
Abstract
The analysis of safety data from spontaneous reporting systems has a proven value for the detection and analysis of the risks of medicines following their placement on the market and use in medical practice. EudraVigilance is the pharmacovigilance database to manage the collection and analysis of suspected adverse reactions to medicines authorised in the European Economic Area. EudraVigilance first operated in December 2001, with access to the database being governed by the EudraVigilance access policy. We performed a literature search including data up to December 2016 to demonstrate how the data from EudraVigilance has been used in scientific publications. We describe the results, including by type of publication, research topics and drugs involved. In 50% of the publications, the data are used to describe safety issues, in 44% to analyse methodologies used in pharmacovigilance activities and in 6% to support clinical perspectives. We also outline a description of the use of the database by the European Union regulatory network. Driven by the full implementation of the 2010 pharmacovigilance legislation, EudraVigilance has undergone further enhancements together with a major revision of its access policy, taking into account the use of the new individual case safety report standard developed by the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use and the International Organization for Standardization. The aim of the broadened access is to facilitate more effective safety monitoring of authorised medicines, to make more data available for research and to provide better access to information on suspected adverse reactions for healthcare professionals and patients. In November 2017, the new full functionalities of EudraVigilance were launched, including the extensive web access to data on suspected adverse drug reactions and the possibilities for academic research institutions to request a more extensive dataset for the purposes of health research. The main objective of this article is to describe the new access to the database together with the opportunities that this new access can bring for research. It is intended to promote an appropriate use of the data to support the safe and effective use of medicines.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
New EudraVigilance functionalities and the increased level of access facilitate more effective monitoring of the safety of medicines. Six different stakeholder groups with different levels of access have been defined |
Since November 2017, academic research institutions have the possibility to request extended access to EudraVigilance data, opening more possibilities for scientific and medical research |
Scientific publications using EudraVigilance data have increased since 2010 and analyse a wide range of safety issues, medicinal products, therapeutics areas, and pharmacovigilance topics and methods |
1 Introduction
EudraVigilance (EV) is the system for collecting, managing and analysing suspected adverse drug reactions (ADRs) to medicines authorised in the European Economic Area (EEA). The European Medicines Agency (EMA) operates the system, which became operational in December 2001, on behalf of the European Union (EU) medicines regulatory network [1].
On 4 February, 2016, 15 years after the pharmacovigilance database was launched, submissions to EV reached 10 million individual case safety reports (ICSRs), making it one of the biggest spontaneous reporting systems in the world. This milestone coincided in time with significant technical enhancements and with a major revision of the EV access policy aimed at increasing the utility and accessibility of the ICSR data in line with the pharmacovigilance legislative requirements [2,3,4], and thereby at further contributing to public health protection.
Since November 2005, the electronic reporting of suspected ADRs is mandatory in the EEA [2, 3]. The submission of ADRs from both marketed use and from trials is based on the standards agreed at the level of the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use [5] and the International Organization for Standardization [6], which allow for a structured and standardised way safety information is to be collected and exchanged.
By the end of 2017, EV held a total of 12,451,826 ICSRs, referring to 7,948,873 individual cases (one case can relate to more than one ICSR owing to follow-up information supplementing and updating initial reports). In total, 402,690 cases were submitted from interventional clinical trials and 7,546,183 from the post-authorisation setting of the medicinal products. Of the cases submitted from post-authorisation, 64% (4,838,460) were submitted from outside the EEA and 36% (2,707,723) from EEA countries. During the course of 2017 alone, 1,076,811 individual cases were transmitted from post-authorisation, 418,383 from EEA countries. Of those, 84,372 were directly submitted by European patients and consumers through national competent authorities (NCAs) in the Member States and marketing authorisation holders (MAHs). The EEA country distribution of patients’ reports analysed by Banovac et al. in 2017 [7] showed that the highest number of reports originated from the Netherlands, followed by the UK, Germany, France and Italy. The same five countries contributed 77% of all healthcare professional reports to EV.
