
Abstract
Survival motor neuron 1 (SMN1), located on chromosome 5q, encodes the survival motor neuron (SMN) protein. A deletion or mutation in SMN1 results in a rare neuromuscular disorder: 5q spinal muscular atrophy (SMA). In such patients, SMN protein production relies solely on SMN2. Nusinersen (Spinraza®) is a modified antisense oligonucleotide approved for the treatment of 5q SMA. Administered intrathecally, it modifies SMN2 pre-messenger RNA splicing, thereby increasing full-length SMN protein levels. Interim analyses from an ongoing phase II study suggest substantial clinical benefits with nusinersen initiation in presymptomatic patients. In phase III studies, nusinersen achieved significant and/or clinically relevant improvements in motor function in symptomatic patients with infantile- and later-onset 5q SMA, and significantly improved event-free survival and overall survival in patients with infantile-onset 5q SMA. Longer term (up to a median of ≈ 6 years of available data), motor function was maintained or improved in symptomatic patients. Nusinersen had a favourable safety profile in clinical studies in presymptomatic and symptomatic patients. Real-world experience supports the effectiveness, safety and tolerability of nusinersen in symptomatic patients of all ages. Thus, nusinersen remains an important treatment option among a broad range of 5q SMA patients.
Plain Language Summary
5q spinal muscular atrophy (SMA) is a rare disease most commonly caused by a defect in the survival motor neuron (SMN) 1 gene, which in a healthy individual produces a protein [spinal motor neuron (SMN) protein] critical to maintaining the nerves that control muscles. Individuals with 5q SMA do not produce this protein in sufficient levels, resulting in muscle weakness and wasting (including the muscles involved in general movement, breathing and swallowing), so increasing the amount of SMN protein by modifying a nearly identical, but low functioning, gene (SMN2) is one way to treat the disease. Nusinersen (Spinraza®) is a treatment that targets SMN2. It is administered via lumbar puncture and is approved for use in presymptomatic and symptomatic individuals with 5q SMA. In both groups of patients, nusinersen increases the amount of SMN protein necessary for the muscles and nerves to work normally, improving motor function. This benefit persists over the longer-term (up to a median of ≈ 6 years of available data), and is well tolerated. Nusinersen continues to be an important treatment option among a broad range of 5q SMA patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Digital Features for this Adis Drug Evaluation can be found at https://doi.org/10.6084/m9.figshare.16571555. |
Modifies SMN2 pre-messenger RNA splicing, thereby increasing full-length SMN protein levels |
Improves motor function in presymptomatic and symptomatic patients, and event-free survival and overall survival in symptomatic patients with infantile-onset disease |
Improvements seen in all age groups, with greater benefits in those receiving earlier treatment |
Most reported adverse events were related to the disease itself or the lumbar puncture procedure |
1 Introduction
Spinal muscular atrophy (SMA) is a rare (incidence of 1 in 8000 to 10,000 individuals [1]) neuromuscular disorder most commonly resulting from a homozygous deletion or mutation in the survival motor neuron 1 (SMN1) gene located on chromosome 5q [1, 2]. SMN1 encodes survival motor neuron (SMN) protein, which is crucial for the maintenance of motor neurons; insufficient levels of SMN protein lead to motor neuron death and subsequently the signs and symptoms of 5q SMA [atrophy and weakness of the skeletal muscles (including those involved in general movement and breathing) and impaired bulbar function (characterized by difficulties in mouth opening, chewing and swallowing)] [1, 3]. Five 5q SMA types have been identified (based on age of symptom onset and maximal acquired motor function) [1, 2, 4, 5]. Type 0, the most severe form, results in death within weeks of birth even with intensive respiratory support. In patients with type 1 (infantile-onset 5q SMA; the most common form of the disease), symptoms are evident at birth or within the first few months of life, individuals are unable to sit independently, and death usually occurs within the first 2 years. Those with type 2 have symptom onset between 6 and 18 months of age. Affected individuals can sit independently but not walk; most live into adulthood with substantial disability. Type 3 is characterized by symptom onset after 18 months of age. Affected individuals can stand and walk independently and have a normal life expectancy, although these abilities will deteriorate over time. Type 4 is the mildest and rarest form of the disease and symptoms often develop in early adulthood. Affected individuals have mild to moderate disability and a normal life expectancy [1, 2, 4, 5].
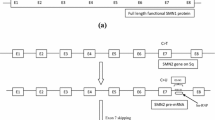
Humans also have SMN2, a nearly identical copy of SMN1 [2]. While SMN2 also encodes SMN protein, a single nucleotide difference in exon 7 of the gene alters the splicing of most pre-messenger RNA (pre-mRNA) transcripts; consequently, ≈ 90% of the SMN protein produced from SMN2 mRNA is truncated (i.e. non-functional) and rapidly degraded [1, 2]. An individual’s SMN2 copy number [which can range from one or two copies (typical) to up to eight copies] is the key determinant of 5q SMA disease severity [1, 6]. While patients with 5q SMA type 1 generally have two SMN2 copies, those with type 2 three copies, those with type 3 three or four copies and those with type 4 four or more copies [1, 6], the number of SMN2 copies does not always correlate directly with the observed clinical phenotype [2].
Only recently have therapeutic approaches beyond symptomatic and supportive care become available for patients with 5q SMA. One of these is the antisense oligonucleotide (ASO) nusinersen (Spinraza®). This article provides an updated overview of pharmacological (summarized in Table 1), therapeutic efficacy and tolerability data relevant to the intrathecal use of nusinersen in patients with 5q SMA, previously reviewed in CNS Drugs [7].
2 Therapeutic Efficacy of Nusinersen
2.1 In Presymptomatic Patients
An open-label, noncomparative, multinational, phase II study (CS5; NURTURE) is evaluating the therapeutic efficacy of nusinersen in preventing or delaying respiratory intervention or death when initiated prior to symptom onset [8, 9]. Eligible patients had a genetic diagnosis of 5q SMA and two or three copies of SMN2, were presymptomatic at screening and were aged ≤ 6 weeks at the time of the first dose. Nusinersen (12 mg dose equivalent) was administered intrathecally on days 1, 15, 29 and 64, and then every 119 days thereafter for a total treatment period of 8 years; the median age at first dose was 22.0 days. An interim analysis was conducted at a data cut-off date of 29 March 2019 (median follow-up duration of 2.9 years). At this timepoint, the 25 enrolled patients had a median (range) age of 34.8 (25.7–45.4) months (i.e. they were past the expected age of symptom onset for 5q SMA type 1 or 2) and had been in the study for 33.9 (25.3–45.1) months. At baseline, 15 and 10 patients had two or three copies of SMN2, respectively [8, 9].
The early (i.e. immediately after establishing a genetic diagnosis of 5q SMA) initiation of nusinersen in presymptomatic infants was associated with substantial clinical benefits [8]. At the time of the interim analysis, the primary endpoint of median time to respiratory intervention (defined as invasive or non-invasive ventilation for ≥ 6 h/day continuously for ≥ 7 days, or tracheostomy) or death could not be estimated owing to too few events. All 25 patients were alive and were able to sit without support, and none required permanent ventilation. Respiratory support for ≥ 6 h/day for ≥ 7 consecutive days was required by four patients owing to acute, reversible illnesses, but at the last study day prior to the data cut-off date was no longer needed by two patients [8].
