
Abstract
Pirfenidone (Esbriet®) is available as capsules containing 267 mg of pirfenidone and, more recently, as bioequivalent tablets containing 267, 534 or 801 mg of pirfenidone. Both formulations are indicated to treat idiopathic pulmonary fibrosis (IPF), with pirfenidone being shown to generally reduce the rate of decline in forced vital capacity in patients with mild to moderate IPF, while prolonging progression-free survival and reducing the risk of IPF-related and all-cause mortality. The availability of the tablet formulation reduces the daily pill burden of pirfenidone, as the recommended daily divided maintenance dose of 2403 mg/day may be administered as one 801 mg tablet three times daily instead of three 267 mg capsules three times daily. Pirfenidone is associated with gastrointestinal and skin-related events, with such events generally being manageable.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Adis evaluation of oral pirfenidone tablets in idiopathic pulmonary fibrosis (IPF)
Tablet formulation reduces the pill burden of maintenance treatment relative to the capsule formulation (one 801 mg tablet 3× daily vs three 267 mg capsules 3× daily to provide maintenance dose of 2403 mg/day) |
Reduces the rate of decline in forced vital capacity in patients with mild to moderate IPF |
Prolongs progression-free survival and reduces the risk of IPF-related and all-cause mortality |
Manageable tolerability profile, with most common adverse events being gastrointestinal and skin-related disorders |
What is the rationale for developing the tablet formulation of pirfenidone?
Idiopathic pulmonary fibrosis (IPF), a highly heterogeneous chronic, fibrosing type of interstitial pneumonia, is characterized by a progressive loss of lung function, potentially leading to acute respiratory decline and death [1,2,3]. Pirfenidone (Esbriet®), a synthetic pyridone compound, inhibits the progression of fibrosis and loss of lung function in patients with IPF via its antifibrotic, anti-inflammatory and antioxidant properties (mechanism of action is not fully established) [4,5,6]. In animal models of pulmonary fibrosis, pirfenidone reduced production of profibrotic and proinflammatory cytokines, such as transforming growth factor-β1 and lung basic fibroblast growth factor, and suppressed both decreases in interferon-γ and elevations in interleukin (IL)-1β, IL-6, IL-12p40, tumour necrosis factor-α and monocyte chemoattractant protein-1 [7]. The antifibrotic properties of pirfenidone appear to involve inhibition of collagen fibril formation, as shown by a recent in vitro study in primary human fibroblasts from patients with IPF and healthy donors [8].
Pirfenidone is available as capsules containing 267 mg of pirfenidone and, more recently, as tablets containing 267, 534 or 801 mg of pirfenidone [4, 5]. The availability of the tablet formulation reduces the daily pill burden of pirfenidone, as the recommended daily divided maintenance dose of 2403 mg/day may be administered as one 801 mg tablet three times daily instead of three 267 mg capsules three times daily. The reduction in pill burden has the potential to improve adherence to pirfenidone therapy [9].
Is the tablet formulation bioequivalent to the capsule formulation?
A single tablet containing 801 mg of pirfenidone is bioequivalent to three capsules containing 267 mg (i.e. a total of 801 mg) of pirfenidone [9]. In a randomized, four-treatment period, four-sequence cross-over study, 44 healthy volunteers received 801 mg of pirfenidone as an 801 mg tablet and as three 267 mg capsules in both the fasted and fed (after a high-fat meal) state [9]. Between-formulation differences in exposure to pirfenidone as measured by the peak plasma concentration (Cmax), area under the plasma concentration versus time curve (AUC) from time 0 to the time of the last quantifiable concentration (AUC0–t) and AUC from time 0 to infinity (AUC0–∞) were used to determine pharmacokinetic bioequivalence; bioequivalence was established if the 90% confidence interval (CI) for the geometric least-squares mean (GLSM) ratio between the two formulations was within the limit of 80.00–125.00% (standard bioequivalence criteria) [9].