In May 2017, following a period of technical development, guideline updates, engagement with the stakeholders and an independent system audit, the EMA Management Board announced that the EV database has achieved full functionality [8]. This triggered the application of both the simplified reporting rules for NCAs and MAHs and the extended access to the database with the goal to further support the safety surveillance of medicinal products in the EU, increase transparency and enable greater use of the data by stakeholders, including researchers.
2 Use of EudraVigilance Data
In this section, we review the use of EV data up to December 2016, including published reviews, with the aim to provide an overview of their use for health research. The launch of the enhanced system functionalities and the new access to data provide the research and medicine safety communities with the opportunity to make a much greater use of this data source, and this review therefore serves both as an inspiration and a challenge.
Use of EV data by the EU regulatory network includes a summary of areas such as signal detection and evaluation, PSUR assessments [9] and referrals. During 2017, the number of signals prioritised and analysed by the Pharmacovigilance Risk Assessment Committee was 82 and overall, the source of 63% included data from EV.
In addition to the use of EV by the EU regulatory network, other groups such as healthcare professionals and patients can obtain information on ADR data submitted to EV via the ‘European Database of Suspected Adverse Drug Reactions Reports’ [10] (hereinafter referred to as the ‘ADR website’). This information is available from the EMA website with guidance on the interpretation of the data. In 2017, the ADR website registered an average of 17,068 visits per month.
To have an overview about how EV data has been used in research and scientific publications, a search in EMBASE [11], PubMed and PubMed Central [12] was conducted using specific keywords [see the Electronic Supplementary Material (ESM)]. This provides an illustration of the use of the data with the aim of informing researchers about how EV can be used. The search included publications up to 31 December, 2016 and retrieved 105 citations referring to publications that contain data from EV.
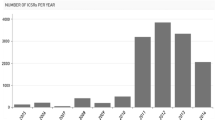
Ninety percent of the publications derived from countries within the EEA and 10% from the rest of the world. Figure 1 shows a distribution of the publications per country of the main author (a) and type of publication (b). Sixty-one percent of the citations from the UK were published by EMA personnel.

Publications using EudraVigilance data up to 31 December, 2016, classified per country of author and type of publication (as per the classification stated in EMBASE). Absolute numbers. EMA European Medicines Agency
The publications were classified in: (1) ‘safety issues’ when they describe a safety signal, characterisation of a risk or characterisation of the general safety profile of the drug; (2) ‘pharmacovigilance topics’ when the data were used to analyse a pharmacovigilance activity, research methodologies (e.g. signal detections methodologies, pharmacovigilance in paediatric population) or ADR reporting patterns (e.g. consumer reports, medication errors); and (3) ‘clinical perspectives’ when the EV data were used to support clinical practice and treatment guidance discussions.
Figure 2 shows the citations distributed by year of publication (a) and the main classification of the publications (b). Within the time distribution, there is a marked increase after 2012–2013 probably driven by the implementation of the 2010 pharmacovigilance legislation and the increased accessibility of the data, including through the release of the public ADR website [10].

Publications per year and the main classification of the publications using EudraVigilance data up to 31 December, 2016. Absolute numbers
Together with the EV data, the publications are supported by other sources of information. These include spontaneous reporting systems such as the World Health Organization (WHO) global database of individual case safety reports (VigiBase) [13], the US Vaccine Adverse Event Reporting System [14], the US FDA Adverse Event Reporting System [15], NCAs databases and safety databases from MAHs. Furthermore, data is provided from electronic health records databases, health insurance companies, legal datasets and patient registries. In 63% of the publications retrieved, EV is the sole spontaneous reporting system used for the research.
The specific drug-related safety issues are summarised in the ESM for this article including the drugs involved and the databases used for the analysis. The classification of drugs for which data are analysed in the 53 (50%) publications related to safety issues shows interests in a wide range of therapeutic areas, from medications administered presumably to relatively healthy populations, such as vaccines, to antineoplastic agents used in patients affected by a significant disease burden. Figure 3 provides the classification of the drugs analysed in these publications classified by Anatomical Therapeutic Chemical code [16] with the exception of the last three sets of drugs (‘drugs prescribed in geriatric setting’, ‘antibiotics’ and ‘all products in the database’) that cannot be allocated to a specific Anatomical Therapeutic Chemical code.

