
Abstract
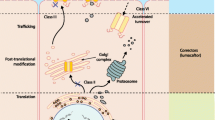
Cystic fibrosis (CF), a severe genetic disease, is caused by mutations that alter the structure and function of CFTR, a plasma membrane channel permeable to chloride and bicarbonate. Defective anion transport in CF irreversibly damages the lungs, pancreas, liver, and other organs. CF mutations cause loss of CFTR function in multiple ways. In particular, class 3 mutations such as p.Gly551Asp strongly decrease the time spent by CFTR in the open state (gating defect). Instead, class 2 mutations impair the maturation of CFTR protein and its transport from the endoplasmic reticulum to the plasma membrane (trafficking defect). The deletion of phenylalanine 508 (p.Phe508del), the most frequent mutation among CF patients (70–90 %), destabilizes the CFTR protein, thus causing both a trafficking and a gating defect. These two defects can be overcome with drug-like molecules generically called correctors and potentiators, respectively. The potentiator Kalydeco™ (also known as Ivacaftor or VX-770), developed by Vertex Pharmaceuticals, has been recently approved by the US FDA and the European Medicines Agency (EMA) for the treatment of CF patients carrying at least one CFTR allele with the p.Gly551Asp mutation (2–5 % of all patients). In contrast, the corrector VX-809, which significantly improves p.Phe508del-CFTR trafficking in vitro, is still under study in clinical trials. Because of multiple defects caused by the p.Phe508del mutation, it is probable that rescue of the mutant protein will require combined treatment with correctors having different mechanisms of action. This review evaluates the status of experimental and clinical research in pharmacotherapy for the CF basic defect.


Similar content being viewed by others


References
Rommens JM, Iannuzzi MC, Kerem B, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245(4922):1059–65.
Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–73.
Kerem B, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245(4922):1073–80.
Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem. 2008;77:701–26.
Kim SJ, Skach WR. Mechanisms of CFTR folding at the endoplasmic reticulum. Front Pharmacol. 2012;3:201.
Poulsen JH, Fischer H, Illek B, et al. Bicarbonate conductance and pH regulatory capability of cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci USA. 1994;91(12):5340–4.
Tabcharani JA, Linsdell P, Hanrahan JW. Halide permeation in wild-type and mutant cystic fibrosis transmembrane conductance regulator chloride channels. J Gen Physiol. 1997;110(4):341–54.
Matsui H, Grubb BR, Tarran R, et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95(7):1005–15.
Donaldson SH, Boucher RC. Sodium channels and cystic fibrosis. Chest. 2007;132(5):1631–6.
Garcia-Caballero A, Rasmussen JE, Gaillard E, et al. SPLUNC1 regulates airway surface liquid volume by protecting ENaC from proteolytic cleavage. Proc Natl Acad Sci USA. 2009;106(27):11412–7.
Garcia MA, Yang N, Quinton PM. Normal mouse intestinal mucus release requires cystic fibrosis transmembrane regulator-dependent bicarbonate secretion. J Clin Invest. 2009;119(9):2613–22.
Gustafsson JK, Ermund A, Ambort D, et al. Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. J Exp Med. 2012;209(7):1263–72.
Pezzulo AA, Tang XX, Hoegger MJ, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487(7405):109–13.
Simmonds NJ, Macneill SJ, Cullinan P, et al. Cystic fibrosis and survival to 40 years: a case-control study. Eur Respir J. 2010;36(6):1277–83.
Hodson ME, Simmonds NJ, Warwick WJ, et al. An international/multicentre report on patients with cystic fibrosis (CF) over the age of 40 years. J Cyst Fibros. 2008;7(6):537–42.
George PM, Banya W, Pareek N, et al. Improved survival at low lung function in cystic fibrosis: cohort study from 1990 to 2007. BMJ. 2011;28(342):d1008.
Atkinson TJ. Cystic fibrosis, vector-mediated gene therapy, and relevance of toll-like receptors: a review of problems, progress, and possibilities. Curr Gene Ther. 2008;8(3):201–7.
Oakland M, Sinn PL, McCray PB Jr. Advances in cell and gene-based therapies for cystic fibrosis lung disease. Mol Ther. 2012;20(6):1108–15.
Illek B, Zhang L, Lewis NC, et al. Defective function of the cystic fibrosis-causing missense mutation G551D is recovered by genistein. Am J Physiol. 1999;277(4 Pt 1):C833–9.
Denning GM, Anderson MP, Amara JF, et al. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358(6389):761–4.
Serohijos AW, Hegedus T, Aleksandrov AA, et al. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci USA. 2008;105(9):3256–61.
Vergani P, Lockless SW, Nairn AC, et al. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature. 2005;433(7028):876–80.
Hwang TC, Sheppard DN. Gating of the CFTR Cl- channel by ATP-driven nucleotide-binding domain dimerisation. J Physiol. 2009;587(Pt 10):2151–61.
Younger JM, Chen L, Ren HY, et al. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126(3):571–82.
Farinha CM, Amaral MD. Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol Cell Biol. 2005;25(12):5242–52.