Under fasted conditions, the tablet formulation met all three bioequivalence criteria for pirfenidone exposure, with GLSM ratios of 101.26% (90% CI 94.41–108.60) for Cmax, 99.63% (90% CI 96.66–102.69) for AUC0–t and 99.61% (90% CI 96.64–102.68) for AUC0–∞ [9]. Under fed conditions, the tablet formulation met the bioequivalence criteria for AUC0–t (103.06%; 90% CI 99.55–106.69) and AUC0–∞ (103.05%; 90% CI 99.54–106.69), but slightly exceeded the upper bound of the 90% confidence interval for the Cmax criterion (116.61%; 90% CI 108.26–125.60) by 0.6%. The median time to Cmax (tmax) was shorter with the tablet formulation than with the capsule formulation [2.05 (range 0.50–6.00) vs 3.00 (range 1.0–6.00) h] [9].
Food reduced exposure to both pirfenidone formulations [9]. As the high-fat diet consumed by volunteers in the ‘fed’ part of the study represents extreme dietary conditions, the differences in Cmax between the two formulations were not expected to be clinically meaningful in the real-world setting where patients with IPF would likely have a more moderate diet [9]. Importantly, both formulations of pirfenidone should be taken with food to improve tolerability (Table 1). Treatment-emergent adverse events (TEAEs) were more commonly reported when pirfenidone was taken in the fasting state (36.4 and 31.8% of subjects when taking the tablet and capsule formulations, respectively) than in the fed state (15.9 and 2.3%) [9]. A relationship between drug exposure and the occurrence of TEAEs was not apparent in this study [9].
For whom are pirfenidone tablets indicated?
Pirfenidone tablets are indicated for the treatment of adults with mild to moderate IPF in the EU [4] and IPF in the USA [5]. Table 1 provides a summary of the EU prescribing information for pirfenidone tablets [4]. Local prescribing information should be consulted for further details.
What potential drug interactions may occur with pirfenidone?
Pirfenidone is predominantly (70–80%) metabolized by cytochrome P450 (CYP) 1A2 (with minor contributions from CYP2C9, CYP2C19, CYP2D6 and CYP2E1) [4]. As a result, clinically significant drug interactions may potentially occur between pirfenidone and CYP1A2 inhibitors/inducers, and the following precautions should be taken.
Interactions between pirfenidone and CYP1A2 inhibitors [4]
-
Do not use fluvoxamine concomitantly (contraindicated as exposure to pirfenidone is increased)
-
Avoid the concomitant use of strong and selective CYP1A2 inhibitors (e.g. enoxacin)
-
Use moderate CYP1A2 inhibitors (e.g. ciprofloxacin, amiodarone, propafenone) with caution (a decrease in pirfenidone dosage may be required)
-
Avoid the concomitant use of agents that inhibit both CYP1A2 and ≥ 1 other CYP enzyme involved in the metabolism of pirfenidone
-
Take special care when CYP1A2 inhibitors are used in combination with potent inhibitors of ≥ 1 other CYP enzymes involved in pirfenidone metabolism, including CYP2C9 (e.g. fluconazole), CYP2C19 (e.g. chloramphenicol) and CYP2D6 (e.g. fluoxetine)
-
Avoid consumption of grapefruit juice.
Interactions between pirfenidone and CYP1A2 inducers [4]
-
Avoid the concomitant use of strong CYP1A2 inducers, including smoking (exposure to pirfenidone is decreased)
-
Use of moderate CYP1A2 inducers (e.g. omeprazole) may decrease plasma concentrations of pirfenidone
-
Avoid agents (e.g. rifampicin) that potently induce both CYP1A2 and other CYP enzymes involved in the metabolism of pirfenidone.
What is the clinical efficacy of pirfenidone?
Clinical trials
As the tablet formulation of pirfenidone is bioequivalent to the capsule formulation [9], clinical trials of the tablet formulation were not necessary for regulatory approval. The following efficacy and tolerability data refer to studies of the pirfenidone capsules.