Anatomical Therapeutic Chemical (ATC) code classification of the drugs analysed in the publications using EudraVigilance data to describe safety issues up to 31 December, 2016. Absolute numbers. excl excluding
Figure 4 provides the MedDRA® System Organ Classes of the 53 publications describing safety issues with the exception of the 11 publications aimed at characterising the entire safety profile of the drugs. Two publications are allocated to two different System Organ Classes.

MedDRA® System Organ Class distribution for the specific safety issues described in the publications using EudraVigilance data up to 31 December, 2016. Absolute numbers. incl including
The 46 (44%) publications describing general pharmacovigilance topics contain research to analyse methodologies, especially on statistical signal detection, together with descriptions of specific situations that should to be taken into consideration when analysing safety data (e.g. paediatric population or ADR reports from consumers). The category related to signal detection methods includes publications under the scope of the IMI PROTECT project for which public funding was allocated [17,18,19].
Figure 5 provides the classification of the pharmacovigilance topics yielded by the literature search.

Classification of the pharmacovigilance topics analysed using EudraVigilance data up to 31 December, 2016. Absolute numbers. ADRs adverse drug reactions
One of the first publications on signal detection methods in EV refers to the validation study performed by the EMA to quantify the benefit that can be obtained by adding signal detection using the proportional reporting ratio to established pharmacovigilance methods. The study determined that statistical analysis has the potential to provide significant early warning on adverse reactions to medicines [20]. Since then, 22 other publications have used EV data to investigate signal detection methods. Choosing the thresholds, comparison of disproportionality measures, development of a score for the prioritisation of torsadogenic signals and analysis of variables that could influence signal detection in well-established products are among the topics covered. Based largely on the research conducted under the auspices of IMI PROTECT Work package 3 (signal detection) [19] [21], the EMA announced in 2015 the revision of the guidance of screening for adverse reactions in EV [17]. The final guideline was published in December 2016 [22]. This represents an important example of research-based improvement in regulatory practice.
The six publications related to clinical perspectives refer to the use of novel oral anticoagulants, in light of the experience with dabigatran [23,24,25,26], information systems for translational pharmacogenomics [27] and the use of sodium-glucose co-transporter 2 inhibitors with insulin for the treatment of type 2 diabetes [28].
3 Changes in Adverse Drug Reaction Reports Management and Access to EudraVigilance Data
The EU pharmacovigilance legislation adopted in 2010 introduced significant changes in the management of reports of suspected ADRs and placed EV as the single European hub for ICSR exchange and access. These changes include the submission of patient’s reports, non-serious ADRs that occurred in the EU, and ADRs arising from all uses of the medicinal products and not only those restricted to authorised use.
In the area of signal detection, EV plays a key role in determining new risks, new aspects of known risks and changes in the benefit-risk balance of medicinal products [2]. This comes with an explicit mandate for the EU regulatory network and the MAHs to monitor the data in EV. During the decision-making process, the Pharmacovigilance Risk Assessment Committee has a central role in scientific assessment in relation to signal management, covering all aspects of risk management. An analysis published to describe signals during the first 18 months of the Pharmacovigilance Risk Assessment Committee confirmed the importance of spontaneous reporting in signal detection [29].
The collaboration with the WHO is also being strengthened by making available all suspected ADRs reported in the EEA to the Uppsala Monitoring Centre and EV has undertaken the pertinent technical and process modifications to achieve this. With the aim of increasing transparency and to facilitate the conduct of safety monitoring and research, the level of access to EV has also been enhanced. In addition to the full data access provided to the EU regulatory network, MAHs are to have access to the extent necessary to comply with their pharmacovigilance obligations and an appropriate level of access is provided to healthcare professionals and the public while guaranteeing personal data protection.
The EMA Management Board endorsed a revision of the EudraVigilance access policy in December 2015 that determines the level of access by stakeholder groups considering the need to protect personal data. This version of the access policy takes into account the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use E2B(R3) [30] standard to determine the level of access for all ICSR data elements.