Penque D, Mendes F, Beck S, et al. Cystic fibrosis F508del patients have apically localized CFTR in a reduced number of airway cells. Lab Invest. 2000;80(6):857–68.
Okiyoneda T, Barrière H, Bagdány M, et al. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science. 2010;329(5993):805–10.
Lukacs GL, Chang XB, Bear C, et al. The deltaF508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem. 1993;268(29):21592–8.
Sharma M, Pampinella F, Nemes C, et al. Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J Cell Biol. 2004;164(6):923–33.
Dalemans W, Barbry P, Champigny G, et al. Altered chloride ion channel kinetics associated with the deltaF508 cystic fibrosis mutation. Nature. 1991;354(6354):526–8.
Drumm ML, Wilkinson DJ, Smit LS, et al. Chloride conductance expressed by deltaF508 and other mutant CFTRs in Xenopus oocytes. Science. 1991;254(5039):1797–9.
Haws CM, Nepomuceno IB, Krouse ME, et al. DeltaF508-CFTR channels: kinetics, activation by forskolin, and potentiation by xanthines. Am J Physiol. 1996;270(5 Pt 1):C1544–55.
Wang F, Zeltwanger S, Hu S, et al. Deletion of phenylalanine 508 causes attenuated phosphorylation-dependent activation of CFTR chloride channels. J Physiol. 2000;1(524 Pt 3):637–48.
Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73(7):1251–4.
Zielenski J, Tsui LC. Cystic fibrosis: genotypic and phenotypic variations. Annu Rev Genet. 1995;29:777–807.
Haardt M, Benharouga M, Lechardeur D, et al. C-terminal truncations destabilize the cystic fibrosis transmembrane conductance regulator without impairing its biogenesis. A novel class of mutation. J Biol Chem. 1999;274(31):21873–7.
Hwang TC, Wang F, Yang IC, et al. Genistein potentiates wild-type and deltaF508-CFTR channel activity. Am J Physiol. 1997;273(3 Pt 1):C988–98.
Verkman AS, Galietta LJ. Chloride channels as drug targets. Nat Rev Drug Discov. 2009;8(2):153–71.
Zegarra-Moran O, Romio L, Folli C, et al. Correction of G551D-CFTR transport defect in epithelial monolayers by genistein but not by CPX or MPB-07. Br J Pharmacol. 2002;137(4):504–12.
Pedemonte N, Sonawane ND, Taddei A, et al. Phenylglycine and sulfonamide correctors of defective delta F508 and G551D cystic fibrosis transmembrane conductance regulator chloride-channel gating. Mol Pharmacol. 2005;67(5):1797–807.
Dérand R, Bulteau-Pignoux L, Mettey Y, et al. Activation of G551D CFTR channel with MPB-91: regulation by ATPase activity and phosphorylation. Am J Physiol. 2001;281(5):C1657–66.
Yang H, Shelat AA, Guy RK, et al. Nanomolar affinity small molecule correctors of defective Delta F508-CFTR chloride channel gating. J Biol Chem. 2003;278(37):35079–85.
Pedemonte N, Diena T, Caci E, et al. Antihypertensive 1,4-dihydropyridines as correctors of the cystic fibrosis transmembrane conductance regulator channel gating defect caused by cystic fibrosis mutations. Mol Pharmacol. 2005;68(6):1736–46.
Pedemonte N, Lukacs GL, Du K, et al. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest. 2005;115(9):2564–71.
Norez C, Noel S, Wilke M, et al. Rescue of functional deltaF508-CFTR channels in cystic fibrosis epithelial cells by the alpha-glucosidase inhibitor miglustat. FEBS Lett. 2006;580(8):2081–6.
Carlile GW, Robert R, Zhang D, et al. Correctors of protein trafficking defects identified by a novel high-throughput screening assay. Chembiochem. 2007;8(9):1012–20.
Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA. 2009;106(44):18825–30.
Hutt DM, Herman D, Rodrigues AP, et al. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat Chem Biol. 2010;6(1):25–33.
Van Goor F, Hadida S, Grootenhuis PD, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci USA. 2011;108(46):18843–8.
Carlile GW, Keyzers RA, Teske KA, et al. Correction of F508del-CFTR trafficking by the sponge alkaloid latonduine is modulated by interaction with PARP. Chem Biol. 2012;19(10):1288–99.
Caputo A, Hinzpeter A, Caci E, et al. Mutation-specific potency and efficacy of cystic fibrosis transmembrane conductance regulator chloride channel potentiators. J Pharmacol Exp Ther. 2009;330(3):783–91.
Yu H, Burton B, Huang CJ, et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J Cyst Fibros. 2012;11(3):237–45.
Pedemonte N, Tomati V, Sondo E, et al. Influence of cell background on pharmacological rescue of mutant CFTR. Am J Physiol Cell Physiol. 2010;298(4):C866–74.
Randell SH, Fulcher ML, O’Neal W, et al. Primary epithelial cell models for cystic fibrosis research. Methods Mol Biol. 2011;742:285–310.
