
Abstract
Background
Oral semaglutide is a novel tablet containing the human glucagon-like peptide-1 (GLP‑1) analogue semaglutide, co-formulated with the absorption enhancer sodium N-(8-[2-hydroxybenzoyl] amino) caprylate (SNAC). The safety and pharmacokinetics of oral semaglutide were investigated in two randomised, double-blind, placebo-controlled trials.
Methods
In a single-dose, first-in-human trial, 135 healthy males received oral semaglutide (2–20 mg semaglutide co-formulated with 150–600 mg SNAC) or placebo with SNAC. In a 10-week, once-daily, multiple-dose trial, 84 healthy males received 20 or 40 mg oral semaglutide (with 300 mg SNAC), placebo, or placebo with SNAC, and 23 males with type 2 diabetes (T2D) received 40 mg oral semaglutide (with 300 mg SNAC), placebo, or placebo with SNAC.
Results
Oral semaglutide was safe and well-tolerated in both trials. The majority of adverse events (AEs) were mild, with the most common AEs being gastrointestinal disorders. In the single-dose trial, semaglutide exposure was highest when co-formulated with 300 mg SNAC. In the multiple-dose trial, semaglutide exposure was approximately twofold higher with 40 versus 20 mg oral semaglutide in healthy males, in accordance with dose proportionality, and was similar between healthy males and males with T2D. The half-life of semaglutide was approximately 1 week in all groups.
Conclusion
The safety profile of oral semaglutide was as expected for the GLP-1 receptor agonist drug class. Oral semaglutide co-formulated with 300 mg SNAC was chosen for further clinical development. The pharmacokinetic results supported that oral semaglutide is suitable for once-daily dosing.
ClinicalTrials.gov identifiers
NCT01037582, NCT01686945.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Oral semaglutide is a novel tablet co-formulation of the human glucagon-like peptide-1 (GLP-1) analogue semaglutide, with the absorption enhancer sodium N-(8-[2-hydroxybenzoyl] amino) caprylate (SNAC). In a first-in-human, single ascending dose trial in healthy males, and in a 10-week, once-daily, multiple-dose trial in healthy males and males with type 2 diabetes, oral semaglutide was safe and well-tolerated, with a safety profile as expected for the GLP-1 receptor agonist drug class. |
The half-life of oral semaglutide was approximately 1 week, which is similar to semaglutide administered subcutaneously, suggesting that the elimination phase of semaglutide administered orally is comparable with that seen with subcutaneous administration. |
Following administration of oral semaglutide with 150–600 mg SNAC, semaglutide plasma exposure levels suggested that co-formulation with 300 mg SNAC is optimal to enhance the absorption of orally administered semaglutide. Oral semaglutide co-formulated with a fixed amount of SNAC (300 mg) was therefore chosen for further clinical development. |
1 Introduction
Significant challenges in obtaining sufficient systemic exposure of orally administered peptide- and protein-based drugs are due to the acidic environment and presence of proteolytic enzymes in the stomach, and the limited permeability of peptides and proteins through the gastrointestinal epithelium [1, 2]. However, many patients prefer oral drug administration compared with alternative delivery routes, likely due to the simplicity and convenience of tablet administration, which could lead to improved compliance [1]. Consequently, several solutions have been developed to facilitate oral peptide and protein delivery [1, 3].
Semaglutide is a human glucagon-like peptide-1 (GLP-1) analogue developed for the treatment of type 2 diabetes (T2D). In addition to a formulation of semaglutide for once-weekly subcutaneous administration [4], a tablet formulation of semaglutide has also been developed, where semaglutide is co-formulated with the absorption enhancer sodium N-(8-[2-hydroxybenzoyl] amino) caprylate (SNAC), thereby providing the first GLP-1 receptor agonist to be administered orally (oral semaglutide). SNAC has been shown to facilitate the absorption of semaglutide across the gastric epithelium in a concentration-dependent manner, primarily via the transcellular route [5]. SNAC has previously been co-formulated with heparin, ibandronate and vitamin B12, leading to increased systemic bioavailability of these compounds after oral administration in humans [6,7,8].