Pirfenidone at the target dosage of 2403 mg/day generally provided beneficial effects on predicted forced vital capacity (FVC) in randomized, placebo-controlled phase 3 trials in adults with mild to moderate IPF [10,11,12,13]. The 52-week ASCEND trial [10] and 72-week CAPACITY 004 and 006 trials [11] included patients aged 40–80 years with a predicted FVC of 50–90% [10] or ≥ 50 [11], a predicted carbon monoxide diffusing capacity (DLCO) of 30–90% [10] or ≥ 35 [11], a 6-min walk distance (6MWD) of ≥ 150 m [10, 11], and either a predicted FVC or DLCO of ≤ 90% [11]. The dosage of pirfenidone was titrated over 2 weeks to 2403 mg/day in three equally divided daily doses.
In ASCEND [10], pirfenidone was significantly (p < 0.001) favoured over placebo with regard to the percentage decline in predicted FVC over 52 weeks (primary endpoint), the proportion of patients with the composite endpoint of ≥ 10% decline in predicted FVC or death (16.5 vs 31.8%; relative change −47.9%) and the proportion of patients with no decline in percentage predicted FVC (22.7 vs 9.7%; relative change +132.5%) [10].
Pirfenidone significantly (p < 0.01) reduced the mean percentage decline in predicted FVC over 72 weeks relative to placebo (primary endpoint) in the pooled CAPACITY 004/006 analysis (−8.5 vs −11.0%) and CAPACITY 004 (−8.0 vs −12.4%), but not in CAPACITY 006 (−9.0 vs −9.6%) [11]. The proportion of patients with a categorical change in predicted FVC of ≥ 10% was also significantly (p < 0.01) smaller with pirfenidone than placebo in the pooled CAPACITY analysis (21 vs 31%) and CAPACITY 004 (20 vs 35%), but not in study 006 (23 vs 27%) [11].
Pirfenidone 2403 mg/day provided treatment benefits with regard to multiple measures of IPF disease status in a prespecified pooled analysis of CAPACITY and ASCEND [14]. The mean decline in FVC from baseline over 1 year was significantly smaller with pirfenidone than placebo [−216 vs −363 mL; p < 0.001; relative difference (RD) 40.7%]. The favourable effects of pirfenidone over placebo with regard to FVC outcomes were seen at each time point from 3 months onwards, and remained generally consistent over 1 year across patient subgroups defined by baseline demographics and disease status. Other treatment benefits for pirfenidone versus placebo over 1 year included a 38% reduction in the risks of death or disease progression, and the proportions of patients who achieved the clinically meaningful composite endpoints of [14]:
-
a ≥ 10% decline in predicted FVC or death [15 vs 27%; p < 0.001; RD 43.8% (95% CI 29.3–55.4)]
-
a ≥ 50 m decline in 6MWD or death [24.8 vs 34.8%; p < 0.001; RD 28.7% (95% CI 15.1–40.2)]
-
a ≥ 20-point increase in University of California at San Diego shortness-of-breath questionnaire score or death [24.0 vs 31.4%; p < 0.05; RD 23.7% (95% CI 8.4–36.4)]
In pooled analysis of mortality in CAPACITY and ASCEND [15], pirfenidone significantly reduced the risk of all-cause mortality [hazard ratio (HR) 0.52 (95% CI 0.31–0.87); p = 0.011], treatment-emergent all-cause mortality [HR 0.45 (95% CI 0.24–0.83); p = 0.009], IPF-related mortality [HR 0.35 (95% CI 0.17–0.72); p = 0.003] and treatment-emergent IPF-related mortality [HR 0.32 (95% CI 0.14–0.76); p = 0.006] at 1 year relative to placebo, with similar results for all-cause mortality in a random-effects meta-analysis [HR 0.50 (95% CI 0.31–0.80); p = 0.004]. The risk of three of these mortality outcomes (treatment-emergent all-cause, IPF-related and treatment-emergent IPF-related mortality) were also reduced at 120 weeks (HR 0.47–0.61; all p < 0.05) [15]. Moreover, in a pooled analysis of hospitalization data in CAPACITY and ASCEND, the risk of non-elective respiratory-related hospitalization over the course of 1 year was significantly lower with pirfenidone than with placebo [7 vs 12% of patients; HR 0.52 (95% CI 0.36–0.77); p = 0.001] [16].