All but two patients were able to walk with support and all but three were able to walk independently; patients unable to achieve these milestones had just two copies of SMN2 [8]. Notably, 84% of patients achieved sitting without support, 65% achieved walking with assistance and 73% achieved walking independently within the window established by the WHO for healthy children (i.e. by the WHO 99th percentile age of achievement). At day 778, all patients could suck and swallow [as assessed by Hammersmith Infant Neurological Examination (HINE) Section 1]. Mean total HINE Section 2 (HINE-2; see Table 2 for definition) scores improved from 2.7 at baseline to 23.9 at the last observed visit (up to and including day 778) in patients with two copies of SMN2 and from 3.2 to 26.0 (the scale maximum) in those with three copies of SMN2. Mean total Children’s Hospital of Philadelphia Infant Test for Neuromuscular Disease (CHOP-INTEND) scores improved steadily from baseline (values of 47.0 and 51.9) until ≈ day 183, after which they remained stable. At the time of the interim analysis, mean total CHOP-INTEND scores were 62.1 and 63.4 in the subgroups of patients with two or three copies of SMN2, with 67% and 100% of patients achieving the maximum score of 64. Protocol-defined symptoms of 5q SMA were reported in 10 patients with two copies of SMN2 and 2 patients with three copies of SMN2 at age 13 months and in 7 and 0 patients at age 24 months. The 7 patients with symptoms of 5q SMA at 24 months of age continued to grow and achieve WHO motor milestones inconsistent with both 5q SMA type 1 and untreated siblings with 5q SMA: all 7 were sitting without support, 5 were walking with support and 4 were walking independently [8].
Preliminary data from another interim analysis (data cut-off date of 19 February 2020) support the WHO motor milestone findings of the earlier analysis, with all NURTURE participants (n = 25; median age of 3.8 years) alive and without permanent ventilation [10]. All of the participants who achieved the motor milestone of being able to walk independently maintained that ability over the 11 months since the previous data cut-off date [10]. In the overall population, swallowing ability (assessed using the Parent Assessment of Swallowing Ability questionnaire) was maintained in 92% of 25 patients [median age at last visit of 3.8 (range 2.8–4.8) years] and only 2 patients (both of whom had two copies of SMN2) required full-time tube feeding [11].
2.2 In Symptomatic Patients
2.2.1 Infantile-Onset 5q SMA
2.2.1.1 In Clinical Studies
The potential of nusinersen in patients with infantile-onset 5q SMA was first demonstrated in an open-label, phase II, dose-escalation study (CS3A) [12]. Results from this study supported the selection of a 12 mg dose for further investigation [7] and informed the design of a subsequent randomized, double-blind, sham-controlled, multinational, phase III study (CS3B; ENDEAR) [13]. ENDEAR enrolled 121 patients with a genetic diagnosis of 5q SMA and two copies of the SMN2 gene who were aged ≤ 6 months at symptom onset and ≤ 7 months at screening [13]. All patients were symptomatic, hypotonic, and weak (features consistent with a phenotype most likely to be 5q SMA type 1). Randomization to nusinersen (12 mg dose equivalent, administered intrathecally on days 1, 15, 29, 64, 183 and 302) or a sham procedure was stratified by disease duration at screening (≤ 12 weeks or > 12 weeks). The median disease duration at screening was 13.1 weeks. Results from the prespecified interim analysis demonstrating a benefit–risk assessment in favour of nusinersen (Table 2) prompted the early termination of ENDEAR, with patients invited to complete an end-of-study visit (used in the final analysis) ≥ 2 weeks after receiving their most recent dose of nusinersen or sham control and transition into an open-label phase III extension study (SHINE; CS11) [13].
Nusinersen improved motor function in patients with infantile-onset 5q SMA, as demonstrated by a significantly higher motor milestone response rate relative to sham control at the time of both the interim and final analyses (co-primary endpoint) (Table 2). At the final analysis, 22% of nusinersen recipients had achieved full head control, 10% were able to roll over, 8% were able to sit independently and 1% were able to stand [13]. Nusinersen also improved the likelihood of event-free survival (EFS) at the time of the final analysis, as indicated by the significant 47% reduction in the risk of death or permanent assisted ventilation in nusinersen compared with sham control recipients (co-primary endpoint) (Table 2). At this timepoint, the median duration of EFS had not yet been reached in the nusinersen group (and was 22.6 weeks in the sham control group) [13].
At the final analysis, nusinersen was associated with a significantly higher CHOP-INTEND response rate than sham control (Table 2). An improvement from baseline in the CHOP-INTEND score of ≥ 1 point was achieved by 73% of nusinersen recipients and 3% of sham control recipients; 7% and 49% of patients demonstrated a worsening from baseline of ≥ 1 point and 3% and 46% demonstrated a worsening from baseline of ≥ 4 points [13]. While the risk of death was significantly reduced (63%) with nusinersen compared with sham control, there was no significant between-group difference in the proportion of patients not requiring permanent assisted ventilation (Table 2) and all subsequent endpoint analyses in the hierarchical testing strategy were considered exploratory. It is worth noting that as data for patients who had died were censored, the effect of nusinersen on the use of permanent ventilation may have been masked by the twofold higher proportion of deaths in the sham control group compared with the nusinersen group. In the subgroup of patients with a disease duration of ≤ 13.1 weeks at screening, the median time to death or permanent assisted ventilation had not yet been reached in nusinersen recipients, but was 25.4 weeks in sham control recipients [13].
A ≥ 5-point increase in the HINE-2 total score was achieved by 28% of 58 nusinersen recipients and 5% of 20 sham control recipients who were alive at the end of ENDEAR and who had been enrolled for ≥ 6 months (prespecified analysis) [13]. Patients receiving nusinersen who achieved HINE-2 or CHOP-INTEND response criteria earlier in ENDEAR generally demonstrated greater overall improvements at the last visit [14]. Among 51 HINE-2 and 71 CHOP-INTEND responders, the respective response criteria were met by 62% and 83% of patients by day 183, by an additional 27% and 13% by day 302 and by an additional 11% and 4% by day ≥ 395 [14].