Fulcher ML, Randell SH. Human nasal and tracheo-bronchial respiratory epithelial cell culture. Methods Mol Biol. 2013;945:109–21.
Mendoza JL, Schmidt A, Li Q, et al. Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell. 2012;148(1–2):164–74.
Rabeh WM, Bossard F, Xu H, et al. Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell. 2012;148(1–2):150–63.
Roxo-Rosa M, Xu Z, Schmidt A, et al. Revertant mutants G550E and 4RK rescue cystic fibrosis mutants in the first nucleotide-binding domain of CFTR by different mechanisms. Proc Natl Acad Sci USA. 2006;103(47):17891–6.
DeCarvalho AC, Gansheroff LJ, Teem JL. Mutations in the nucleotide binding domain 1 signature motif region rescue processing and functional defects of cystic fibrosis transmembrane conductance regulator ∆F508. J Biol Chem. 2002;277(39):35896–905.
Thibodeau PH, Richardson JM 3rd, Wang W, et al. The cystic fibrosis-causing mutation deltaF508 affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. J Biol Chem. 2010;285(46):35825–35.
Grove DE, Rosser MF, Ren HY, et al. Mechanisms for rescue of correctable folding defects in CFTRDelta F508. Mol Biol Cell. 2009;20(18):4059–69.
Accurso FJ, Rowe SM, Clancy JP, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363(21):1991–2003.
De Boeck K, Derichs N, Fajac I, et al. New clinical diagnostic procedures for cystic fibrosis in Europe. J Cyst Fibros. 2011;10(Suppl 2):S53–66.
Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–72.
Flume PA, Liou TG, Borowitz DS, et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest. 2012;142(3):718–24.
Clancy JP, Rowe SM, Accurso FJ, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax. 2012;67(1):12–8.
Wilschanski M, Yahav Y, Yaacov Y, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349(15):1433–41.
Welch EM, Barton ER, Zhuo J, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447(7140):87–91.
Sermet-Gaudelus I, Boeck KD, Casimir GJ, et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med. 2010;182(10):1262–72.
Wilschanski M, Miller LL, Shoseyov D, et al. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur Respir J. 2011;38(1):59–69.
Linde L, Boelz S, Nissim-Rafinia M, et al. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J Clin Invest. 2007;117(3):683–92.
Rowe SM, Sloane P, Tang LP, et al. Suppression of CFTR premature termination codons and rescue of CFTR protein and function by the synthetic aminoglycoside NB54. J Mol Med (Berl). 2011;89(11):1149–61.
Hirsh AJ, Sabater JR, Zamurs A, et al. Evaluation of second generation amiloride analogs as therapy for cystic fibrosis lung disease. J Pharmacol Exp Ther. 2004;311(3):929–38.
Hirsh AJ, Zhang J, Zamurs A, et al. Pharmacological properties of N-(3,5-diamino-6-chloropyrazine-2-carbonyl)-N′-4-[4-(2,3-dihydroxypropoxy)phenyl]butyl-guanidine methanesulfonate (552-02), a novel epithelial sodium channel blocker with potential clinical efficacy for cystic fibrosis lung disease. J Pharmacol Exp Ther. 2008;325(1):77–88.
Rowe SM, Reeves G, Hathorne H, et al. Reduced sodium transport with nasal administration of the prostasin inhibitor camostat in cystic fibrosis subjects. Chest. 2013;. doi:10.1378/chest.12-2431.
Ratjen F, Durham T, Navratil T, et al. Long term effects of denufosol tetrasodium in patients with cystic fibrosis. J Cyst Fibros. 2012;11(6):539–49.
Moss RB. Pitfalls of drug development: lessons learned from trials of denufosol in cystic fibrosis. J Pediatr. 2013;162(4):676–80.
Yang YD, Cho H, Koo JY, et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455(7217):1210–5.
Schroeder BC, Cheng T, Jan YN, et al. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134(6):1019–29.
Caputo A, Caci E, Ferrera L, et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322(5901):590–4.
Scudieri P, Caci E, Bruno S, et al. Association of TMEM16A chloride channel overexpression with airway goblet cell metaplasia. J Physiol. 2012;590(Pt 23):6141–55.
Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem. 2011;286(3):2365–74.
Davies JC, Wainwright CE, Canny GJ, et al. Efficacy and safety of Ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013 (Epub ahead of print).
Boyle MP, Bell S, Konstan M, et al. The investigational CFTR corrector, VX-809 (Lumacaftor) co-administered with the oral potentiator Ivacaftor improved CFTR and lung function in F508del homozygous patients: phase II study results. Pediatr Pulmonol. 2012;47(S35):315.
Acknowledgments
No sources of funding were used to assist in the preparation of this review. Luis Galietta has no conflicts of interest that are directly relevant to the content of this report. Thanks to Dr. Anna Capurro for critically reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Galietta, L.J.V. Managing the Underlying Cause of Cystic Fibrosis: A Future Role for Potentiators and Correctors. Pediatr Drugs 15, 393–402 (2013). https://doi.org/10.1007/s40272-013-0035-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40272-013-0035-3