Continued treatment with pirfenidone provided clinical benefits in patients who had clinically meaningful declines in FVC during earlier treatment [17]. In the pooled CAPACITY and ASCEND trials, 34 pirfenidone and 68 placebo recipients had a ≥ 10% absolute decline in FVC between baseline and month 6 [17]. In these patients during the next 6-month treatment period, fewer patients who continued to receive pirfenidone than continued to receive placebo experienced a second ≥ 10% decline in FVC or death (5.9 vs 27.9%; RD −78.9%; p = 0.0009). This suggests that continued treatment with pirfenidone may be beneficial even in those patients who displayed disease progression during early treatment [17].
The beneficial effects of pirfenidone versus placebo were generally maintained for up to 72 weeks of treatment, with pirfenidone significantly reducing the risks of the composite endpoints of a ≥ 10% decline in predicted FVC or death, death or disease progression, ≥ 50 m decline in 6MWD or death, and worsening dyspnoea or death (risk reduced by 52, 38, 34 and 25%, respectively; all p < 0.01) [14].
Patients with IPF who completed one of the CAPACITY or ASCEND trials could enrol in an open-label extension study (RECAP); 79.3% (1058/1334) of eligible patients entered this safety study [18]. The dosage of pirfenidone was titrated over 15 days to 2403 mg/day (or the highest tolerated dose of ≤ 2403 mg/day) in three equally divided daily doses in all patients (regardless of whether they had been previously receiving pirfenidone or placebo in CAPACITY or ASCEND), with a mean daily dosage in RECAP of 2091 mg/day [18]. In patients who were entered RECAP from CAPACITY 004/006, the following FVC-related and survival outcomes at 180 weeks were consistent with those previously observed [18]:
-
Slow decrease in FVC over the 180-week period (annualized rate of FVC decline 144.3 mL);
-
Mean change in percentage predicted FVC of −9.6% from RECAP baseline to 180 weeks;
-
Median duration of on-treatment survival of 77.2 months (6.4 years) from the first dose of pirfenidone 2403 mg/day in RECAP.
Real-world studies
The efficacy of pirfenidone in stabilizing lung function or reducing the decline of predicted FVC in the clinical practice setting was generally consistent with that in clinical trials [19,20,21,22]. For example, in the largest (n > 100) of the multicentre observational studies in patients with IPF, the following results were reported:
-
Study in Germany and Italy (n = 197) [21] Pirfenidone provided significant improvements with regards to the annual decline from baseline in FVC, total lung capacity, DLCO and oxygenation relative to the pretreatment year; patients who were stable remained stable, whereas those with progressive disease generally showed substantial improvements.
-
Study in Italy (n = 128) [22] The rate of annual decline in percentage FVC over 1 year decreased in pirfenidone recipients, with more pronounced effects in patients with moderate to severe disease.
What is the tolerability profile of pirfenidone?
Pirfenidone has a manageable tolerability profile in patients with IPF [10, 11, 14, 18, 20,21,22,23,24]. This section focuses on the rates of TEAEs in the pooled CAPACITY and ASCEND phase 3 trials, in which the median duration of exposure was 1.0 years (range > 0 to 2.3 years) in both the pooled pirfenidone (n = 623) and placebo (n = 624) groups [14], and in an integrated analysis of safety data in 1299 pirfenidone recipients in five studies (CAPACITY, ASCEND and two open-label studies), in which the median exposure to pirfenidone was 1.7 years (range > 0 to 9.9 years) [23]. Both analyses are descriptive in nature.
Over 1 year of treatment, treatment-emergent adverse events (TEAEs) were reported in 98.7 and 96.5% of pirfenidone and placebo recipients, with serious TEAEs in 20.5 and 22.3% of patients in the corresponding groups, and TEAEs that led to early treatment discontinuation in 11.9 and 8.7%. Over the longer-term in the integrated analysis, TEAEs were reported in 97.6% of pirfenidone recipients, serious TEAEs in 49.2% (most commonly IPF in 17.5% of patients and pneumonia in 7.9%), and TEAEs led to early treatment discontinuation in 38.1% (most commonly IPF in 11.5% of patients) [23]. When adjusted for patient exposure, the rates of overall and serious TEAEs were comparable across the pirfenidone and placebo groups over 1 year and the pirfenidone group over the longer-term [41.7, 44.2 and 49.8 serious TEAEs per 100 person-exposure years (PEY), respectively] [23]. Corresponding rates of treatment-emergent death were 3.7, 5.9 and 7.4 per 100 PEY [23].