Interim results (data cut-off date of 27 August 2019) from SHINE (in which all patients were receiving nusinersen) demonstrated improvements in motor function outcomes in both patients originally treated with nusinersen and those originally treated with sham control in ENDEAR, with the greatest benefits seen in patients with earlier treatment initiation [15, 16]. In patients originally treated with nusinersen in ENDEAR, the mean change from baseline in the CHOP-INTEND score at day 1058 was + 19.8 points in those aged ≤ 5.5 months (n = 32) and + 13.1 points in those aged > 5.5 to ≤ 8 months (n = 26) at their first dose of nusinersen [15]. In those originally treated with sham control in ENDEAR who commenced nusinersen therapy in SHINE aged ≥ 14 to ≤ 23 months (n = 11), the mean change from baseline in the CHOP-INTEND score at day 1058 was + 5.1 points [15]. At this timepoint, 40% of 105 ENDEAR/SHINE participants were able to sit independently [16]. Sitting independently was first achieved by 1, 2, 3 or 4 years of age, respectively, by 37%, 10%, 10% and 2% of patients aged 1.7 to ≤ 5.5 months at their first dose (n = 41) and by 8%, 10%, 23%, and 3% of those aged > 5.5 to ≤ 8.0 months at their first dose (n = 40). Only one of the 24 patients aged > 8.0 to ≤ 23.0 months at their first dose achieved sitting independently at 4 years of age. Walking independently was achieved by one patient (aged 1.7 to ≤ 5.5 months at their first dose) at 3 years of age [16]. In a multivariate analysis, weight-by-age percentile (≥ 5% vs < 5%), baseline CHOP-INTEND score and disease duration were identified as prognostic for independent sitting [17].
2.2.1.2 In Real-World Studies
Real-world experience supports the effectiveness of nusinersen in improving motor function in patients with infantile-onset 5q SMA. In the largest (n = 104) real-world study in this patient population, nusinersen for 6 months was associated with significant (p < 0.001) changes from baseline in both CHOP-INTEND and HINE-2 scores [18]. Patients in this study were aged 0–19 years, had mean baseline CHOP-INTEND and HINE-2 scores of 15.08 points and 0.82 points, and were enrolled in the Italian expanded access programme (EAP). Improvements did not appear to be related to the number of SMN2 copies, with significant changes (p < 0.001) in these endpoints seen in both patients with two and three SMN2 copies (n = 65 and 24). At month 6, 55.7% and 20.2% of patients achieved improvements from baseline of > 2 points in CHOP-INTEND and HINE-2 scores, respectively; such improvements occurred in 36.6% of 71 patients aged > 2 years and 35.0% of 20 patients aged > 10 years [18]. No significant change from baseline in body mass index was seen at month 6 (n = 84) [19]. Among patients in the Italian EAP cohort with 12-month follow-up data (mean baseline CHOP-INTEND and HINE-2 scores of 15.66 points and 0.69 points) [20], there were significant (p < 0.05) improvements from baseline in both the CHOP-INTEND and HINE-2 scores for the entire cohort (n = 85), and for the subgroups with two and three SMN2 copies (n = 61 and 18). Significant (p < 0.05) improvements from baseline at month 12 were also seen in both scores in patients aged < 210 days (n = 6) and those aged < 2 years (n = 23) at baseline, and in the CHOP-INTEND score in those aged 2–4 years at baseline (n = 20); improvements from baseline at month 12 were not significant in either score in patients aged > 5 years [i.e. 5–11 years and > 12 years (n = 29 and 5)] at baseline [20].
Improvements in motor function may occur beyond the first year of nusinersen treatment, according to 24-month follow-up data from the Italian EAP cohort (n = 68), with improvements more obvious in patients younger than 2 years of age [21]. For both CHOP-INTEND and HINE-2 scores, significant (p < 0.001) improvements were seen from baseline at 12 months, from baseline at 24 months and from 12 months at 24 months. Patient age (but not SMN2 copy number) was predictive of changes in both the CHOP-INTEND and HINE-2 scores, with the improvement from baseline to 24 months significant (p < 0.05) across all age subgroups (i.e. < 210 days, < 2 years, 2–4 years, 5–11 years and 12–18 years) for the CHOP-INTEND score and for all age subgroups before the age of 4 years for the HINE-2 score [21].
2.2.2 Later-Onset 5q SMA
2.2.2.1 In Clinical Studies
The efficacy of nusinersen in 126 patients with later-onset 5q SMA aged 2–9 years was evaluated in a randomized, double-blind, sham-controlled, multinational, phase III study (CS4; CHERISH) [22]. Patients with a genetic diagnosis of 5q SMA who were aged > 6 months at symptom onset (most likely to be classified as 5q SMA type 2 or 3) were eligible [22]. All patients were non-ambulatory and 88% had three copies of the SMN2 gene [22, 23]. Randomization to treatment arms [nusinersen (12 mg intrathecally on days 1, 29, 85 and 274) or a sham procedure] was stratified by age at screening (< 6 or ≥ 6 years) [22]. CHERISH was terminated early following the results of a prespecified interim analysis, which showed the benefits of nusinersen over sham control (Table 3), with patients who had not already had a 15-month assessment invited to complete an end-of study visit. Patients who completed CHERISH were invited to enrol in SHINE [22].
Therapy with nusinersen in CHERISH (Table 3) was associated with a statistically significant and clinically meaningful improvement in motor function [as assessed by the total Hammersmith Functional Motor Scale Expanded (HFMSE) score; primary endpoint] compared with sham control [between-group least-squares mean (LSM) difference of 5.9 points] at the time of the prespecified interim analysis [22]. At the time of the final analysis, the LSM between-group difference in this endpoint was 4.9 points (no formal statistical analysis undertaken as significance was established at the time of the interim analysis) [22]. Among nusinersen and sham control recipients with observed values at month 15 (n = 66 and 34), 73% and 41% demonstrated an improvement (not specified) while 23% and 44% demonstrated a worsening (not specified) in the total HFMSE score [22, 23].
At the final analysis (Table 3), nusinersen demonstrated a significant advantage over sham control in the proportion of patients achieving a ≥ 3-point increase from baseline in the total HFMSE score, reflecting a six-fold higher likelihood of achieving this endpoint with nusinersen than sham control at month 15 [22]. However, there was no significant between-group difference in the proportion of patients achieving ≥ 1 new WHO motor milestone; all subsequent endpoint analyses in the hierarchical testing strategy were considered exploratory [22].
Interim results (data cut-off date of 27 August 2019) from SHINE (in which all patients were receiving nusinersen) demonstrated improved motor function [as assessed by total HFMSE and Revised Upper Limb Module (RULM) scores] in both patients originally treated with nusinersen and those originally treated with sham control in CHERISH [24]. Greater benefits were seen in patients with earlier treatment initiation (i.e. those who received nusinersen throughout CHERISH and SHINE). The mean change from baseline in the total HFMSE score was 4.6 at day 1650 in patients treated with nusinersen in both CHERISH and SHINE (n = 20) and 1.7 at day 930 (n = 35) in those who commenced nusinersen in SHINE. In the respective groups, the mean change from baseline in the total RULM score was 6.4 at day 1650 (n = 20) and 3.4 at day 930 (n = 36). At the time of this analysis, the median time on nusinersen was 4.1 years for patients treated with nusinersen in CHERISH and SHINE and 2.8 years for those receiving sham control in CHERISH and nusinersen in SHINE [24].
