, Koo V, Ohashi E, et al. Everolimus-based therapy versus chemotherapy among patients with HR+/HER2- metastatic breast cancer: comparative effectiveness from a chart review study. Int J Breast Cancer. 2015;2015:240750.
Lloyd A, Nafees B, Narewska J, Dewilde S, Watkins J. Health state utilities for metastatic breast cancer. Br J Cancer. 2006;95(6):683–90.
Peasgood T, Ward SE, Brazier J. Health-state utility values in breast cancer. Expert Rev Pharmacoecon Outcomes Res. 2010;10(5):553–66.
Commercial Medicines Unit (CMU), Department of Health and Social Care. Drugs and pharmaceutical electronic market information tool (eMIT). Last updated 5 January 2018. https://www.gov.uk/government/publications/drugs-and-pharmaceutical-electronic-market-information-emit. Accessed 25 May 2018.
Joint Formulary Committee. British National Formulary. 75. London: BMJ Group and Pharmaceutical Press; 2018.
National Institute for Health and Care Excellence. Advanced breast cancer: diagnosis and treatment. NICE clinical guideline 81. Last updated August 2017. https://www.nice.org.uk/guidance/cg81. Accessed 25 May 2018.
National Institute for Health and Care Excellence. Everolimus in combination with exemestane for treating advanced HER2-negative hormone-receptor-positive breast cancer after endocrine therapy. NICE technology appraisal guidance 421. https://www.nice.org.uk/guidance/ta421. Accessed 30 Mar 2017.
Büyükkaramikli NC, Blommestein HM, Riemsma R, Armstrong N, Clay F, Ross J, et al. Ramucirumab for treating advanced gastric cancer or gastro-oesophageal junction adenocarcinoma previously treated with chemotherapy: an evidence review group perspective of a NICE single technology appraisal. Pharmacoeconomics. 2017;35(12):1211–21.
Büyükkaramikli NC, de Groot S, Fayter D, Wolff R, Armstrong N, Stirk L, et al. Pomalidomide with dexamethasone for treating relapsed and refractory multiple myeloma previously treated with lenalidomide and bortezomib: an evidence review group perspective of an NICE single technology appraisal. Pharmacoeconomics. 2018;36(2):145–59.
Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16(1):25–35.
National Institute for Health and Care Excellence. Fulvestrant for the treatment of locally advanced or metastatic breast cancer. NICE technology appraisal guidance 239. https://www.nice.org.uk/guidance/ta239. Accessed 30 Mar 2017.
Pennington B. Ribociclib in combination with an aromatose inhibitor for previously untreated advanced or metastatic hormone receptor-positive, HER2-negative breast cancer: a review of the model structure, inputs and assumptions: report by the decision support unit. http://www.nicedsu.org.uk/. Accessed 24 Jul 2018.
National Institute for Health and Care Excellence. Position statement on use of the EQ-5D-5L valuation set. https://www.nice.org.uk/Media/Default/About/what-we-do/NICE-guidance/NICE-technology-appraisal-guidance/eq5d5l_nice_position_statement.pdf. Accessed 25 May 2018.
Mitra D, Wood R, de Courcy J, Iyer S. Patient reported health utility in HR+/HER2− advanced/metastatic breast cancer. Value Health. 2016;19(7):A749.
Author information
Authors and Affiliations
Contributions
Nasuh C. Büyükkaramikli acted as a health economist on this assessment, critiqued the manufacturer’s economic evaluation, wrote the manuscript and acted as the overall guarantor for the overall content of this article. Saskia de Groot and Nigel Armstrong acted as health economists on this assessment and critiqued the manufacturer’s economic evaluation. Rob Riemsma acted as a project lead and systematic reviewer on this assessment and critiqued the clinical effectiveness methods and evidence. Debra Fayter and Piet Portegijs acted as systematic reviewers and critiqued the clinical effectiveness methods and evidence. Steven Duffy acted as an information specialist, and critiqued the manufacturer’s clinical and economic literature searches. Jos Kleijnen critiqued the manufacturer’s definition of the decision problem and their description of the underlying health problem and current service provision. Maiwenn J. Al acted as a health economic project lead and critiqued the manufacturer’s economic evaluation. All authors contributed to the conception and planning of the work, and critically revised and approved the final submitted version of the manuscript. This summary has not been externally reviewed by PharmacoEconomics.
Corresponding author
Ethics declarations
Funding
This project was funded by the National Institute for Health Research Health Technology Assessment (HTA) Program (project number 16/112/03 STA) [see the HTA program website for further information, http://www.hta.ac.uk]. This summary of the Evidence Review Group’s report was compiled after the Appraisal Committee’s review. The views and opinions expressed are the authors’ and do not necessarily reflect those of the HTA Programme, National Institute for Health and Care Excellence, National Institute for Health Research, National Health Service or the Department of Health.
Conflict of interest
Nasuh C. Büyükkaramikli, Saskia de Groot, Rob Riemsma, Debra Fayter, Nigel Armstrong, Piet Portegijs, Steven Duffy, Jos Kleijnen and Maiwenn J. Al have no conflicts of interest that are directly relevant to the contents of this review.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Büyükkaramikli, N.C., de Groot, S., Riemsma, R. et al. Ribociclib with an Aromatase Inhibitor for Previously Untreated, HR-Positive, HER2-Negative, Locally Advanced or Metastatic Breast Cancer: An Evidence Review Group Perspective of a NICE Single Technology Appraisal. PharmacoEconomics 37, 141–153 (2019). https://doi.org/10.1007/s40273-018-0708-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40273-018-0708-4