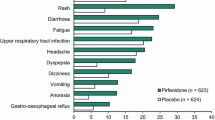
The incidences of respiratory events (with the exception of IPF, which favoured active treatment) were generally comparable in the placebo and pirfenidone groups in the pooled phase 3 trials (Fig. 1) [14]. As expected given its longer observation period, as well as the chronic, progressive nature of IPF, the rates of respiratory events in pirfenidone recipients in the integrated analysis [23] were higher than those in the pooled phase 3 trials [14].

Tolerability of oral pirfenidone 2403 mg/day in adults with IPF. Most common (reported in ≥ 10% of patients in either treatment group in the pooled phase 3 trials) treatment-emergent adverse events at 1 year in a pooled analysis of three phase 3 trials [14] and at 1.7 years in an integrated analysis of five trials [23]. GORD gastro-oesophageal reflux disease, IPF idiopathic pulmonary fibrosis, URTI upper respiratory tract infection
The following adverse events may occur with pirfenidone and are of special interest. To ameliorate their risk and/or minimize their impact, it may be necessary to adjust the dosage of pirfenidone, interrupt or discontinue treatment, monitor patients and/or take other measures (Table 1).
-
Gastrointestinal disorders Nausea, diarrhoea, dyspepsia, vomiting and gastro-oesophageal reflux disease were more common with pirfenidone than with placebo over 1 year (Fig. 1), with most events being of mild-to-moderate severity and rarely leading to treatment discontinuation [14]. Where reported, the rates of events with longer-term treatment were generally consistent with those with 1-year treatment (Fig. 1), suggesting the risks of these events does not increase over time [23].
-
Weight loss and anorexia Both were more common with pirfenidone than with placebo over 1 year (Fig. 1) [14], with an increase in weight loss over the longer-term (Fig. 1) [23].
-
Rash More common with pirfenidone than with placebo over 1 year (Fig. 1) [14], generally mild to moderate in severity and seldom resulted in treatment discontinuation [14]; incidence with longer-term treatment is comparable (Fig. 1) [23].
-
Dizziness and fatigue More common with pirfenidone than with placebo over 1 year (Fig. 1) [14]; slight increase over the longer term (Fig. 1) [23].
-
Liver function abnormalities Aminotransferase elevations of ≥ 3× the upper limit of normal (ULN) were more common with pirfenidone than with placebo (3.2 and 0.6% of patients; one pirfenidone recipient also experienced a > 2× ULN increase in serum total bilirubin) over 1 year [14]; no evidence of an increased risk of liver function abnormalities over the longer-term (3.1% of patients had ALT or AST elevations of > 3× ULN; generally transient, reversible with dosage adjustment, and not associated with any clinically relevant effects) [23].
-
Angioedema Reported in the post-marketing setting and may be serious [4]; the use of pirfenidone in patients who have a history of or who develop pirfenidone-related angioedema is contraindicated (Table 1).
The safety profile of pirfenidone in patients aged > 80 years was generally consistent with that in the overall population of patients with IPF, according to pooled data from the open-label extension study [18] and real-world data from the EU and USA [25]. Among the 370 pirfenidone recipients aged > 80 years, TEAEs, mostly mild or moderate in severity, were reported in 74.1% of patients, serious TEAEs in 4.1% and TEAEs led to treatment discontinuation in 25.9% (33.4 per 100 PEY) [25].
What is the current clinical positioning of pirfenidone?
Pirfenidone reduces the rate of decline in predicted FVC, improves other clinically meaningful outcomes and has a manageable tolerability profile in patients with IPF. It is, therefore, a valuable option for the treatment of this severe and very challenging to treat lung disease [1,2,3, 26,27,28]. The recent development of bioequivalent tablet formulation of pirfenidone reduces the daily pill burden associated with its capsule formulation; the recommended daily pirfenidone maintenance dosage of 2403 mg/day may be administered as one 801 mg tablet three times daily, instead of three 267 mg capsules three times daily. In theory, the reduction in pill burden should help improve adherence to pirfenidone treatment; however, adherence studies have yet to be conducted.
