
Abstract
Oral siponimod (Mayzent®), a next-generation, selective sphingosine 1-phosphate receptor (S1PR) 1 and 5 modulator, is approved in several countries for the treatment of secondary progressive multiple sclerosis (SPMS), with specific indications varying between individual countries. In the pivotal EXPAND trial (median duration double-blind treatment 18 months) in a broad spectrum of patients with SPMS, once-daily oral siponimod 2 mg (initial dose titration over 6 days) was significantly more effective than placebo in reducing clinical and MRI-defined outcomes of disease activity and disability progression, including 3-month confirmed disability progression on the Expanded Disability Status Scale (EDSS), and was generally well tolerated in the core phase of the study. These beneficial effects of siponimod appeared to be sustained during up to 5 years of treatment in the ongoing open-label extension phase of EXPAND. The safety profile of siponimod is similar to that of other agents in its class, including adverse events of special interest (i.e. those known to be associated with S1PR modulators). No new safety signals were identified during up to 5 years’ treatment in the open-label extension phase. Albeit further long-term efficacy and safety data from the real-world setting are required to fully define its role, given the paucity of current treatment options and its convenient dosage regimen, siponimod represents an important emerging option for the treatment of adult patients with SPMS with active disease evidenced by relapses or imaging-features of inflammatory activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Next-generation, highly-selective S1PR modulator; exhibits highly affinity binding at S1PR1 and S1PR5 |
Reduces the risk of disability progression on EDSS; improves other clinical and MRI-defined outcomes of disability progression and disease activity (vs placebo) |
Beneficial effects of treatment sustained |
Generally well tolerated, with a safety profile similar to that of other S1PR modulators |
1 Introduction
Multiple sclerosis (MS) is a common, autoimmune-mediated disease of the CNS, characterized by multifocal localized inflammation, demyelination/remyelination, neurodegenerative changes and glial scar formation [1, 2]. The disease is the leading cause of non-traumatic disability in young adults, with a mean age at diagnosis of ≈ 30 years and an estimated ≈ 2.2 million individuals affected worldwide in 2016 [3]. The disease has a significant impact on quality of life and imposes a considerable burden from a societal and healthpayer perspective [1,2,3]. Although its exact course is highly heterogeneous in nature, most individuals (≈ 85%) present with relapsing-remitting MS (RRMS) at onset, which is characterized by recurrent relapses and remission of neurological symptoms. Within 10 to 20 years, about two-thirds of patients will progress to secondary progressive MS (SPMS), which is associated with gradual worsening of disability, progressive neurological deterioration, and no or few relapses (≈ 30% of patients experience relapses after progression has started [2]) [1,2,3,4]. To date, although MS is treatable, there is no curative therapy. An improved understanding of the pathophysiology of the disease has resulted in the development of several disease-modifying therapies (DMTs) that slow the progression of symptoms, reduce the frequency of relapses and limit disability progression in patients with RRMS [1,2,3]. However, management of progressive MS, including SPMS, remains challenging, with limited options available for treating these forms of the disease [2, 4,5,6].
Extensive evidence has firmly established the pivotal role of sphingosine 1-phosphate (S1P) in several physiological systems, including immune, cardiovascular and CNS processes, with S1P exerting its effects via five G protein-coupled receptors [S1P receptors (S1PR1–5)] that are differentially expressed in various cell types and tissues [7,8,9]. Given the key role of S1P in mediating multiple pathways, including lymphocyte trafficking, vascular homeostasis, microglial activation, neuronal interactions, axonal growth, oligodendrocyte survival, myelination and integrity of the blood-brain barrier, the S1PR axis has been implicated in several immune-mediated disorders, including MS (as reviewed by Chaudhry et al. [8]) [8, 9]. Thus, modulation of S1PRs offers a novel targeted approach to treating MS, with fingolimod the first S1PR functional antagonist (i.e. the S1PR modulators) approved for the treatment of RRMS. However, fingolimod is a non-selective functional antagonist with activity at S1PR1, 3, 4, 5 and, as such, is associated with known adverse events such as bradycardia, atrioventricular nodal block and macular edema [8]. Fingolimod activity at S1PR3 is considered to be responsible for the Mobitz type 1 atrioventricular nodal block [8] and inducing macular edema (via increasing vascular permeability in the retina) [8, 10]. Hence, there is a drive to develop more selective S1PR modulators, optimized for potency against S1PR1 and lacking activity against S1PR3 [8, 9].
Oral siponimod (Mayzent®), a S1PR3-sparing, next-generation S1PR modulator, is approved in several countries, including EU countries [11] and the USA [12], for the treatment of SPMS/active SPMS (featured indication); specific indications vary between individual countries. This review, written from an EU perspective, focuses on therapeutic efficacy and tolerability data relevant to the use of oral siponimod in adults with SPMS and summarizes its pharmacological properties.
2 Pharmacodynamic Properties of Siponimod
Siponimod, a novel alkoxyimino derivative, is a highly selective S1PR1 and S1PR5 modulator, with both receptors (mainly S1PR1) pivotal in pathways regulating lymphocyte egress from lymph nodes, and S1PR1 (on astrocytes) and S1PR5 (on oligodendrocytes) playing a key role in myelination and CNS repair [13,14,15]. In vitro, the half maximal effective concentrations for siponimod at human S1PR1 and S1PR5 were 0.39 nmol/L and 0.98 nmol/L (vs > 10,000 nmol/L at S1PR2, > 1000 nmol/L at S1PR3 and 750 nmol/L at S1PR4) [14]. Relative to fingolimod, structural modifications in siponimod abrogate its activity at S1PR3 (potentially minimizing the risk of adverse effects) and negate the need for in vivo phosphorylation (results in a shorter elimination half-life) [13, 14].