These findings are generally supported by data from ≈ 3 years of nusinersen therapy (six or seven total doses) in 28 patients with later-onset 5q SMA participating in an open-label, multicentre, phase 1b/2a, dose-escalation study (CS2) and its extension (CS12) [25]. In patients with 5q SMA type 2 (symptom onset 7–18 months; n = 11), clinically meaningful improvements from baseline in both the total HFMSE score and the Upper Limb Module score (a change of ≥ 3 and ≥ 2 points, respectively, was considered clinically meaningful) were seen at day 1150. In patients with 5q SMA type 3 (symptom onset > 18 months; n = 17), total HFMSE scores were stable while the 6-min walk test (6MWT) distance was improved by a clinically meaningful extent (a change of ≥ 30 m) from baseline at day 1150. Such changes were not seen in comparable natural history cohorts [25]. Interim results (data cut-off date of 27 August 2019; median time on study of 6.2 and 6.3 years for patients with 5q SMA type 2 or type 3) from SHINE for these patients further support the maintenance of or improvements in motor function seen with longer-term nusinersen therapy [26]. At day 2010, the total HFMSE score had improved in patients with 5q SMA type 2 (n = 7) and were stable in those with 5q SMA type 3 (n = 9) compared with baseline. At this timepoint, one patient with 5q SMA type 2 had gained the ability to walk independently, while the mean change from baseline in the 6MWT distance had improved at day 2010 in patients with 5q SMA type 3 (n = 7). At day 1770, 83% of 12 patients with 5q SMA type 3 had experienced a clinically meaningful improvement (≥ 30 m) in the 6MWT distance [26].
2.2.2.2 In Real-World Studies
Data from the real-world setting support the effectiveness of nusinersen in improving motor function in children with later-onset 5q SMA. For example, in the largest (n = 73–77) of these studies, which enrolled patients with 5q SMA type 2 who were aged > 2.5 to < 18 years, nusinersen significantly (p < 0.001) improved mean HFMSE and RULM scores from baseline (mean baseline values of 10.32 and 13.50) by a mean of 1.90 and 1.59 at month 12 [27]. According to a multivariate linear regression analysis, patient age and baseline score, but not SMN2 copy number, appear to be predictive of changes in HFMSE and RULM scores [27]. These findings are generally supported by those from a cohort of 5q SMA type 3 patients aged < 18 years participating in a recent observational study [28]. Mean HFMSE and RULM scores were significantly (p < 0.05) improved, while the mean 6MWT distance was stable after 12 months’ nusinersen therapy (respective mean change from baseline of 1.53, 1.26, and 10.32 m; mean baseline values of 43.05, 31.90, and 319.87 m) in these patients (n = 59, 45 and 34) [28].
Results from various real-world studies [29,30,31,32,33] also support the effectiveness of nusinersen in improving motor function in adults with later-onset 5q SMA. For example, in the largest of these (a multicentre, observational study [29]), the treatment of patients (aged 16–65 years with a genetic diagnosis of 5q SMA; 98% of 124 evaluable patients had 5q SMA type 2 or 3) with nusinersen for ≥ 6 months was associated with significant (p ≤ 0.0014) mean changes from baseline in the mean total HFMSE score (primary endpoint) [n = 124, 92 and 57] and the RULM score (n = 120, 90 and 58) at the 6-, 10- and 14-month assessments. Baseline total HFMSE score was significantly (p = 0.0006) correlated with an improvement in total HFMSE score at 6 months (r = 0.3); there was no correlation between motor function improvement and patient age. Clinically meaningful improvements in the total HFMSE score were seen in 28% of patients at month 6, 35% at month 10 and 40% at month 14. These improvements were seen at the 6-, 10- and 14-month assessments in more patients with 5q SMA type 3 (n = 77, 60 and 37) than type 2 (n = 45, 30 and 20) [month 6: 30% vs 2%; month 10: 32% vs 7%; month 14: 41% vs 5%). There were also significant (p ≤ 0.0022) mean improvements from baseline in the 6MWT distance (baseline values of 321.76, 353.03 and 371.43 m; n = 124, 92 and 57) of 22.1 m at month 6 (n = 47), 31.1 m at month 10 (n = 37) and 46.0 m at month 14 (n = 25) [29]. Likewise, a retrospective study (nusinersen therapy commenced at > 18 years of age for ≥ 6 months) reported significant (p ≤ 0.012) median improvements from baseline at months 6, 10 and 14 in the total HFMSE score and at months 10 and 14 in the RULM score in patients with 5q SMA type 3 (n = 103), but not in those with 5q SMA type 2 (n = 13) [30]. In patients with 5q SMA type 3 who were able to take at least a few steps independently or with aids (e.g. a cane), but without the assistance of others, there were significant (p ≤ 0.016) median improvements from baseline in the 6MWT distance (baseline value of 322 m) of 11 m at month 6 (n = 48), 25 m at month 10 (n = 35) and 20 m at month 14 (n = 24) [30]. Bulbar dysfunction did not appear to deteriorate during nusinersen treatment in a prospective German study in 22 adults with 5q SMA type 2 or 3 who had evidence of bulbar dysfunction prior to commencing therapy [3]. Bulbar function was assessed using the Sydney Swallow Questionnaire (SSQ) and the bulbar subscore of the Amyotrophic Lateral Sclerosis Functional Rating Scale Revised (ALSFRS-R) before initiating nusinersen, and after 6 and 14 months of treatment [3].
Patient satisfaction with nusinersen was assessed using the Treatment Satisfaction Questionnaire for Medication Version 1.4 in 91 patients (aged 10–65 years with a genetic diagnosis of 5q SMA; 96% of patients had 5q SMA type 2 or 3) participating in a multicentre observational study [34]. Following a median treatment duration of 10 months, most patients were at least ‘somewhat satisfied’ with nusinersen (≈ 91%), its ability to prevent or treat the disease (> 90%) and the way it relieves symptoms (≈ 80%). However, over half (≈ 55%; estimated from a figure) confirmed nusinersen was difficult to use, which likely reflects the need for multiple lumbar punctures [34].
2.2.3 Mixed Population
A randomized, double-blind, sham-controlled, phase II study (CS7; EMBRACE) [35] evaluated nusinersen in patients with infantile- or later-onset 5q SMA who did not meet the eligibility criteria for ENDEAR or CHERISH. EMBRACE enrolled patients with a genetic diagnosis of 5q SMA and one of the following: three copies of SMN2 who were aged ≤ 6 months at symptom onset; two copies of SMN2 who were aged ≤ 6 months at symptom onset and > 7 months at screening; or two or three copies of SMN2 who were aged > 6 months at symptom onset and ≤ 18 months at screening. Randomization to treatment arms [nusinersen (12 mg dose equivalent, administered intrathecally on days 1, 15, 29, 64, 183 and 302) or a sham procedure] was stratified by age at symptom onset (≤ 6 or > 6 months). The 14-month double-blind period of EMBRACE (hereafter referred to as part 1) was halted (data cut-off date of 30 March 2017) following the early termination of ENDEAR, with eligible EMBRACE participants (those who had completed the final part 1 evaluation among other criteria) transferred to a 28-month open-label extension (part 2; consisting of a 24-month treatment period and an ≈ 4-month follow-up period). In part 2, patients originally treated with nusinersen in part 1 continued to receive nusinersen every 4 months while those originally treated with sham control in part 1 commenced nusinersen therapy, receiving doses on days 1, 15, 29, 64, and then every 4 months thereafter. Overall, a median of 10 (range 8–12) nusinersen doses were administered. Part 2 of EMBRACE was terminated early to permit participants to transition to the SHINE study [35].