Based on the evidence of their overall favourable effectiveness and tolerability profiles in randomized, clinical trials, three-times-daily pirfenidone and twice-daily nintedanib (a multi-target tyrosine kinase inhibitor) are recommended to treat IPF according to the most recent international guidelines (conditional recommendation for both based on moderate confidence in effect estimates); the use of antacid therapy is also conditionally recommended; however, the quality of evidence supporting its use is very low [1]. Recent (2017) Nordic [26], Spanish [27] and French [28] guidelines also recommend pirfenidone and nintedanib as first-line options for the treatment of mild to moderate IPF (Fig. 2).

Individualized treatment with pirfenidone or nintedanib based on the clinical characteristics of patients with idiopathic pulmonary fibrosis, as suggested by recent (2017) Nordic [26], Spanish [27] and French [28] guidelines. Consult local prescribing information and guidelines for further information
IPF should be treated as soon as the diagnosis is established, with the choice of first-line treatment (i.e. pirfenidone or nintedanib) being individualized, taking into account the expected benefits and risks of treatment [26,27,28]. The effectiveness, tolerability and pharmacological profiles of the drug, and the characteristics of the patient (e.g. presence of comorbidities and use of concomitant drugs) should be considered (Fig. 2) [26,27,28]. For example, clinically relevant drug interactions may potentially occur between pirfenidone and CYP1A2 inhibitors and inducers (concomitant use of fluvoxamine is contraindicated, and the use of tobacco is strongly discouraged), and between nintedanib and P-glycoprotein inhibitors and inducers [26,27,28]. The use of pirfenidone is contraindicated in patients with severe hepatic or renal failure, whereas the use of nintedanib is contraindicated in patients with hypersensitivity to soy or peanuts or severe hepatic insufficiency, and should be avoided or used with caution in patients receiving anticoagulant treatment or high-dose antiplatelet therapy, and those with ischaemic heart disease, cerebrovascular stroke, abdominal surgery (< 4 weeks) or other risks of haemorrhage [26,27,28]. As IPF is a highly heterogenous condition with a varied clinical course, it is important that treatment is continued even when disease stabilization or progression occurs (Fig. 2) [3, 26,27,28]. Head-to-head clinical, adherence and pharmacoeconomic studies would help clarify the relative position of the available IPF therapies.
Change history
26 June 2018
The article Pirfenidone tablets in idiopathic pulmonary fibrosis: a profile of their use , written by Katherine A. Lyseng-Williamson, was originally published Online First without open access.
References
Raghu G, Rochwerg B, Zhang Y, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192(2):e3–19.
Raghu G. Pharmacotherapy for idiopathic pulmonary fibrosis: current landscape and future potential. Eur Respir Rev. 2017;26(145). https://doi.org/10.1183/16000617.0071-2017
Clarke DL, Murray LA, Crestani B, et al. Is personalised medicine the key to heterogeneity in idiopathic pulmonary fibrosis? Pharmacol Ther. 2017;169:35–46.
Esbriet (pirfenidone) tablets and capsules: summary of product characteristics. London: European Medicines Agency; 2017.
Esbriet (pirfenidone) tablets: US prescribing information. South San Francisco: Genetech USA, Inc.; 2017.
Kim ES, Keating GM. Pirfenidone: a review of its use in idiopathic pulmonary fibrosis. Drugs. 2015;75(2):219–30.
Oku H, Shimizu T, Kawabata T, et al. Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Pharmacol. 2008;590(1–3):400–8.
Knüppel L, Ishikawa Y, Aichler M, et al. A novel antifibrotic mechanism of nintedanib and pirfenidone: inhibition of collagen fibril assembly. Am J Respir Cell Mol Biol. 2017;57(1):77–90.
Pan L, Belloni P, Ding HT, et al. A pharmacokinetic bioequivalence study comparing pirfenidone and capsule dosage forms in healthy adult volunteers. Adv Ther. 2017;34:2071–82.
King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–92.
Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–9.