In preclinical studies, siponimod dose dependently induced functional antagonism at S1PR1, leading to a reduction in peripheral lymphocytes consequent to inhibition of lymphocyte egress from lymph nodes; functional antagonism at S1PR1 results in long-lasting internalization of S1PR1 [13, 14]. Inhibition of egress from lymph nodes reduces circulating lymphocytes, thereby preventing T cells from entering the CNS and limiting central inflammation [11]. Preclinical studies also indicated that siponimod is associated with S1PR1-mediated anti-inflammatory effects on CNS lymphocytes and glial cells, and S1PR5-mediated repair effects on oligodendrocytes, thereby sparing S1PR3,4-mediated pro-inflammatory effects on astrocytes [16]. Other effects of siponimod included modulation of glial cell function and attenuation of demyelination [15], and reducing circulating monocytes independently of S1PR3 activity [17]. In a transgenic tadpole model, siponimod was associated with effective remyelination of the optic nerve [18, 19], with this promyelinating effect mediated via activity at S1PR5 [18]. In rodent models of experimental autoimmune encephalomyelitis (EAE; model for human MS), siponimod completely suppressed EAE, as evidenced by a reduction in disease scores [14, 20] (associated with down regulation of microglial activity [20]), and provided neuroprotective effects in the CNS and reduced grey matter inflammation [21]. Siponimod treatment was associated with recovery of GABAergic transmission alterations in the striatum of EAE mice, promoted parvalbumin-positive interneuron survival and reduced astrogliosis and microgliosis [21]. In EAE mice, siponimod demonstrated a more favourable CNS/blood drug exposure ratio than fingolimod (ratios 6–7 vs 20–30) [16]. In a mouse model of EAE-optic neuritis, siponimod treatment resulted in prophylactic and therapeutic effects, with an ≈ 80–95% reduction in clinical EAE scores [19, 22] and reduced degeneration of the inner retinal nerve fiber layers [22].
In healthy adults, siponimod was associated with dose-dependent (dose range 0.3–10 mg) reduction in peripheral CD4+ T cells, naive T cells, central memory T cells and B cells within 4–6 h due to the reversible sequestration of lymphocytes in lymphoid tissues [11, 14], with maximum reductions sustained during 28 days’ treatment and levels returning to normal within 1 week of stopping treatment [14]. With continued daily dosing, the lymphocyte counts continue to decrease, reaching a nadir median lymphocyte count of ≈ 0.560 cells/nL in non-Japanese patients with SPMS with a typical CYP2C9*1*1 or CYP2C9*1*2 genotype, corresponding to 20–30% of baseline values [11]. Low lymphocyte counts are maintained with daily dosing. In the vast majority of patients (90%), lymphocyte counts return to the normal range within 10 days of stopping therapy. After stopping siponimod treatment, residual lowering effects on peripheral lymphocyte count may persist for up to 3–4 weeks after the last dose [11].
The AMS04 study evaluated immunological profile changes with siponimod 2 mg/day (recommended maintenance dosage; Sect. 6 ) in a subgroup of 33 participants from the pivotal EXPAND trial (Sect. 4) [23]. At 6 and 12 months, siponimod treatment was associated with a lower percentage of CD4+ and B cells and a higher percentage of monocytes and natural killer cells than placebo; there was no clinically meaningful change in the percentage of CD8+ cells at these timepoints [23]. In another subgroup of patients from EXPAND (n = 525), siponimod treatment (> 21 months duration) was associated with reduced blood levels of neurofilament light chain (Nfl), a biomarker of neuronal damage [24]. Nfl levels in the blood were reduced by 5.7% in the siponimod group versus an increase of 9.2% in the placebo group (p = 0.0004), with similar beneficial effects of siponimod treatment observed irrespective of whether or not patients had experienced relapses in the 2 years prior to the study. Elevated Nfl levels are detected in the cerebrospinal spinal fluid (CSF) and blood of patients with MS [24].
There was a transient reduction in heart rate and atrioventricular conduction on treatment initiation of siponimod [25]. Based on in vitro human atrial myocyte studies, this effect is considered to be mechanistically related to siponimod-induced S1PR1 activation of G-protein-coupled inwardly rectifying potassium channels leading to cellular hyperpolarization and reduced excitability [14]. Siponimod therapy could be reinitiated within 96 h of treatment discontinuation without a need for retitration of the drug [25]. If maintenance treatment is interrupted for ≥ 4 days, siponimod treatment needs to be reinitiated with a new titration pack (Sect. 6) [11].
In healthy volunteers, concomitant treatment with siponimod and steady-state propranolol was associated with a more pronounced negative chronotropic effect than adding propranolol to steady-state siponimod [26].
In a thorough QT study in healthy volunteers, therapeutic (2 mg) and supratherapeutic (10 mg) doses of siponimod resulted in QTC prolongation of 7.8 ms and 7.2 ms [27]; these results did not suggest an arrhythmogenic potential related to QT prolongation with siponimod [11]. Categorical analysis revealed no treatment-emergent QTc values above 480 ms, no QTc increases from baseline of more than 60 ms and no QTc or QT values exceeded 500 ms [11].
3 Pharmacokinetic Properties of Siponimod
After multiple once-daily doses, siponimod exhibits dose-proportional pharmacokinetics across a dose range of 0.3–20 mg [11]. Siponimod is extensively absorbed (> 70%), with maximum plasma concentrations attained ≈ 4 h after multiple oral doses. Steady-state plasma concentrations are reached after ≈ 6 days, with levels at steady state ≈ 2- to 3-fold higher than after the initial dose. There was no clinically relevant effect of food on the absorption of siponimod. The mean volume of distribution of siponimod is 124 L, with the drug extensively bound (> 99%) to proteins in healthy volunteers and in patients with hepatic or renal impairment. In humans, the siponimod fraction found in plasma is 68% [11]. Siponimod readily crosses the blood-brain barrier [11, 28], with all five evaluated adults with SPMS participating in the EXPAND trial (Sect. 4) having detectable siponimod levels (nanomolar concentrations) in their CSF [28].
Siponimod is extensively metabolized by CYP2C9 (79.3%) and, to a lesser extent, by CYP3A4 (18.5%) [11, 29]. The pharmacological activity of the main metabolites M3 and M17 is not expected to contribute to the clinical efficacy or safety of siponimod in humans. Elimination of siponimod from the systemic circulation occurs primarily via oxidative metabolism and subsequent biliary excretion, with an apparent systemic clearance of 3.11 L/h in patients with MS and an elimination half-life (t½) of ≈ 30 h. Unchanged siponimod was not detected in the urine [11].