The purpose of the present investigation was to generate the first safety, pharmacokinetic and pharmacodynamic data for oral semaglutide in healthy subjects and subjects with T2D. In a first-in-human trial in healthy subjects, single ascending doses of oral semaglutide were tested (‘single-dose trial’). Variable amounts of SNAC in the formulation were used to gain information on the optimal amount of SNAC. Subsequently, a 10-week, once-daily, multiple-dose trial in healthy males and males with T2D was conducted to evaluate the safety, pharmacokinetics and pharmacodynamics of oral semaglutide at steady state (‘multiple-dose trial’).
2 Methods
2.1 Trial Design
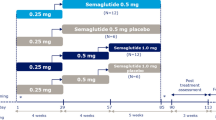
Both trials were randomised, placebo-controlled, double-blind trials (Fig. 1), each conducted at single sites (Parexel International, Harrow, UK, and Parexel International GmbH, Berlin, Germany, respectively). The trial protocols and subject information/informed consent forms were reviewed and approved by independent ethics committees (Capenhurst Independent Research Ethics Committee and Ethik-Kommission des Landes Berlin, respectively), and by appropriate health authorities according to local regulations. The trials were performed in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice. Written informed consent was provided by all subjects prior to any trial-related activities. The trials were registered at ClinicalTrials.gov (trial identifiers: NCT01037582 and NCT01686945, respectively).

Trial design of the a single-dose trial and b multiple-dose trial. SNAC sodium N-(8-[2-hydroxybenzoyl] amino) caprylate, T2D type 2 diabetes
2.2 Participants
In the single-dose trial, eligible subjects were healthy males, aged 18–50 years with a body weight of 65.0–95.0 kg and a body mass index (BMI) of 18.5–27.5 kg/m2. In the multiple-dose trial, eligible subjects were healthy males, aged 18–64 years, with a BMI of 20.0–29.9 kg/m2, and males with T2D diagnosed within the last 10 years, treated with diet and exercise and/or metformin, aged 18–64 years, with a BMI of 20.0–37.0 kg/m2 and glycosylated haemoglobin (HbA1c) of 6.5–9.0%.
Subjects were excluded if they had clinically significant concomitant diseases or disorders, clinically significant abnormal values in laboratory screening tests, any history of gastrointestinal surgery (except uncomplicated surgical procedures such as appendectomy and hernia surgery), or if they smoked more than five cigarettes or the equivalent per day.
2.3 Procedures
The single-dose trial consisted of parts 1a, 1b and 2 (Fig. 1). In part 1a, four ascending dose groups were tested in a sequential design. The decision to proceed to the next dose level was made by a safety group with access to a blinded clinical safety report prepared by the investigator and blinded preliminary pharmacokinetic data for semaglutide. In part 1b, three additional dose groups were tested in a parallel design, and, in part 2, three of the doses from part 1 were selected (by the safety group) to be repeated in a parallel design in another three groups. Within each dose group, subjects were randomised to receive either oral semaglutide or placebo containing matching amounts of SNAC. Part 2 also included two additional dose groups (intravenous and subcutaneous semaglutide) in order to investigate the absolute and relative bioavailability of oral semaglutide. However, bioavailability could not be reliably estimated since the chosen dose level for intravenous administration was too low for the terminal part of the concentration-time curve to be apparent, and the plasma semaglutide exposure levels in this single-dose trial were generally insufficient for a robust estimation of bioavailability. Therefore, these data are not shown.
Subjects came to the clinic the day before dosing and fasted overnight for ≥10 h before dosing (water ad libitum was allowed until 1 h before dosing). Subjects received a single tablet of oral semaglutide or placebo with SNAC administered with 50 mL water and followed by 5 h of post-dose fasting. Subjects stayed at the clinic until 8 days after dosing. Oral semaglutide tablets used in the single-dose trial differed from those that have subsequently been used in the oral semaglutide development programme (including the multiple-dose trial). The tablets used in the single-dose trial contained synthetic semaglutide and the manufacturing included a wet granulation step, while those in subsequent trials contained recombinant semaglutide and the manufacturing included a dry granulation step.