The revision of the access policy categorises stakeholders into six groups incorporating as new stakeholders the WHO–Uppsala Monitoring Centre and medicines regulatory authorities in third countries (outside the EEA). Nevertheless, the most significant update of the access policy is in the level of access, the number of data fields available, and the availability of new tools to access and analyse the data. Table 1 outlines the access provided and the tools for the different stakeholder groups.
4 Research Organisations
In the context of EV access, academic research organisations should be understood as public or private higher education establishments awarding academic degrees, public or private non-profit research organisations whose primary mission is to pursue research, and international European interest organisations whose principal objective is to promote scientific and technological cooperation in Europe [32]. Until November 2017, the level of access to EV data provided to academia and research organisations was limited to the same set of data elements publicly available in the ADR website. This is reflected in some of the publications that analysed EV data for safety assessments. The revision of the access policy extends the access to data for research to provide the possibility for a more thorough analysis of the data, hence contributing to the promotion and protection of public health and fostering innovation of scientific and medical research. Research institutions can directly access level 1 data via the ADR website and can also request the EMA level 2A data sets. Figures 6 and 7 provide an overview of how research institutions can access EV data.

Access to Level 1 EudraVigilance data by research organisations. ADR adverse drug reaction, ICSR individual case safety report

Access to Level 2A EudraVigilance data by research organisations. EMA European Medicines Agency
The data provided in the ADR website include aggregated information based on numbers of spontaneous cases stratified by different parameters (e.g. number of cases by age group, sex, geographic origin, reactions groups, time distribution). From November 2017, the ADR website provides the possibility to retrieve data for the individual cases based on some core data elements (level 1) within the ‘patient characteristics’, ‘reactions/events’ and ‘drugs information’ sections.
However, the data provided to research institutions upon request contain a significant increase in the potential number of data elements (level 2A) provided and the access is extended to all cases submitted to the EudraVigilance post-authorisation module, therefore not restricted to spontaneous cases, and includes reports from non-interventional post-authorisation studies.Footnote 1 Within the patient characteristics section and compared with level 1, level 2A provides 83 extra data elements including, amongst others, body weight and height, medical and family history, past drug history, death details and information concerning the parent for the parent–child reports. The reaction section contains ten extra data elements including the start and end dates of the reaction and the identification of the country where the reaction occurred. The drug section includes 49 extra data elements including the product name for non-centrally authorised medicinal products, strength, dates for the first and last drug administration, cumulative dose to first reaction, gestation period at the time of exposure and batch/lot numbers. Furthermore, the data set contains 13 additional data elements for the results of tests and procedures relevant to the investigation of the patient. To comply with data protection requirements, the case narratives, containing free text, are not provided to research organisations.
To obtain level 2A access, the interested institutions should submit a request via the EMA website [33] (http://www.ema.europa.eu/ema/index.jsp?curl=pages/about_us/landing/ask_ema_landing_page.jsp&mid=) with details on the research methodology, potential impact on public health, a signed copy of the confidentiality undertaking [32] and a copy of the approval of the research protocol by an ethics committee/review body. Following the analysis of the data, researchers should make all possible efforts to publish the outcomes of the research and for information purposes only, should provide a copy of the articles to the EMA ahead of publication.
5 Data Protection
The need to maintain the confidentiality of the identity of individuals in compliance with EU personal data protection legislation [34, 35] is one of the key drivers of the EV access policy and the levels of access have been developed considering data protection in conjunction with the public health needs and pharmacovigilance obligations for the different stakeholders. Users of EV should comply with the data protection requirements and should have appropriate measures in place to protect the information [32].
In this context, it is especially important to avoid the identification of individual patients from the data available in the ADR website published by the EMA. Level 1 access is based on this principle while providing meaningful information on suspected ADR reports. Table 2 outlines additional measures taken in the ADR website to avoid the identification of individual patients.
6 Discussion
Particularly since 2011, there has been a constant increase in the number of research publications that considered data from EV and this likely reflects the growing recognition of how the database can contribute to drug safety. The suspected ADR data in EV have provided a foundation to identify safety signals and characterise risks. Furthermore, the data have allowed the investigation of new methodologies for signal detection as well as specific situations such as paediatrics, vaccines, pandemics, consumer reports and traceability of biological products.