CYP2C9 is polymorphic and the genotype influences the fractional contributions of the two oxidative metabolic pathways to overall elimination of siponimod. Physiologically based pharmacokinetic (PBPK) modelling indicated a differential CYP2C9 genotype-dependent inhibition and induction of CYP3A4 pathways (Sect. 6) [11, 30]. In healthy volunteers, after a single 0.25 mg dose of siponimod, mean exposure was ≈ 2- and 4-fold higher in those who were poor metabolizers (i.e. with the CYP2C9*2*3 and CYP2C9*3*3 genotypes) than in those who were extensive metabolizers (i.e. CYP2C9*1*1 genotype), with a clinically relevant prolongation of t½ by 51 h and 126 h, respectively (vs 28 h) [31]. Hence, the CYP2C9 genotype of the patient should be determined prior to initiating siponimod treatment (Sect. 6) [11]. With decreased CYP2C9 metabolic activity in poor metabolizers, a larger effect of drugs that are CYP3A4 inhibitors or inducers on siponimod exposure is anticipated [11].
Using PBPK modelling, a maximum twofold increase in exposure to siponimod is predicted across genotypes with any type of CYP3A4 and CYP2C9 inhibitors, except for patients with a CYP2C9*2*2 genotype. In patients with a CYP2C9*2*2 genotype, a 2.7-fold increase in exposure to siponimod is expected in the presence of moderate CYP2C9 and CYP3A4 inhibitors. Given this increase in exposure, the concomitant use of siponimod and medicinal products that cause moderate CYP2C9 and moderate or strong CYP3A4 inhibition is not recommended, including the use of a drug that is a moderate CYP2C9/CYP3A4 dual inhibitor (e.g. fluconazole [31, 32]) or a moderate CYP2C9 inhibitor in combination with a separate moderate or strong CYP3A4 inhibitor (e.g. itraconazole [33]) [11].
Siponimod may be coadministered with most types of CYP2C9 and CYP3A4 inducers. However, because of an expected reduction in exposure to siponimod, the appropriateness and possible benefit of the treatment should be considered when siponimod is combined with strong CYP3A4/moderate CYP2C9 inducers (e.g. rifampicin [34], carbamazepine) in all patients regardless of genotype; and with moderate CYP3A4 inducers (e.g. modafinil) in patients with a CYP2C9*1*3 or CYP2C9*2*3 genotype. PBPK modelling predicts a 76% and 51% reduction in exposure to siponimod under these respective conditions [11].
No clinically relevant effects on the pharmacokinetics and pharmacodynamics of siponimod were observed when the drug was coadministered with oral ethinylestradiol/levonorgestrel contraceptive [11, 35]. No interaction studies have been conducted with other oral contraceptives containing progestogens; however, an effect of siponimod on the efficacy of oral contraceptives is not expected [11].
Race/ethnicity [11], gender [11], renal impairment [11, 36] and hepatic impairment [11, 37] had no clinically meaningful effects on the pharmacokinetics of siponimod.
4 Therapeutic Efficacy of Siponimod
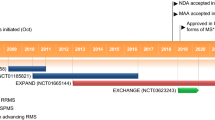
The efficacy of oral siponimod was investigated in the randomized, event- and exposure-driven, double-blind, placebo-controlled, phase 3 EXPAND trial in adult patients (aged ≥ 18 years) with SPMS (n = 1099 randomized) [38]. The core placebo-controlled phase of the trial (median 18 months’ duration) was followed by an open-label extension (OLEX) phase [39], with data collection ongoing for up to 10 years.
Eligible patients had an Expanded Disability Status Scale (EDSS) score of 3.0–6.5, documented EDSS progression in the 2 years prior to the study, a history of RRMS and no evidence of relapse in the 3 months prior to randomization [38]. Key exclusion criteria included the presence of substantial immunological, cardiac or pulmonary conditions, uncontrolled diabetes, ongoing macular oedema or CYP2C9*3/*3 genotype. Demographics were generally similar across treatment groups; the median time since MS diagnosis was 11.6 years and to conversion to SPMS was 2.6 years, 60% of patients were females, 82% were aged > 41 years, 55% had an EDSS score of 6.0–6.5 and 78% had not had a relapse in the year prior to screening. Participants received siponimod 2 mg (maintenance dosage; dosage titrated from 0.25–2 mg/day from day 1 to 6) or placebo once daily [38].
The primary efficacy analysis was planned when a minimum of 374 3-month confirmed disability progression (CDP) events had occurred; as more CDP events occurred than expected (potentially shortening the follow-up period), a protocol amendment also required that ≥ 95% of patients had to have been randomly assigned to treatment for ≥ 12 months before the core phase of the study was stopped. The primary endpoint of the time to 3-month CDP was defined as a 1-point increase in EDSS score if the baseline score was 3.0–5.0 or a 0.5-point increase if the baseline score was 5.5–6.5, confirmed at a scheduled visit ≥ 3 months later. The primary endpoint and two key secondary endpoints (defined in Table 1) were assessed using hierarchical statistical testing [38]. For EDSS, a score of 3–4 indicates moderate disability but no limitation in walking; 4.5–5.5 indicates increasing limitation in walking but can walk without aids; 6.0 indicates requires unilateral aid to walk; 6.5 indicates requires bilateral support; and > 6.5 indicates wheelchair restricted [40].
4.1 Core Double-Blind Phase
At the time of the primary time-to-event analysis (using Kaplan-Meier methods), siponimod treatment was associated with a significantly lower (reduced by 21%) risk of 3-month CDP than placebo in the overall population (primary endpoint), with 26% and 32% of patients having 3-month CDP (Table 1). Results for 3-month CDP in prespecified subgroups of patients defined by disease characteristics [i.e. superimposed relapse activity in previous 2 years (yes or no), disease progression (rapid progression yes or no) and disease severity (MS Severity Score ≥ 4 yes or no)] were consistent with those in the overall population, with hazard ratios (HRs) favouring (i.e. all HR < 1.0) siponimod over placebo (no formal statistical analyses performed). Exploratory analyses of 3-month CDP in other subgroups of patients were also consistent with the overall population, including based on baseline age, disease duration since onset, EDSS score, the number of gadolinium-enhancing T1 lesions, and previous treatment with interferon (IFN) β-1b or DMTs [38].
For key secondary endpoints, there was no statistically significant difference between the siponimod and placebo groups for the time to 3-month confirmed worsening of ≥ 20% from baseline in the timed 25-foot walk (T25FW) test in the overall population (Table 1). Given this lack of statistical significance in hierarchical testing, adjusted mean changes from baseline in total lesion volume on T2-weighted (T2W) images at 12 months and 24 months in the siponimod group were considered nominally significant versus placebo (both timepoints p < 0.0001) (Table 1), with the adjusted average mean increase from baseline over both timepoints lower in the siponimod than placebo group (183.9 vs 879.2 mm3; nominal p < 0.0001) [38].