Findings from EMBRACE are consistent with those from ENDEAR and CHERISH, and support the use of nusinersen among a broad population of infants and children with 5q SMA (i.e. not just those fulfilling ENDEAR and CHERISH enrolment criteria) [35]. Despite the small sample size [n = 14 (nusinersen) and 7 (sham control)] and the shortened duration of part 1, 79% of nusinersen recipients and 29% of sham control recipients were motor milestone responders at the time of the part 1 final assessment. At the time of the final assessment in part 2, 93% of 14 patients originally randomized to nusinersen and 83% of 6 patients originally randomized to sham control were responders. Outcomes were independent of age at the onset of SMA [35].
These findings are generally supported by data from 12 ± 2 months of nusinersen therapy in a multicentre retrospective study (n = 123; 34 and 89 patients had 5q SMA type 1 or 2), which reported significant (p < 0.001) improvements from baseline in motor function (HINE-2 scores in patients aged < 2 years and Motor Function Measure scores in patients aged > 2 years), but not in the number of patients requiring ventilatory or nutritional support [36].
3 Safety and Tolerability of Nusinersen
Nusinersen demonstrated a favourable safety profile in clinical studies in presymptomatic [8] and symptomatic [24, 35, 37, 38] patients with 5q SMA, and in real-world studies in symptomatic patients with later-onset 5q SMA [29, 30]. Most adverse events (AEs) associated with nusinersen administration via lumbar puncture (e.g. back pain, headache, vomiting) occur within 72 h of the procedure [23]. Serious lumbar puncture complications (e.g. serious infection) have been reported in the post-marketing setting [23, 39].
Results from an integrated analysis [37] of the CS1, CS2, CS3A, CS10, CS12, ENDEAR and CHERISH studies in symptomatic patients with infantile- (n = 141) or later-onset (n = 182) 5q SMA found that most AEs and serious AEs (SAEs) were consistent with the nature and frequency of events commonly seen with the disease itself or the lumbar puncture procedure. At least one AE occurred in 96% of 240 nusinersen recipients (total nusinersen exposure of 375.9 patient–years) and 99% of 83 sham control recipients, with the most common (occurring with a higher incidence in the nusinersen group) being pyrexia (48% vs 47%), upper respiratory tract infection (URTI; 38% vs 34%), nasopharyngitis (25% vs 23%), vomiting (24% vs 16%), headache (22% vs 4%) and constipation (20% vs 17%) [37].
One AE (procedural nausea) in a nusinersen recipient was considered by the study investigators to be treatment related [37]. Possibly treatment-related AEs were reported in 17% of nusinersen recipients (vs 12% of sham control recipients), with headache (n = 9), pyrexia (n = 8), back pain (n = 7), post-lumbar puncture syndrome (n = 4), vomiting (n = 3) and tachycardia (n = 2) occurring in more than one patient. SAEs were reported in 41% of nusinersen recipients and 61% of sham control recipients [37].
Several toxicities associated with ASOs, including increased alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, proteinuria and thrombocytopenia, have been seen in clinical studies [37]. They appear to result from the chemical structure of the ASO and the individual nucleic acid sequence, are related to the dose (i.e. systemic exposure) and are usually self-limiting following treatment cessation. In the integrated analysis, there was no systemic evidence of such toxicities. In nusinersen recipients, there were no cases of severe, sustained thrombocytopenia, no reports of glomerulonephritis, nephrotic syndrome or renal failure, and median alkaline phosphatase, ALT, AST, and direct and indirect bilirubin levels were stable over time [37]. According to a recent retrospective analysis of laboratory parameter data from 50 adults with 5q SMA type 2 or 3 receiving nusinersen, there was no evidence of clinically relevant thrombocytopenia, coagulopathies, or renal or hepatic toxicities [40].
The nature and frequency of AEs with nusinersen in presymptomatic patients in NURTURE was consistent with those reported in the integrated analysis [8].
Preliminary data from SHINE suggest that the safety findings of nusinersen over the longer term are consistent with previously reported results [24, 38, 41]. Among patients with infantile-onset 5q SMA participating in SHINE [data cut-off date of 27 August 2019; median time on study of 3.7 years for patients aged ≤ 7 months at screening (n = 100) and 3.0 years for patients aged > 7 months at screening (n = 37)], SAEs occurred in 91% of patients in the ≤ 7-months’ screening group and 84% of those in the > 7-months’ screening group [41]. Most SAEs were related to the disease; none were considered to be related to nusinersen therapy [41]. Among patients with later-term onset 5q SMA who started nusinersen therapy in the CS1, CS2, EMBRACE, CHERISH and/or SHINE studies (data cut-off date of 27 August 2019; median time of observation of 4.0 years), the incidence of AEs typically associated with the lumbar puncture procedure appeared to decrease/stabilize over time [38]. Post-lumbar puncture syndrome was reported in 13% of 190 patients in year 1 and in 6–23% of 35–182 patients in years 2–7. Most events were mild to moderate in severity, with serious post-lumbar puncture syndrome seen in 2% of patients in year 1 and in 3% in each subsequent year. In general, the incidence of headache (26% in year 1 and 10–17% in years 2–7) and vomiting (20% in year 1 and 7–14% in years 2–7) decreased over time; most cases were mild in severity. No cases of meningitis or hydrocephalus were reported [38].
3.1 Immunogenicity
As with all oligonucleotides, there is a potential for immunogenicity with nusinersen [39]. In 346 patients with baseline and post-baseline anti-drug antibody (ADA) assessments, 15 (4%) patients developed ADAs [23]. In four patients the ADAs were transient and in five they were persistent; the ADA response in six patients had not been classified by the data cut-off date. Although the immunogenicity impact on safety has not been formally analysed (as the number of patients with ADAs is low), no AEs of interest were identified among treatment-emergent ADA-positive cases [23].
4 Dosage and Administration of Nusinersen
Nusinersen is approved for the treatment of 5q SMA in numerous countries worldwide, including those of the EU [23], as well as the USA [39]. The recommended dose is 12 mg, administered intrathecally [23, 39]. Therapy should be initiated with four loading doses (with the first three administered at 14-day intervals and the fourth administered 30 days [39] or 35 days [23] after the third), with maintenance doses administered once every 4 months thereafter. A clinically meaningful benefit may not be experienced by patients with profound hypotonia and respiratory failure at birth (where nusinersen has not been studied) due to severe SMN protein deficiency, and the need for continued therapy should be regularly reviewed based on the patient’s clinical presentation and therapeutic response [23].
Local prescribing information should be consulted for detailed information regarding administration procedures, missed or delayed doses, contraindications, use in special patient populations, and warnings and precautions.