In 2016, Fouretier et al. [36] performed an analysis of 11 pharmacovigilance databases with open access to characterise the accessibility of the relevant data in those systems. In their study, the EV access was considered to be ‘medium’, mainly based on the fact that EV did not provide public access at the level of the individual cases in the ADR website. This assessment and the perception of EV will change drastically with the recent implementation of the new access specified in the revised access policy.
The main implication for MAHs following the release of the new EV system and extended access is the obligation to monitor the data for their substances and products. The extended access provided at the level of the substance brings the opportunity for MAHs to fully assess the data and provides greater transparency during assessment and decision making. The applied statistical analysis follows an evidence-based methodology [22] and this provides a common understanding about how statistical signal detection has been implemented in EV.
Apart from the power demonstrated by statistical methods within large spontaneous reporting systems [37], statistical signal detection should be complemented by scientific assessment and medical judgement, taking into consideration what is known about the product. The detailed analysis of individual cases remains one of the key activities during assessments and can provide significant information when determining causal associations.
At the same time as the new access is in place, the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use E2B(R3) standard is implemented in EV with the subsequent advantages derived from the new data structure and data elements. These will bring advantages for data analysis (e.g. in the old format, seriousness criteria are reported at the level of the case whereas these are reported at the level of the individual reactions in the new format). Safety assessors should be well aware of the differences between the old and the new format when analysing the data.
The enhanced access to the data is also expected to trigger more engagement with academia. Up to November 2017, the information from EV publicly available could have been considered acceptable for patient use but not optimally for research purposes [36]. The extended access provides research institutions with a rich data source and there is therefore a unique opportunity to conduct research for public health.
7 Conclusion
Pharmacovigilance is an essential discipline for the promotion and protection of patient and public health. In the EEA, the EU regulatory network formed by the European Commission, the NCAs and the EMA has a key role together with the MAHs on the safety surveillance on medicinal products.
The EU legislation places the EV database in a core role in the management of reports of suspected adverse reactions and in determining changes in the safety profile of the medicinal products once they are used in everyday medical practice. For over 16 years, EV has demonstrated its value especially in the area of signal detection, providing a tool to analyse the safety data submitted.
Although EV has been fully accessible to the EMA and the NCAs in the countries that belong to the EEA, the access provided in the past to other stakeholders has been more limited. The new extended EV access provides a unique platform for all stakeholders to use the data according to their needs, interest and pharmacovigilance obligations. This enhanced access brings opportunities for a broader involvement in safety monitoring, for increased transparency, for collective collaborations, for methods improvement and for innovation in safety research.
We call on patients, healthcare professionals, MAHs, research institutions, the WHO, non-EEA regulatory authorities, NCAs in the EEA Members States and the EMA to embrace the opportunities provided by the new EudraVigilance system to be better informed, to better manage product risks, to better understand the science of pharmacovigilance and, ultimately, to better promote public health.
Notes
Non-interventional studies are considered studies where the medicinal product(s) is (are) prescribed in the usual manner in accordance with the terms of the marketing authorisation. The assignment of the patient to a particular therapeutic strategy is not decided in advance by a trial protocol but falls within current practice and the prescription of the medicine is clearly separated from the decision to include the patient in the study. Non-interventional post-authorisation studies are identified in the database by the mandatory fields ‘type of report’ and ‘study type where the reactions events occurred’.
References
European Medicines Agency. EudraVigilance. http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000679.jsp&mid=WC0b01ac05800250b5. Accessed 31 Jan 2018.
European Commission. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use. https://ec.europa.eu/health/documents/eudralex/vol-1_en. Accessed 31 Jan 2018.
European Commission. Regulation (EC) No. 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency. https://ec.europa.eu/health/documents/eudralex/vol-1_en. Accessed 31 Jan 2018.
European Commission. Commission Implementing Regulation (EU) No. 520/2012 of 19 June 2012 on the performance of pharmacovigilance activities provided for in Regulation (EC) No. 726/2004 of the European Parliament and of the Council and Directive 2001/83/EC of the European Parliament and of the Council. https://ec.europa.eu/health/documents/eudralex/vol-1_en. Accessed 31 Jan 2018.