For other secondary clinical outcomes, siponimod treatment was associated with improvements (all nominal p ≤ 0.0058) in 6-month CDP, annualized relapse rates and time to first confirmed relapse compared with placebo (Table 1). In post hoc analyses, the time to 6-month CDP in prespecified subgroups of patients were consistent with that in the overall population, favouring siponimod treatment over placebo [38]. Siponimod treatment also reduced the risk of 3- and 6-month CDP compared with placebo, irrespective of whether patients had or did not have relapses in the 1- or 2-year periods prior to study entry and in patients who would not have relapsed on study regardless of study treatment [41]. In non-relapsing patients, at all assessed timepoints (12, 18 and 24 months), siponimod reduced 3-month CDP (by 14–20%) and 6-month CDP (by 29–33%) compared with placebo [41]. The efficacy of siponimod on disability progression was supported by analyses utilizing the novel motor integration (ambulation and brainstem/pyramidal functions) and collateral (bowel and bladder, brainstem, cerebral, sensory and visual functions) subscales of EDSS, with effect sizes favouring siponimod over placebo from 6 months onwards over a 36-month period for EDSS and the motor integration and collateral subscales [42]. In post hoc analyses, siponimod treatment also delayed the time to wheelchair dependence compared with placebo in a subgroup of patients with a baseline EDSS score of 6.5 who were at high-risk of reaching wheelchair dependency [43]. Using a multistate model, there was a 28% reduction in the risk of transitioning from an EDSS score of 6.5 to a sustained EDSS score of ≥ 7 in the overall population, with the median time to reach an EDSS score of ≥ 7 prolonged in the siponimod group under the assumption of stable effect over time (12.0 vs 16.3 years with placebo) [43]. There were no between-group differences for adjusted mean changes in 12-point MS Walking Scale (MSWS-12) scores at 12 or 24 months or for the adjusted mean overall MSWS-12 score over all visits (up to and including 30 month visits) [score 2.69 vs 4.46 in the placebo group] [38].
Secondary MRI-related outcomes were generally improved (based on nominal p values; all p ≤ 0.02) in the siponimod compared with the placebo group. Percentage decreases in brain volume from baseline at 12 and 24 months and averaged over both timepoints (− 0.5% vs − 0.65%; nominal p = 0.0002) were smaller in the siponimod than placebo group, with a lower adjusted mean cumulative number of gadolinium-enhancing lesions on T1W images (0.08 vs 0.60; rate ratio 0.14; nominal p < 0.0001) and new or enlarging lesions on T2W images (0.70 vs 3.60; rate ratio 0.19; nominal p < 0.0001). These outcomes, in turn, were reflected in numerically more siponimod than placebo recipients being free from gadolinium-enhancing lesions on T1W images (89% vs 67%) and free from new and enlarging lesions on T2W images (57% vs 37%) on all post-baseline images (assessed in patients with ≥ 1 scan post-baseline) [38]. At 12 and 24 months, siponimod treatment (vs placebo) significantly reduced cortical grey matter volume loss (all p < 0.0001) and thalamic volume loss (all p < 0.05) in the overall population and subgroups of patients, including based on age, disease activity, EDSS score and prior use of DMT [44].
Relative to placebo (n = 263), siponimod treatment (n = 516) significantly (p < 0.05) delayed disability progression and in an exploratory analysis improved cognitive processing speed [CPS; assessed using the Symbol Digit Modalities Test (SDMT)] in a subgroup of patients with active disease (i.e. presence of relapses in the 2 years prior to screening and/or ≥ 1 gadolinium-enhancing TW1 lesion at baseline) [45]. There was a > 30% reduction in the risk of 3-month (24.9% vs 34.6%; HR 0.69; 95% CI 0.53–0.91; p = 0.0094) and 6-month (19.0% vs 28.1%; HR 0.63; 95% CI 0.47–0.86; p = 0.004) CDP in siponimod versus placebo recipients. In terms of CPS, siponimod treatment increased the risk of a sustained improvement in CPS by 51% (34% vs 23% of patients; HR 1.51; 95% CI 1.12–2.04; p = 0.007) and reduced the risk of sustained worsening in CPS by 28% (27% vs 38%; HR 0.72; 95% CI 0.56–0.94; p = 0.0166). A sustained effect on CPS was defined as a change that continued until the end of the core part of the study or occurred at last assessment, with worsening and improvement defined by a ≥ 4-point change in SDMT score [45]. In addition, relative to placebo, siponimod treatment was associated with functional benefits on CPS in the overall population in terms of sustained improvements and deterioration in SDMT scores (both p < 0.05) [46].
Siponimod treatment also improved myelination compared with placebo in the EXPAND MRI substudy, based on the magnetization transfer ratio (MTR; a putative marker of changes in myelin density in the brain) [47]. Compared with placebo, siponimod treatment reduced the decrease in mean normalized MTR (nMTR) at 12 months by 38% (nonsignificant) and at 24 months by 55% (p < 0.05), corresponding reductions in cortical grey matter tissue at these timepoints were 27% (not significant) and 55% (p = 0.0468) and those for normal-appearing white matter were 105% and 98% (both p < 0.05) [47].
4.2 Open-Label Extension Phase
The beneficial effects of siponimod on CDP, CPS and relapses observed during the core study appeared to be sustained during the subsequent OLEX phase after ≤ 5 years of siponimod treatment [39]. At this timepoint, of the 1224 patients who entered the OLEX study, 72% are ongoing. For the continuous siponimod group (siponimod 2 mg/day during core and OLEX phases), the time to 6-month CDP on EDSS score was delayed by 54% for the 25th percentile compared with the switch group (placebo switched to siponimod during the OLEX phase) [21.0 vs 13.6 months], with the time to 6-month CDP based on SDMT score delayed by 62% (29.6 vs 18.3 months). The absolute risk reduction for the time to 6-month CDP on EDSS in the continuous siponimod group relative to the switch group was 22% (59% vs 52% of patients with an event up to month 48; HR 0.78; 95% CI 0.66–0.92; p = 0.0048), with a 23% reduction in the absolute risk of the time to 6-month CDP on SDMT (68% vs 58%; HR 0.77; 95% CI 0.65–0.92; p = 0.0014) [39].