5 Place of Nusinersen in the Management of 5q SMA
While symptomatic and supportive care continue to be important components of the ongoing management of patients with 5q SMA, the approval of disease modifying agents has significantly changed the treatment landscape. Nusinersen was the first to be approved for the treatment of 5q SMA [7, 23, 39], followed by onasemnogene abeparvovec (an adeno-associated viral vector-based gene therapy that delivers a functional copy of SMN1 to the motor neuron cells of patients with 5q SMA) [42,43,44] and the small molecule risdiplam (which is available as a once-daily oral solution) [45,46,47]. Like nusinersen (Table 1), risdiplam [45, 46] acts by modifying SMN2 pre-mRNA splicing, resulting in an increase in the production of full-length SMN protein, although nusinersen binds to one site (in intron 7) while risdiplam binds to two sites (one in intron 7 and one in exon 7) on SMN2 pre-mRNA [48]. Both intrathecal nusinersen [23, 39] and oral risdiplam [45, 46] require repeated administrations (with nusinersen maintenance doses administered once every 4 months and risdiplam once daily), while onasemnogene abeparvovec requires only a single intravenous infusion [42, 43].
In presymptomatic infants with 5q SMA and two or three copies of SMN2, substantial clinical benefits were associated with the early (i.e. immediately after establishing a genetic diagnosis) initiation of nusinersen (Sect. 2.1). At a data cut-off date of 19 February 2020, all NURTURE participants (n = 25; median age at last visit of 3.8 years) were alive without permanent ventilation, and most were able to walk independently and swallow. Such milestones are incongruent with the natural history of this patient population and highlight the value of an early diagnosis and treatment [8]. Further data from studies in presymptomatic patients are awaited with interest.
In clinical studies in symptomatic patients with infantile- or later-onset 5q SMA (Sect. 2.2), nusinersen improved motor function relative to sham control, with the likelihood of EFS (i.e. survival without the use of permanent assisted ventilation) and overall survival also improved in the former patient population. Interim longer-term data showed that motor function outcomes were maintained or improved in patients with infantile- and later-onset 5q SMA regardless of their initial treatment regimen, although the greatest benefits were seen in infantile-onset 5q SMA patients in whom therapy was initiated early. Eligibility criteria for these studies (ENDEAR and CHERISH) were strictly prescribed; however, findings consistent with these were seen in a study in a symptomatic patient population who did not meet ENDEAR and CHERISH eligibility criteria (Sect. 2.2.3), supporting the use of nusinersen in a broad population of infants and children with 5q SMA.
Limitations (e.g. natural disease course of 5q SMA, observational or retrospective design, lack of a comparator, missing data, and/or floor and ceiling effects on evaluating scales) notwithstanding, real-world studies in symptomatic patients with infantile- and later-onset 5q SMA (Sect. 2.2) support the effectiveness of nusinersen on motor function outcomes, including in adults with later-onset 5q SMA. With evidence still emerging on the efficacy of nusinersen in adults with 5q SMA [31,32,33], caution is advised when counselling these patients and tailoring their expectations in relation to their 5q SMA phenotype (type 2 or 3) and ambulatory level [49]. Further natural history data in this patient population would be of interest [49]. Also of interest is the effect of disease modifying agents on impaired bulbar function. Recent data from a small retrospective analysis suggest that bulbar dysfunction (assessed using the Paediatric Functional Oral Intake Scale) persists in children with 5q SMA type 1 [50], and did not deteriorate in adults with 5q SMA types 2 and 3 (Sect. 2.2.2.2) treated with nusinersen. Further studies, particularly those of a longer duration, evaluating bulbar function would be of value [3].
In patients with 5q SMA, respiratory problems are the major cause of hospitalization, morbidity and mortality [51]. Although the strict eligibility criteria of CHERISH excluded patients with respiratory insufficiency [22], the effects of nusinersen on lung function (Table 1) may improve survival and reduce the need for respiratory support [52]. Further studies assessing the effect of nusinersen on respiratory function would be of interest.
Nusinersen demonstrated a favourable safety profile in clinical studies in presymptomatic and symptomatic patients and in real-world studies in symptomatic patients with later-onset 5q SMA (Sect. 3). Most AEs were consistent with the disease itself or the lumbar puncture procedure, with AEs associated with the administration of nusinersen mostly occurring within 72 h. Preliminary longer-term data suggest that the safety findings of nusinersen in patients with infantile and later-onset 5q SMA are consistent with previously reported results (Sect. 3). In the post-marketing setting, hydrocephalus has been reported in patients receiving nusinersen [23, 39]. According to a recent retrospective analysis of US electronic health record data from patients with 5q SMA (n = 5354) and matched non-5q SMA controls prior to the approval of nusinersen, patients with 5q SMA had an approximately fourfold increased risk of hydrocephalus compared with controls, regardless of sex and age [53]. This suggests that hydrocephalus may be part of the natural history of 5q SMA in some patients, although whether there is a causal relationship between 5q SMA and hydrocephalus is not yet known [53]. Further studies are awaited with interest.
5q SMA is associated with a substantial economic burden, and pharmacoeconomic considerations play an important role in determining pricing and reimbursement in a contemporary healthcare system [54]. Pharmacoeconomic analyses using standard willingness-to-pay thresholds indicate that none of the currently available treatments for 5q SMA are cost effective [55,56,57]. Interpreting such estimates is difficult because of the limited evidence supporting longer-term benefits, the difficulty in clearly distinguishing between the SMA subtypes and the difference in what can be achieved for these various patients without treatment [58]. Economic modelling in a rare disease is often challenging and frequently does not portray the full picture of the unmet medical needs of the community or adequately address how to objectively assign monetary value to quality of life [59]. The UK’s National Institute for Health and Care Excellence (NICE) also acknowledged that cost-effectiveness estimates for nusinersen were above the range normally considered cost effective by NICE, initially recommending it for a prescribed 5q SMA patient population as part of a Managed Access Agreement [58]. Following a review of data collected as part of this Managed Access Agreement, NICE has extended the clinical eligibility criteria for nusinersen [60].
In conclusion, nusinersen improved motor function in presymptomatic and symptomatic patients with 5q SMA, has maintained this benefit in those who have received treatment longer term and showed a favourable safety profile in these patient populations. Thus, nusinersen continues to represent an important treatment option among a broad range of 5q SMA patients, with its early initiation associated with clinically meaningful benefits.
Data Selection Nusinersen: 647 records identified | |
|---|---|
Duplicates removed | 134 |
Excluded during initial screening (e.g. press releases; news reports; not relevant drug/indication; preclinical study; reviews; case reports; not randomized trial) | 243 |
Excluded during writing (e.g. reviews; duplicate data; small patient number; nonrandomized/phase I/II trials) | 203 |
Cited efficacy/tolerability articles | 29 |
Cited articles not efficacy/tolerability | 38 |
Search Strategy: EMBASE, MEDLINE and PubMed from 2018 to present. Previous Adis Drug Evaluation published in 2018 was hand-searched for relevant data. Clinical trial registries/databases and websites were also searched for relevant data. Key words were nusinersen, Spinraza, spinal muscular atrophy. Records were limited to those in English language. Searches last updated 19 October 2021 | |
Change history
27 December 2021
A Correction to this paper has been published: https://doi.org/10.1007/s40263-021-00889-8
References
US Library of Medicine. Genetics Home Reference: spinal muscular atrophy. 2020. http://ghr.nlm.nih.gov/condition/spinal-muscular-atrophy. Accessed 19 Apr 2021.