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). http://www.ich.org/home.html. Accessed 31 Jan 2018.
International Organization for Standardization. https://www.iso.org/home.html. Accessed 31 Jan 2018.
Banovac M, Candore G, Slattery J, Houÿez F, Haerry D, Genov G, et al. Patient reporting in the EU: analysis of EudraVigilance data. Drug Saf. 2017;40(7):629–45. https://doi.org/10.1007/s40264-017-0534-1.
European Medicines Agency. Announcement of the EMA Management Board: confirmation of full functionality of the EudraVigilance database. http://www.ema.europa.eu/docs/en_GB/document_library/Other/2017/05/WC500228158.pdf. Accessed 31 Jan 2018.
Arlett P, Postigo R, Janssen H, Spooner A. Periodic benefit-risk evaluation report: a European Union regulatory perspective. Pharm Med. 2014;28(6):309–15. https://doi.org/10.1007/s40290-014-0071-5.
European Medicines Agency. European database of adverse reactions reports. http://www.adrreports.eu/en/index.html. Accessed 31 Jan 2018.
EMBASE. https://www.embase.com. Accessed 13 Feb 2017.
National Center for Biotechnology Information. https://www.ncbi.nlm.nih.gov. Accessed 14 Feb 2017.
World Health Organization. VigiBase. https://www.who-umc.org/vigibase/vigibase/. Accessed 31 Jan 2018.
Vaccine Adverse Event Reporting System. https://vaers.hhs.gov/. Accessed 31 Jan 2018.
US Food and Drug Administration. FDA Adverse Event Reporting System (FAERS). https://www.fda.gov/drugs/informationondrugs/ucm135151.htm. Accessed 31 Jan 2018.
World Health Organization Collaborating Centre for Drug Statistics Methodology. ATC/DDD Index 2017. https://www.whocc.no/atc_ddd_index/. Accessed 15 May 2017.
Candore G, Slattery J, Kurz X, Arlett P. Revision of guidance on screening for adverse drug reactions in eudravigilance. Drug Saf. 2015;38(10):964–5. https://doi.org/10.1007/s40264-015-0346-0.
Kurz X, Swain EJ, Blackburn S, Prilla S, Arlett P. The PROTECT project: an innovative public-private partnership for new methodologies in pharmacovigilance and pharmacoepidemiology. Drug Saf. 2010;33(10):895. https://doi.org/10.2165/11532470-000000000-00000.
The Innovative Medicines Initiative (IMI). The pharmacoepidemiological research on outcomes of therapeutics by a European consortium. http://www.imi-protect.eu/. Accessed 31 Jan 2018.
Alvarez Y, Hidalgo A, Maignen F, Slattery J. Validation of statistical signal detection procedures in eudra vigilance post-authorization data: a retrospective evaluation of the potential for earlier signalling. Drug Saf. 2010;33(6):475–87. https://doi.org/10.2165/11534410-000000000-00000.
Wisniewski AFZ, Bate A, Bousquet C, Brueckner A, Candore G, Juhlin K, et al. Good signal detection practices: evidence from IMI PROTECT. Drug Saf. 2016;39(6):469–90. https://doi.org/10.1007/s40264-016-0405-1.
European Medicines Agency. Screening for adverse reactions in EudraVigilance. http://www.ema.europa.eu/docs/en_GB/document_library/Other/2016/12/WC500218606.pdf. Accessed 31 Jan 2018.
Malmström RE, Godman BB, Diogene E, Baumgärtel C, Bennie M, Bishop I, et al. Dabigatran: a case history demonstrating the need for comprehensive approaches to optimize the use of new drugs. Front Pharmacol. 2013;14(4):39. https://doi.org/10.3389/fphar.2013.00039.
Shehab A, Elnour AA, Bhagavathula AS, Erkekoglu P, Hamad F, Al Nuaimi S, et al. Novel oral anticoagulants and the 73rd anniversary of historical warfarin. J Saudi Heart Assoc. 2016;28(1):31–45. https://doi.org/10.1016/j.jsha.2015.05.003.