5 Tolerability of Siponimod
Oral siponimod was generally well tolerated in the pivotal EXPAND trial in patients with SPMS, with the safety analysis set including 1099 siponimod and 546 placebo recipients (median exposure to study drug was 18 months) [38]. Although the majority of patients in the siponimod and placebo groups experienced ≥ 1 treatment-emergent adverse event (TEAE; 89 vs 82%), most of these were mild to moderate in severity and relatively few patients discontinued treatment because of these TEAEs (4% vs 3%). The most common TEAEs (i.e. incidence ≥ 5% and at rate ≥ 1% higher incidence than placebo) occurring in the siponimod and placebo groups were headache (15% vs 13%), fall (12% vs 11%), hypertension (10% vs 8%), dizziness (7% vs 5%), nausea (7% vs 4%), diarrhoea (6% vs 4%), increased alanine aminotransferase levels (6% vs 4%) and pain in extremity (6% vs 4%). Serious TEAEs occurred in 18% of patients in the siponimod group and 15% in the placebo group, with 3% and 2% of these events leading to discontinuation of study drug. There were four deaths in each group (< 1% vs 1% in the placebo group); in the siponimod group, there was one case each of death due to metastatic gastrointestinal melanoma within 4 months of commencing siponimod, septic shock in a patient with terminal colon cancer, urosepsis > 10 weeks after discontinuing siponimod treatment and after two doses of rituximab, and a suicide [38].
The most common (incidence ≥ 2% and at rate ≥ 1% higher incidence than placebo) adverse events of special interest (AESI; i.e. those previously associated with SIP-receptor modulation) occurring in the siponimod and placebo group were liver-related investigations (signs and symptoms; 12% vs 4%), hypertension (10% vs 8%), peripheral oedema (5% vs 2%), bradycardia during treatment initiation (4% vs 3%), bradyarrhythmia during treatment initiation (3% vs 0.1%), macular oedema (2% vs < 1%), herpes zoster reactivation (2% vs 1%) and convulsions (2% vs <1%). There was no between-group difference in the rates of malignancies (2% vs 3% in the placebo group), including basal cell carcinoma (1% in both groups), or of TEAEs and serious TEAEs related to infection (except for herpes zoster reactivation), with infections and infestations occurring in 49% of patients in both groups. A case of herpes zoster meningitis occurred in the siponimod group [38].
No new safety signals were identified during the OLEX phase of EXPAND after up to 5 years’ follow-up, with the incidence rates of AEs/100 patient-years’ exposure consistent with those in the double-blind phase of the trial [39].
As siponimod dose-dependently reduces lymphocyte counts (Sect. 2), siponimod may increase the risk of infections, some of which may be serious in nature [11]. Siponimod is contraindicated in patients with immunodeficiency syndrome or a history of progressive multifocal leukoencephalopathy or cryptococcal meningitis. Prior to initiation of siponimod treatment, a recent complete blood count (CBC; i.e. within the last 6 months or after discontinuation of prior therapy) should be available, with the siponimod dosage adjusted and/or treatment interrupted based on absolute lymphocyte counts. CBC should also be monitored periodically during treatment [11]. The use of live attenuated vaccines may carry a risk of infection and should therefore be avoided during siponimod treatment and for ≤ 4 weeks after treatment [11, 48]. During and for ≤ 4 weeks after treatment with siponimod, vaccinations may be less effective; the efficacy of vaccination is not considered to be compromised if siponimod is paused 1 week prior to vaccination until 4 weeks after vaccination [11].
Patients with the following cardiac conditions should be monitored for a 6-h period after the first dose of siponimod for signs and symptoms of bradycardia: sinus bradycardia (heart rate < 55 bpm), history of first- or second degree (Mobitz type I) atrioventricular block, history of myocardial infection or a history of heart failure (patients with NYHA class I or II). Siponimod is contraindicated in patients with a history of symptomatic bradycardia or recurrent syncope, uncontrolled hypertension or severe untreated sleep apnoea. Based on a thorough QT study, siponimod is not associated with significant direct QT-prolongation and is not associated with an arrhythmogenic potential related to QT prolongation (Sect. 2) [11].
With regard to liver function, although there are no data to establish that patients with pre-existing liver disease are at increased risk of developing elevated liver function test values when taking siponimod, caution is advised in patients with a history of significant liver disease. Recent (i.e. within the last 6 months) transaminase and bilirubin levels should be available prior to initiating siponimod treatment. In EXPAND, treatment was discontinued if the elevation in transaminase levels exceeded a threefold increase and the patient showed symptoms related to hepatic function or if the elevation exceeded a fivefold increase; 1% of all discontinuations met one of these criteria [11].
6 Dosage and Administration of Siponimod
Oral siponimod is approved in several countries for the treatment of SPMS/active SPMS (featured indication); specific indications vary between individual countries. In the EU, siponimod is indicated for the treatment of adult patients with SPMS with active disease evidenced by relapses or imaging-features of inflammatory activity [11]. Treatment with siponimod should be started with a 5-day dose titration pack (0.25 mg once daily on day 1 and 2, with the dosage incremented by 0.25 mg/day on day 3 and 4, and then to 1.25 mg/day on day 5). The maintenance dosage from day 6 onwards is 2 mg once daily, with the drug taken without regard to food. Prior to initiation of siponimod treatment, patients must be genotyped for CYP2C9 to determine their CYP2C9 metabolizer status (Sect. 3). Siponimod should not be used in patients with a CYPC9*3*3 genotype; the recommended maintenance dosage is 1 mg once daily in those with a CYP2C9*2*3 genotype. The recommended maintenance dosage of siponimod in patients with all other CYP2C9 genotypes is 2 mg once daily [11]. Consult local prescribing information for detailed information, including specific indications, contraindications, precautions and warnings, potential drug interactions and use in special patient populations.