Groen EJN, Talbot K, Gillingwater TH. Advances in therapy for spinal muscular atrophy: promises and challenges. Nat Rev Neurol. 2018;14(4):214–24.
Brakemeier S, Stolte B, Thimm A, et al. Assessment of bulbar function in adult patients with 5q-SMA Type 2 and 3 under treatment with nusinersen. Brain Sci. 2021;11(9):1244.
Wadman RI, van der Pol WL, Bosboom WM, et al. Drug treatment for spinal muscular atrophy type I. Cochrane Database Syst Rev. 2019;12(Cd006281):1–46.
Wadman RI, van der Pol WL, Bosboom WM, et al. Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst Rev. 2020;1(Cd006282):1–97.
Volpe JJ. Infantile spinal muscular atrophy: the potential for cure of a fatal disease. J Neonatal Perinatal Med. 2021;14(2):153–7.
Hoy SM. Nusinersen: a review in 5q spinal muscular atrophy. CNS Drugs. 2018;32(7):689–96.
De Vivo DC, Bertini E, Swoboda KJ, et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: interim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. 2019;29(11):842–56.
US National Institutes of Health. ClinicalTrials.gov identifier NCT02386553. 2021. https://clinicaltrials.gov/. Accessed 23 Aug 2021.
Biogen Inc. New results from landmark NURTURE study show that pre-symptomatic SMA patients treated with SPINRAZA® (nusinersen) continue to demonstrate sustained benefit from treatment. 2020. https://www.biogen.com/. Accessed 8 Oct 2021.
Swoboda KJ, Sansone VA, De Vivo DC, et al. Preserved swallowing function in infants who initiated nusinersen treatment in the presymptomatic stage of SMA: results from the NURTURE study [abstract]. In: 2021 MDA Virtual Clinical & Scientific Conference. 2021.
Finkel RS, Chiriboga CA, Vajsar J, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388(10063):3017–26.
Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723–32.
Kuntz N, Tizzano E, Topaloglu H, et al. Time to motor function response among nusinersen-treated infants from the ENDEAR study [abstract no. 141]. Ann Neurol. 2018;84(Suppl 22):S389–90.
Finkel R, Kirschner J, Mercuri E, et al. Longer-term effects of nusinersen on motor function outcomes based on age at treatment initiation [abstract no P.264]. Neuromuscul Disord. 2020;30(Suppl 1):S123–4.
Servais L, Finkel RS, Kirschner J, et al. Longer-term effects of nusinersen on motor function outcomes based on age at treatment initiation [abstract no. 29]. Dev Med Child Neurol. 2021;63(Suppl 1):23.
Servais L, Finkel RS, Castro D, et al. Predictive factors of nusinersen treatment response in infantile-onset spinal muscular atrophy: results from the ENDEAR/SHINE studies [abstract]. In: 2021 MDA Virtual Clinical & Scientific Conference. 2021.
Pane M, Palermo C, Messina S, et al. Nusinersen in type 1 SMA infants, children and young adults: preliminary results on motor function. Neuromuscul Disord. 2018;28(7):582–5.
Messina S, Sframeli M, Bruno C, et al. Nutritional status in SMA type 1 over a 6 month follow-up in nusinersen expanded access program (EAP) [abstract]. Acta Myol. 2018;37(1):53–4.
Pane M, Coratti G, Sansone VA, et al. Nusinersen in type 1 spinal muscular atrophy: twelve-month real-world data. Ann Neurol. 2019;86(3):443–51.
Pane M, Coratti G, Sansone VA, et al. Type I SMA “new natural history”: long-term data in nusinersen-treated patients. Ann Clin Transl Neurol. 2021;8(3):548–57.
Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378(7):625–35.
Biogen Netherlands B.V. Spinraza 12 mg solution for injection: EU summary of product characteristics. 2021. http://www.ema.europa.eu/. Accessed 16 Mar 2021.
Mercuri E, Darras BT, Chiriboga CA, et al. Longer-term treatment with nusinersen: results in later-onset spinal muscular atrophy from the SHINE study [abstract no. 265]. J Neuromuscular Dis. 2021;8(S1):S67.
Darras BT, Chiriboga CA, Iannaccone ST, et al. Nusinersen in later-onset spinal muscular atrophy: long-term results from the phase 1/2 studies. Neurology. 2019;92(21):e2492–506.
Tulinius M, Darras B, Chiriboga C, et al. Longer-term improved/maintained motor function in nusinersen-treated children with later-onset SMA in CS2/CS12 and SHINE [abstract no EPR-279]. Eur J Neurol. 2021; 28(Suppl 1):394.
Coratti G, Pane M, Lucibello S, et al. Age related treatment effect in type II spinal muscular atrophy pediatric patients treated with nusinersen. Neuromuscul Disord. 2021;31(7):596–602.
Pera MC, Coratti G, Bovis F, et al. Nusinersen in pediatric and adult patients with type III spinal muscular atrophy. Ann Clin Transl Neurol. 2021;8(8):1622–34.
Hagenacker T, Wurster CD, Günther R, et al. Nusinersen in adults with 5q spinal muscular atrophy: a non-interventional, multicentre, observational cohort study. Lancet Neurol. 2020;19(4):317–25.
Maggi L, Bello L, Bonanno S, et al. Nusinersen safety and effects on motor function in adult spinal muscular atrophy type 2 and 3. J Neurol Neurosurg Psychiatry. 2020;91(11):1166–74.
Duong T, Wolford C, McDermott MP, et al. Nusinersen treatment in adults with spinal muscular atrophy. Neurol Clin Pract. 2021;11(3):e317–27.
Elsheikh B, Severyn S, Zhao S, et al. Safety, tolerability, and effect of nusinersen treatment in ambulatory adults with 5q-SMA. Front Neurol. 2021;12:650635.
Elsheikh B, Severyn S, Zhao S, et al. Safety, tolerability, and effect of nusinersen in non-ambulatory adults with spinal muscular atrophy. Front Neurol. 2021;12:65032.
Osmanovic A, Ranxha G, Kumpe M, et al. Treatment satisfaction in 5q-spinal muscular atrophy under nusinersen therapy. Ther Adv Neurol Disord. 2021;14:1756286421998902.
Acsadi G, Crawford TO, Muller-Felber W, et al. Safety and efficacy of nusinersen in spinal muscular atrophy: the EMBRACE study. Muscle Nerve. 2021;63:668–77.
Audic F, de la Banda MGG, Bernoux D, et al. Effects of nusinersen after one year of treatment in 123 children with SMA type 1 or 2: a French real-life observational study. Orphanet J Rare Dis. 2020;15(148):1–10.
Darras BT, Farrar MA, Mercuri E, et al. An integrated safety analysis of infants and children with symptomatic spinal muscular atrophy (SMA) treated with nusinersen in seven clinical trials. CNS Drugs. 2019;33(9):919–32.