Godman B, Sakshaug S, Berg C, Wettermark B, Haycox A. Combination of prescribing restrictions and policies to engineer low prices to reduce reimbursement costs. Exp Rev Pharmacoecon Outcomes Res. 2011;11(1):121–9. https://doi.org/10.1586/erp.10.87.
Madan S, Shah S, Partovi S, Parikh SA. Use of novel oral anticoagulant agents in atrial fibrillation: current evidence and future perspective. Cardiovasc Diagn Ther. 2014;4(4):314–23. https://doi.org/10.3978/j.issn.2223-3652.2014.08.01.
Lakiotaki K, Kartsaki E, Kanterakis A, Katsila T, Patrinos GP, Potamias G. ePGA: a web-based information system for translational pharmacogenomics. PLoS One. 2016;11(9):e0162801. https://doi.org/10.1371/journal.pone.0162801.
John M, Gopinath D, Jagesh R. Sodium-glucose cotransporter 2 inhibitors with insulin in type 2 diabetes: clinical perspectives. Indian J Endocrinol Metab. 2016;20(1):22–31. https://doi.org/10.4103/2230-8210.172268.
Pacurariu AC, Coloma PM, van Haren A, Genov G, Sturkenboom MC, Straus SM. A description of signals during the first 18 months of the EMA pharmacovigilance risk assessment committee. Drug Saf. 2014;37(12):1059–66. https://doi.org/10.1007/s40264-014-0240-1.
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Clinical safety data management: data elements for transmission of individual case safety reports. http://www.ich.org/products/guidelines/efficacy/article/efficacy-guidelines.html. Accessed 31 Jan 2018.
European Medicines Agency. Guideline on good pharmacovigilance practices (GVP) module VI: collection, management and submission of reports of suspected adverse reactions to medicinal products (Rev 2). http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2017/08/WC500232767.pdf. Accessed 31 Jan 2018.
European Medicines Agency. European Medicines Agency policy on access to EudraVigilance data for medicinal products for human use: revision 3. http://www.ema.europa.eu/docs/en_GB/document_library/Other/2016/12/WC500218300.pdf. Accessed 31 Jan 2018.
European Medicines Agency. Available from: http://www.ema.europa.eu/ema/. Accessed 31 Jan 2018.
European Union. Directive 95/46/EC of the European Parliament and of the Council of 24 October 1995 on the protection of individuals with regard to the processing of personal data and on the free movement of such data. http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex:31995L0046. Accessed 31 Jan 2018.
European Union. Regulation (EC) No. 45/2001 of the European Parliament and of the Council of 18 December 2000 on the protection of individuals with regard to the processing of personal data by the institutions and bodies of the Community and on the free movement of such data. http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex:32001R0045. Accessed 31 Jan 2018.
Fouretier A, Malriq A, Bertram D. Open access pharmacovigilance databases: analysis of 11 databases. Pharm Med. 2016;30(4):221–31. https://doi.org/10.1007/s40290-016-0146-6.
Evans SJ, Waller PC, Davis S. Use of proportional reporting ratios (PRRs) for signal generation from spontaneous adverse drug reaction reports. Pharmacoepidemiol Drug Saf. 2001;10(6):483–6. https://doi.org/10.1002/pds.677.
Acknowledgements
The authors gratefully acknowledge the contribution made by Luis Pinheiro.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were received for the preparation of this review.
Conflict of interest
Rodrigo Postigo, Sabine Brosch, Jim Slattery, Xavier Kurz, Gianmario Candore, Francois Domergue and Peter Arlett are employees of the European Medicines Agency. Jean-Michel Dogné is a member of the Pharmacovigilance Risk Assessment Committee, as well as an employee of the Federal Agency for Medicines and Health Products of Belgium. Anja van Haren is an employee of the Medicines Evaluation Board of the Netherlands and she acts as a co-chair of the EudraVigilance Expert Working Group.
Disclaimer
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the agencies or organisations with which the authors are affiliated.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Postigo, R., Brosch, S., Slattery, J. et al. EudraVigilance Medicines Safety Database: Publicly Accessible Data for Research and Public Health Protection. Drug Saf 41, 665–675 (2018). https://doi.org/10.1007/s40264-018-0647-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-018-0647-1