7 Place of Siponimod in the Management of Secondary Progressive Multiple Sclerosis
Given the heterogeneity and complexity of MS, its management requires comprehensive individualized treatment with medical and rehabilitation therapies targeted at controlling symptoms, reducing the frequency of relapses and preventing/slowing disability progression. Several DMTs have been approved for RRMS in the EU, including subcutaneous interferon-β (IFNβ)-1a, IFNβ-1b and pegIFNβ-1a, subcutaneous glatiramer acetate, small-molecule oral agents (cladribine, dimethyl fumarate, fingolimod, teriflunomide), intravenous mitoxantrone and intravenous monoclonal antibodies (alemtuzumab, natalizumab, ocrelizumab) [1]. Treatment options for SPMS are more limited making its management more challenging. In patients with active SPMS, current ECTRIMS/EAN guidelines recommend (weak recommendation for all of these DMTs) the following DMTs: IFNβ-1a or -1b taking into account the efficacy, safety and tolerability profiles of these drugs; mitoxantrone taking into account its efficacy and specifically considering the safety and tolerability of the drug (cardiotoxicity, delayed congestive heart failure, myelosuppression and acute treatment related leukemia); and ocrelizumab or cladribine [1]. EU approval of siponimod for the treatment of adult patients with active SPMS (Sect. 6) is too recent for its consideration in these guidelines. A recent UK NICE appraisal has recommended the use of siponimod for the treatment of adult patients with SPMS with active disease evidenced by relapses or imaging-features of inflammatory disease [49]. Ultimately the choice of treatment will depend upon a number of factors, including patient preference, disease characteristics and pharmacoeconomic considerations.
In the large, multinational EXPAND trial in adult patients with SPMS (≈ two-thirds of whom had not relapsed in the previous 2 years and more than 50% required walking assistance), siponimod significantly reduced the risk of disability progression on EDSS compared with placebo during the double-blind core phase, including in prespecified subgroups of patients defined by disease characteristics such as severity of disease, history of relapse in the previous 2 years and the rate of disease progression (Sect. 4.1). These changes were considered clinically relevant as even small deteriorations in EDSS score in SPMS are likely to have a clinically relevant impact, given changes in EDSS score in this population are less likely to be reversible than in the RRMS population. Siponimod also provided better efficacy than placebo for most other clinical and MRI-defined outcomes, including slowing disability progression based on other measures, improving CPS, improving myelination (EXPAND MRI substudy) and reducing losses in brain volume (an objective marker of permanent CNS damage), cortical grey matter and thalamic volume (Sect. 4.1). These beneficial effects of siponimod in terms of delaying disability progression and improving CPS and relapses appeared to be sustained after up to 5 years of treatment in the ongoing OLEX study (Sect. 4.2). Ongoing clinical experience will help to define the long-term efficacy and safety of siponimod in the real-world setting, with a multicentre, non-interventional real-world study (AMASIA) planned in Germany [50].
Of note, at the time of the primary efficacy analysis, there was no significant between-group difference for the first key secondary endpoint (T25FW test; a gold standard for assessing ambulation in MS, along with EDSS [51]), with the hierarchical statistical testing scheme meaning that subsequent significant between-group differences for individual outcomes were considered nominally significant (Sect. 4.1). This lack of between-group difference for the T25FW test may, at least in part, reflect that the T25FW test assesses gait velocity rather than a person’s ability to vary their gait for different walking tasks and secondly, it may reflect the high degree of variability in its sensitivity in patients with progressive MS [51]. Hence, the sensitivity of the T25FW test may have been markedly reduced, given that more than 50% of patients already required assistance with walking prior to study enrolment. Additionally, protocol-defined switching to rescue medication (i.e. active open-label treatment) was more common in the placebo than siponimod group (17 vs 11% of patients), which may also have reduced the power of the study to detect a between-group difference in this and some other secondary outcomes [38]. Of interest for rescue therapy, no DMT was given a strong recommendation for treating SPMS in current ECTRIMS/EAN guidelines [1]. To date, there have been no direct head-to-head randomized controlled trials of siponimod versus other DMTs (e.g. IFNβ-1a or -1b) in SPMS.
Oral siponimod was generally well tolerated in the EXPAND trial in patients with SPMS (Sect. 5), with a similar safety profile to that of other S1PR modulators [38]. The majority of TEAEs were of mild to moderate severity and relatively few (3%) patients discontinued treatment because of these events (Sect. 5). The most common AESI (i.e. AEs associated with the S1PR modulator class) that occurred more frequently in siponimod than placebo recipients were increases in liver transaminase levels, hypertension, peripheral oedema and bradycardia at treatment initiation. The use of a dose-titration schedule at treatment initiation mitigated the first-dose effects of siponimod on heart rate and conduction [38]. Reflecting its mechanism of action (Sect. 2), siponimod treatment is associated with dose-dependent reductions in lymphocytes resulting in lymphopenia. In EXPAND, there were no differences between the siponimod
and placebo group for infection (except for herpes zoster reactivation) or malignancies rates (Sect. 5). However, as siponimod may increase the risk of infection, including serious infection, CBC monitoring is required prior to initiating treatment and periodically during treatment. Local prescribing information should be consulted for the management of AESI in patients with specified pre-existing conditions (Sect. 5). Alterations in the chemical structure of siponimod relative to fingolimod (Sect. 2) offer an advantage in terms of a lower risk of bradycardia with siponimod and a faster restoration of lymphocytes counts after discontinuation of treatment (within 1 week [11] vs 2 months for fingolimod [52]) [9].
In conclusion, once-daily oral siponimod was significantly more effective than placebo in reducing clinical and MRI-defined outcomes of disease activity and disability progression, including 3-month CDP on EDSS, and was generally well tolerated in the core phase of the EXPAND trial in patients with active SPMS. These beneficial effects appeared to be sustained during up to 5 years of siponimod treatment in the ongoing OLEX. Albeit further long-term efficacy and safety data are required to fully define its role, given the paucity of current treatment options and its convenient dosage regimen, siponimod represents an important emerging option for the treatment of adult patients with SPMS with active disease evidenced by relapses or imaging-features of inflammatory activity.
Data Selection Siponimod: 205 records identified
Duplicates removed | 24 |
Excluded during initial screening (e.g. press releases; news reports; not relevant drug/indication; preclinical study; reviews; case reports; not randomized trial) | 88 |
Excluded during writing (e.g. reviews; duplicate data; small patient number; nonrandomized/phase I/II trials) | 42 |
Cited efficacy/tolerability articles | 9 |
Cited articles not efficacy/tolerability | 43 |
Search Strategy: EMBASE, MEDLINE and PubMed from 1946 to present. Clinical trial registries/databases and websites were also searched for relevant data. Key words were siponimod, Mayzent, secondary progressive multiple sclerosis, SPMS. Records were limited to those in English language. Searches last updated 30 September 2020 | |
Change history
18 January 2021
A Correction to this paper has been published: https://doi.org/10.1007/s40263-020-00785-7
References
Montalban X, Gold R, Thompson AJ, et al. ECTRIMS/EAN guideline on the pharmacological treatment of people with multiple sclerosis. Mult Scler J. 2018;24(2):96–120.