Farrar M, Castro D, Finkel R, et al. Longer-term safety data in individuals with later-onset SMA support the favourable tolerability profile of nusinersen [abstract no. EPR-123]. Eur J Neurol. 2021;28(Suppl 1):287.
Biogen Inc. SPINRAZA (nusinersen) injection, for intrathecal use: US prescribing information. 2020. http://www.fda.gov/. Accessed 16 Mar 2021.
Stolte B, Nonnemacher M, Kizina K, et al. Nusinersen treatment in adult patients with spinal muscular atrophy: a safety analysis of laboratory parameters. J Neurol. 2021;268(12):4667–79.
Sansone V, Finkel R, Tulinius M, et al. Integrated analysis of annualized incidence of serious adverse events in infantile-onset SMA treated with nusinersen [abstract no. 765]. J Neuromuscular Dis. 2021;8(Suppl 1):S152–3.
Novartis Gene Therapies EU Limited. Zolgensma 2 × 1013 vector genomes/mL solution for infusion: EU summary of product characteristics. 2021. http://www.ema.europa.eu/. Accessed 17 Aug 2021.
AveXis Inc. ZOLGENSMA® (onasemnogene abeparvovec-xioi): US prescribing information. 2021. https://www.fda.gov/vaccines-blood-biologics/zolgensma. Accessed 25 May 2021.
Hoy SM. Onasemnogene abeparvovec: first global approval. Drugs. 2019;79(11):1255–62.
Roche Registration GmbH. Evrysdi 0.75 mg/mL powder for oral solution: EU summary of product characteristics. 2021. http://www.ema.europa.eu/. Accessed 25 May 2021.
Genentech Inc. EVRYSDI® (risdiplam) for oral solution: US prescribing information. 2021. https://www.fda.gov/. Accessed 25 May 2021.
Dhillon S. Risdiplam: first approval. Drugs. 2020;80(17):1853–8.
Messina S, Sframeli M. New treatments in spinal muscular atrophy: positive results and new challenges. J Clin Med. 2020;9(7):13.
Mercuri E, Sansone V. Nusinersen in adults with spinal muscular atrophy: new challenges. Lancet Neurol. 2020;19(4):283–4.
Weststrate H, Stimpson G, Thomas L, et al. Bulbar function evolution in SMA type 1 children treated with nusinersen [abstract no. EP.276]. Neuromuscul Disord. 2021;31(Suppl 1):S133–4.
LoMauro A, Mastella C, Alberti K, et al. Effect of nusinersen on respiratory muscle function in different subtypes of type 1 spinal muscular atrophy. Am J Respir Crit Care Med. 2019;200(12):1547–50.
Chacko A, Sly PD, Ware RS, et al. Effect of nusinersen on respiratory function in paediatric spinal muscular atrophy types 1-3. Thorax. 2021. https://doi.org/10.1136/thoraxjnl-2020-216564.
Viscidi E, Wang N, Juneja M, et al. The incidence of hydrocephalus among patients with and without spinal muscular atrophy (SMA): results from a US electronic health records study. Orphanet J Rare Dis. 2021;16(1):207.
Dangouloff T, Botty C, Beaudart C, et al. Systematic literature review of the economic burden of spinal muscular atrophy and economic evaluations of treatments. Orphanet J Rare Dis. 2021;16(1):47.
Pearson SD, Thokala P, Stevenson M, et al. The effectiveness and value of treatments for spinal muscular atrophy: a summary from the Institute for Clinical and Economic Review’s New England Comparative Effectiveness Public Advisory Council. J Manag Care Spec Pharm. 2019;25(12):1300–6.
Dean R, Jensen I, Cyr P, et al. An updated cost-utility model for onasemnogene abeparvovec (Zolgensma(R)) in spinal muscular atrophy type 1 patients and comparison with evaluation by the Institute for Clinical and Effectiveness Review (ICER). J Mark Access Health Policy. 2021;9(1):1889841.
Broekhoff TF, Sweegers CCG, Krijkamp EM, et al. Early cost-effectiveness of onasemnogene abeparvovec-xioi (Zolgensma) and nusinersen (Spinraza) treatment for spinal muscular atrophy I in the Netherlands with relapse scenarios. Value Health. 2021;24(6):759–69.
UK National Institute for Health and Care Excellence. Nusinersen for treating spinal muscular atrophy: technology appraisal guidance [TA588]. 2019. https://www.nice.org.uk/guidance/ta588. Accessed 27 May 2021.
Ollendorf DA, Chapman RH, Pearson SD. Evaluating and valuing drugs for rare conditions: no easy answers. Value Health. 2018;21(5):547–52.
UK National Institute for Health and Care Excellence. NICE announces more people eligible for nusinersen following review of Managed Access Agreement. 2021. https://www.nice.org.uk/. Accessed 31 May 2021.
Darras BT, Crawford TO, Finkel RS, et al. Neurofilament as a potential biomarker for spinal muscular atrophy. Ann Clin Transl Neurol. 2019;6(5):932–44.
Finkel R, Sumner C, Darras B, et al. Association between plasma phosphorylated neurofilament heavy chain and efficacy endpoints in the nusinersen NURTURE study [abstract no. 173]. Ann Neurol. 2019;86(Suppl 23):S122.
Sumner CJ, Darras BT, Muntoni F, et al. Association of phosphorylated neurofilament heavy chain (pNF-H) levels with motor function achievement in individuals with spinal muscular atrophy (SMA) treated with nusinersen [abstract no. S27.009]. Neurology. 2019;92(15 Suppl 1).
Darras BT, Sumner CJ, Muntoni F, et al. Plasma phosphorylated neurofilament heavy chain levels over time in participants with infantile-and later-onset SMA: data from the SHINE study [abstract no. 1692]. Neurology. 2020;94(15 Suppl).
Bonanno S, Marcuzzo S, Malacarne C, et al. Circulating myomiRs as potential biomarkers to monitor response to nusinersen in pediatric SMA patients. Biomedicines. 2020;8(2):21.
Gómez-Garcia de la Banda M, Amaddeo A, Khirani S, et al. Assessment of respiratory muscles and motor function in children with SMA treated by nusinersen. Pediatr Pulmonol. 2020;56(1):299–306.
Sansone VA, Pirola A, Albamonte E, et al. Respiratory needs in patients with type 1 spinal muscular atrophy treated with nusinersen. J Pediatr. 2020;219:223-8.e4.
Acknowledgments
During the peer review process, the manufacturer of nusinersen was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of interest
Sheridan M. Hoy is a salaried employee of Adis International Ltd/Springer Nature, and declares no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Additional information
The manuscript was reviewed by: C. Campbell, Department of Paediatrics, University of Western Ontario, London, ON, Canada; T. Hagenacker, Department of Neurology, University Hospital Essen, Essen, Germany.
The original online version of this article was revised due to a retrospective Open Access order.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Hoy, S.M. Nusinersen: A Review in 5q Spinal Muscular Atrophy. CNS Drugs 35, 1317–1328 (2021). https://doi.org/10.1007/s40263-021-00878-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-021-00878-x