Azuma A, Nukiwa T, Tsuboi E, et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171(9):1040–7.
Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35(4):821–9.
Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J. 2016;47(1):243–53.
Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Resp Med. 2017;5(1):33–41.
Ley B, Swigris J, Day DM, et al. Pirfenidone reduces respiratory-related hospitalizations in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196(6):756–61.
Nathan SD, Albera C, Bradford WZ, et al. Effect of continued treatment with pirfenidone following clinically meaningful declines in forced vital capacity: analysis of data from three phase 3 trials in patients with idiopathic pulmonary fibrosis. Thorax. 2016;71(5):429–35.
Costabel U, Albera C, Lancaster LH, et al. An open-label study of the long-term safety of pirfenidone in patients with idiopathic pulmonary fibrosis (RECAP). Respiration. 2017;94(5):408–15.
Greig SL, Lyseng-Williamson KA, Kim ES, et al. Pirfenidone in idiopathic pulmonary fibrosis: a guide to its use in the EU. Drugs Ther Perspect. 2016;32(8):323–9.
Cottin V, Maher T. Long-term clinical and real-world experience with pirfenidone in the treatment of idiopathic pulmonary fibrosis. Eur Respir Rev. 2015;24(135):58–64.
Loeh B, Drakopanagiotakis F, Bandelli GP, et al. Intraindividual response to treatment with pirfenidone in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2015;191(1):110–3.
Harari S, Caminati A, Albera C, et al. Efficacy of pirfenidone for idiopathic pulmonary fibrosis: an Italian real life study. Respir Med. 2015;109(7):904–13.
Lancaster L, Albera C, Bradford WZ, et al. Safety of pirfenidone in patients with idiopathic pulmonary fibrosis: integrated analysis of cumulative data from 5 clinical trials. BMJ Open Respir Res. 2016;3(1):e000105.
Anderson A, Shifren A, Nathan SD. A safety evaluation of pirfenidone for the treatment of idiopathic pulmonary fibrosis. Expert Opin Drug Saf. 2016;15(7):975–82.
Lancaster L, Morrison LD, Auais A, et al. Pirfenidone in patients aged 80 years and older with idiopathic pulmonary fibrosis (ipf): safety findings from pooled trial databases [abstract]. Am J Respir Crit Care Med. 2017;95:A5385.
Sköld CM, Bendstrup E, Myllärniemi M, et al. Treatment of idiopathic pulmonary fibrosis: a position paper from a Nordic expert group. J Intern Med. 2017;281(2):149–66.
Xaubet A, Molina-Molina M, Acosta O, et al. Guidelines for the medical treatment of idiopathic pulmonary fibrosis. Arch Bronconeumol. 2017;53(5):263–9.
Cottin V, Crestani B, Cadranel J, et al. French practical guidelines for the diagnois and management of idiopathic pulmonary fibrosis - 2017 update: full-length version. Rev Mal Respir. 2017;34(8):900–68.
Acknowledgements
The article was reviewed by: J. Sellares, Interstitial Lung Disease Program, Servei de Pneumologia, Institut Clínic de Respiratori, Hospital Clínic, Institut d’Investigacions Biomediques August Pi i Sunyer (IDIBAPS), Barcelona, Spain; A. Xaubet, Servicio de Neumología, Hospital Clínic and Centro de Investigación Biomédica en Red de Enfermedades Respiratorias (CIBERES), Universitat de Barcelona, Barcelona, Spain. During the peer review process, the manufacturer of pirfenidone was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
K.A. Lyseng-Williamson is an employee of Adis/Springer, is responsible for the article content and declares no relevant conflicts of interest.
Funding
The preparation of this review was not supported by any external funding. Additional information about this Adis Drug Review can be found here http://www.medengine.com/Redeem/26CCF06034D49A5C.
Additional information
The original version of this article was revised due to a retrospective Open Access request.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
About this article

Cite this article
Lyseng-Williamson, K.A. Pirfenidone tablets in idiopathic pulmonary fibrosis: a profile of their use. Drugs Ther Perspect 34, 8–15 (2018). https://doi.org/10.1007/s40267-017-0459-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40267-017-0459-x