Oh J, Alikhani K, Bruno T, et al. Diagnosis and management of secondary-progressive multiple sclerosis: time for change. Neurodegener Dis Manag. 2019;9(6):301–17.
Wallin MT, Culpepper WJ, Nichols E, et al. Global, regional, and national burden of multiple sclerosis 1990–2016: a systematic analysis for the Global Burden of Disease study 2016. Lancet Neurol. 2019;18:269–85.
Inojosa H, Proschmann U, Akgün K, et al. A focus on secondary progressive multiple sclerosis (SPMS): challenges in diagnosis and definition. J Neurol. 2019. https://doi.org/10.1007/s00415-019-09489-5.
De Angelis F, Plantone D, Chataway J. Pharmacotherapy in secondary progressive multiple sclerosis: an overview. CNS Drugs. 2018;32(6):499–526.
Baldassari LE, Fox RJ. Therapeutic advances and challenges in the treatment of progressive multiple sclerosis. Drugs. 2018;78(15):1549–66.
Hla T, Brinkmann V. Sphingosine 1-phosphate (S1P): physiology and the effects of S1P receptor modulation. Neurology. 2011;76(Suppl 3):S3–8.
Chaudhry BZ, Cohen JA, Conway DS. Sphingosine 1-phosphate receptor modulators for the treatment of multiple sclerosis. Neurotherapeutics. 2017;14(4):859–73.
Dumitrescu L, Constantinescu CS, Tanasescu R. Siponimod for the treatment of secondary progressive multiple sclerosis. Expert Opin Pharmacother. 2019;20(2):143–50.
Cugati S, Chen CS, Lake S, et al. Fingolimod and macular edema: pathophysiology, diagnosis, and management. Neurol Clin Pract. 2014;5:402–7.
Novartis Europharm Ltd. Siponimod (Mayzent®): EU summary of product characteristics. 2020. https://www.ema.europa.eu/. Accessed 9 Jul 2020.
Novartis Pharamceuticals Corporation. MAYZENT® (siponimod) tablets, for oral use: US prescribing information. 2019. https://www.fda.gov/. Accessed 9 Jul 2020.
Pan S, Gray NS, Gao W, et al. Discovery of BAF312 (siponimod), a potent and selective S1P receptor modulator. ACS Med Chem Lett. 2013;4(3):333–7.
Gergely P, Nuesslein-Hildesheim B, Guerini D, et al. The selective shingosine 1-phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate. Br J Pharmacol. 2012;167:1035–47.
O’Sullivan C, Schubart A, Mir AK, et al. The dual S1PR1/S1PR5 drug BAF312 (siponimod) attenuates demyelination in organotypic slice cultures. J Neuroinflammation. 2016;13:31.
Bigaud M, Tisserand S, Albrecht P, et al. Siponimod: from understanding mode of action to differentiation versus fingolimod [abstract no. 1536]. Neurology. 2020; 94(15 Suppl).
Lewis ND, Haxhinasto SA, Anderson SM, et al. Circulating monocytes are reduced by sphingosine-1-phosphate receptor modulators independently of SIP3. J Immunol. 2013;190:3533–40.
Mannioui A, Vauzanges Q, Fini JB, et al. The Xenopus tadpole: an in vivo model to screen drugs favoring remyelination. Mult Scler J. 2018;24(11):1421–32.
Dietrich M, Hecker C, Martin E, et al. Protective and remyelinating potential of siponimod in a Xenopus model and a mouse model of experimental autoimmune encephalomyelitis [poster no. P0991]. In: 8th Joint ACTRIMS-ECTRIMS Virtual Meeting. 2020.
Husseini L, Geladaris A, Steinleitnes M, et al. Siponimod treatment leads to a dose-dependent reduction of EAE severity associated with down regulation of microglial activity [poster no. P0394]. In: 8th Joint ACTRIMS-ECTRIMS Virtual Meeting. 2020.
Gentile A, Musella A, Bullitta S, et al. Siponimod (BAF312) prevents synaptic neurodegeneration in experimental multiple sclerosis. J Neuroinflammation. 2016;13(1):207.
Dietrich M, Hecker C, Ramseier P, et al. Neuroprotective potential for siponimod (BAF312) revealed by visual system readouts in a model of experimental autoimmune encephalomyelitis-optic neuritis (EAEON) [abstract no. P844 plus poster]. Mult Scler J. 2019;25(Suppl 2):434–5.
Wu Q, Mills EA, Wang Q, et al. Siponimod enriches regulatory T and B lymphocytes in secondary progressive multiple sclerosis. JCI Insight. 2020;5(3):1–14.
Kuhle J, Kropshofer H, Barro C, et al. Siponimod reduces neurofilament light chain blood levels in secondary progressive multiple sclerosis patients [abstract no. S8.006]. Neurology. 2018;90(15 Suppl).
Legangneux E, Shakeri-Nejad K, Aslanis V, et al. Cardiac effects of siponimod (BAF312) re-initiation after variable periods of drug discontinuation in healthy subjects. Clin Ther. 2016;38(3):631-45.e1.
Biswal S, Polus F, Pal P, et al. Pharmacokinetic and pharmacodynamic interaction of siponimod (BAF312) and propranolol in healthy subjects. Int J ClinPharmacol Ther. 2015;53(10):855–65.
Shakeri-Nejad K, Aslanis V, Veldandi UK, et al. Effects of therapeutic and supratherapeutic doses of siponimod (BAF312) on cardiac repolarization in healthy subjects. Clin Ther. 2015;37(11):2489–505.
Bigaud M, Rudolph B, Briard E, et al. Siponimod penetrates, distributes and acts on the central nervous system: translational insights [abstract no. 3973]. Neurology. 2020;94(15 Suppl).
Glaenzel U, Jin Y, Nufer R, et al. Metabolism and disposition of siponimod, a novel selective S1P1/S1P5 agonist, in healthy volunteers and in vitro identification of human cytochrome P450 enzymes involved in its oxidative metabolism. Drug Metab Dispos. 2018;46(7):1001–13.
Huth F, Gardin A, Umehara K, et al. Prediction of the impact of cytochrome P450 2C9 genotypes on the drug-drug interaction potential of siponimod with physiologically-based pharmacokinetic modeling: a comprehensive approach for drug label recommendations. Clin Pharmacol Therap. 2019;106(5):1113–24.
Gardin A, Ufer M, Legangneux E, et al. Effect of fluconazole coadministration and CYP2C9 genetic polymorphism on siponimod pharmacokinetics in healthy subjects. Clin Pharmacokinet. 2019;58(3):349–61.
Jin Y, Borell H, Gardin A, et al. In vitro studies and in silico predictions of fluconazole and CYP2C9 genetic polymorphism impact on siponimod metabolism and pharmacokinetics. Eur J Clin Pharmacol. 2018;74(4):455–64.
Gardin A, Shakeri-Nejad K, Feller A, et al. Siponimod pharmacokinetics, safety, and tolerability in combination with the potent CYP3A4 inhibitor itraconazole in healthy subjects with different CYP2C9 genotypes. Eur J Clin Pharmacol. 2019;75(11):1565–74.
Gardin A, Gray C, Neelakantham S, et al. Siponimod pharmacokinetics, safety, and tolerability in combination with rifampin, a CYP2C9/3A4 inducer, in healthy subjects. Eur J Clin Pharmacol. 2018;74(12):1593–604.
Biswal S, Veldandi UK, Derne C, et al. Effect of oral siponimod (BAF312) on the pharmacokinetics and pharmacodynamics of a monophasic oral contraceptive in healthy female subjects. Int J Clin Pharmacol Ther. 2014;52(11):996–1004.
Gardin A, Dodman A, Kalluri S, et al. Pharmacokinetics, safety, and tolerability of siponimod (BAF312) in subjects with severe renal impairment: a single-dose, open-label, parallel-group study. Int J Clin Pharmacol Ther. 2017;55(1):54–65.
Shakeri-Nejad K, Aslanis V, Veldandi UK, et al. Pharmacokinetics, safety, and tolerability of siponimod (BAF312) in subjects with different levels of hepatic impairment: a single-dose, open-label, parallel-group study. Int J Clin Pharmacol Ther. 2017;55(1):41–53.
Kappos L, Bar-Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet. 2018;391(10127):1263–73.
Kappos L, Giovannoni G, Gold R, et al. Long-term efficacy and safety of siponimod treatment for up to 5 years in patients with secondary progressive multiple sclerosis: analysis of the EXPAND extension study [abstract no. EPR2128]. Eur J Neurol. 2020;27(Suppl 1):337.
Cinar BP, Yorgun G. What have we learned from the history of multiple sclerosis measurement: expanded disability status scale. Arch Neuropsychiatry. 2018;55(Suppl 1):S69-75.
Cree B, Fox R, Giovannoni G, et al. Siponimod affects disability progression in patients with SPMS independent of relapse activity: results from the phase 3 EXPAND study [abstract]. In: American Academy of Physical Medicine and Rehabiltation Annual Meeting. 2019.
Cutter G, Meng X, Bar-Or A, et al. Effect of siponimod on disability in EXPAND re-examined with two new subscales of the expanded disability status scale in patients with SPMS [abstract no. 4051]. Neurology. 2020;94(15 Suppl).
Vermersch P, Gold R, Kappos L, et al. Siponimod delays the time to wheelchair in patients with SPMS: results from the EXPAND study [abstract no. 158]. Mult Scler J. 2019;25(Suppl 2):55–6.
Vermersch P, Fox RJ, Arnold DL, et al. Effect of siponimod on grey matter atrophy in patients with secondary progressive multiple sclerosis: subgroup analyses from the EXPAND study [abstract no. EPR3098]. Eur J Neurol. 2020;27(Suppl 1):478.
Gold R, Kappos L, Benedict RHB, et al. Siponimod slows physical disability progression and decline in cognitive processing speed in SPMS patients with active disease: a post hoc analysis of the EXPAND study [abstract no. EPR2118]. Eur J Neurol. 2020;27(Suppl 1):328–9.
Benedict R, Fox R, Tomic D, et al. Siponimod improves cognitive processing speed in patients with secondary progressive multiple sclerosis: EXPAND subgroup analyses [abstract no. EPO3212]. Eur J Neurol. 2019;26(Suppl 1):868.
Arnold DL, Vermersch P, Cree BAC, et al. Evidence for improved myelination in patients treated with siponimod: results from the phase 3 EXPAND MRI substudy [abstract no. EPR1147]. Eur J Neurol. 2020;27(Suppl 1):194–5.
Ufer M, Shakeri-Nejad K, Gardin A, et al. Impact of siponimod on vaccination response in a randomized, placebo-controlled study. Neurol Neuroimmunol Neuroinflamm. 2017;4:1–10.
National Institute for Health and Care Excellence. Final appraisal document: siponimod for treating secondary progressive multiple sclerosis. 2020. http://www.nice.org.uk/guidance/gid-ta10436/documents/final-appraisal-determination-document. Accessed 19 Oct 2020.
Ziemssen T, Hoffmann O, Klotz L, et al. Study protocol of AMASIA, a non-interventional study to gain first insights on SPMS patients treated with siponimod in clinical routine. JIMR Res Protocols. 2020. https://doi.org/10.2196/19598.
Bennett SE, Bromley LE, Fisher NM, et al. Validity and reliability of four gait measures in patients with multiple sclerosis. Int J MS Care. 2017;19:247–52.
FDA. Gilenya (fingolimod) capsules: US prescribing information. 2010. https://www.fda.gov. Accessed 28 July 2020.
Acknowledgements
During the peer review process, the manufacturer of the agent under review was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of interest
Lesley Scott is a salaried employee of Adis International Ltd/Springer Nature, and declares no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Additional information
The original online version of this article was revised due to a retrospective Open Access order.
Enhanced material for this Adis Drug Evaluation can be found at https://doi.org/10.6084/m9.figshare.12725414.
The manuscript was reviewed by: S. de Biase, Neurology Unit, Ospedale Dell’Angello, Venezia-Mestre, Italy; D. N. Bourdette, Department of Neurology, Oregon Health and Science University, Portland, OR, USA; M. S. Freedman, Multiple Sclerosis Research Clinic, University of Ottawa and the Ottawa Hospital Research Institute, Ottawa, ON, Canada; E. R. Comini-Frota, Neurologist, Neuroimmunologist, Belo Horizonte, Brazil
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Scott, L.J. Siponimod: A Review in Secondary Progressive Multiple Sclerosis. CNS Drugs 34, 1191–1200 (2020). https://doi.org/10.1007/s40263-020-00771-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-020-00771-z