In the multiple-dose trial, subjects were originally allocated to four cohorts in a semi-parallel design: healthy subjects receiving oral semaglutide maintenance doses of 20, 40 or 60 mg, and subjects with T2D receiving a maintenance dose of 60 mg. Within each cohort, subjects were randomised to once-daily treatment for 10 weeks with either oral semaglutide tablets (co-formulated with 300 mg SNAC), placebo tablets or placebo tablets with SNAC (300 mg). In order to mitigate gastrointestinal adverse events (AEs), the semaglutide dose level was step-wise escalated starting with 5 mg during week 1, increasing to 10 mg during week 2 and 20 mg from week 3. Depending on the decision of the safety group to proceed to a higher dose level, it was planned to escalate to 40 mg from week 5 (in the 40 and 60 mg cohorts), and to 60 mg from week 7 (in the 60 mg cohort). However, after escalation to 40 mg, it was decided not to escalate further since it was assessed that the 60 mg dose level would lead to an unacceptable number of AEs. The two cohorts of healthy subjects to receive 40 and 60 mg were combined and the cohort of subjects with T2D to receive 60 mg was adjusted to 40 mg, thus resulting in three cohorts: healthy oral semaglutide 20 and 40 mg, and T2D 40 mg. On each day of the 10-week treatment period, oral administration was performed in the morning after an overnight fast of ≥10 h (water was allowed until 2 h before dosing) followed by 2-h post-dose fasting where subjects were not allowed to take any other oral concomitant medications. Dosing was overseen by trial staff during visits to the clinic, but otherwise handled by the subjects.
Blood samples for determination of semaglutide and SNAC concentrations in plasma were drawn at predefined time points, as shown in electronic supplementary Tables S1 and S2.
2.4 Assessments
Safety assessments included AEs, hypoglycaemic episodes, laboratory safety parameters, physical examination, vital signs and electrocardiogram. Hypoglycaemic episodes were defined as ‘confirmed’ when they were either ‘severe’ as according to the 2005 American Diabetes Association definition, i.e. requiring third-party assistance [9], or verified by a plasma glucose level < 3.1 mmol/L.
In the single-dose trial, semaglutide was measured using a validated luminescence oxygen channelling immunoassay (LOCI); however, the results from this assay revealed that semaglutide plasma concentrations appeared to be underestimated due to a matrix effect. Therefore, semaglutide concentration-time profiles are presented using an arbitrary scale. In the multiple-dose trial, semaglutide was measured by means of a validated assay using plasma protein precipitation followed by liquid chromatography tandem mass spectrometry detection, with a lower limit of quantification (LLOQ) of 0.73 nmol/L. In both trials, SNAC was measured by a validated method using a cohesive turbulent flow system equipped with a triple quadruple mass spectrometer, with an LLOQ of 5.0 ng/mL.
2.5 Endpoints
The primary endpoint in the single-dose trial comprised all safety assessments, while the primary endpoint in the multiple-dose trial was the number of AEs.
Pharmacokinetic endpoints were derived after a single dose of trial product (single-dose trial) and at steady state after 10 weeks of once-daily dosing (multiple-dose trial). Area under the semaglutide plasma concentration-time curve from 0 to 24 h at steady state (AUC0–24 h,semaglutide,SS) was determined using a standard non-compartmental method applying the trapezoidal rule on observed concentrations and the actual sampling time points. Maximum plasma semaglutide concentration at steady state (Cmax,semaglutide,SS) was derived from the observed pharmacokinetic profiles. Both AUC0–24 h,semaglutide,SS and Cmax,semaglutide,SS were derived on days 68, 69 and 70 of the 10-week treatment period. The terminal half-life of semaglutide (t½,semaglutide,SS) was calculated as ln(2)/λz, where the terminal elimination rate constant λz was estimated by log-linear regression on the terminal part of the pharmacokinetic profiles. No pharmacokinetic endpoints are presented for semaglutide in the single-dose trial due to the apparent underestimation of semaglutide concentrations with the applied pharmacokinetic assay (see Sect. 2.4).
The AUC and Cmax endpoints for SNAC in the single-dose (AUC0–5 h,SNAC,SD and Cmax,SNAC,SD) and multiple-dose (AUC0–24 h,SNAC,SS and Cmax,SNAC,SS) trials were derived as described above for semaglutide endpoints. The time to maximum plasma SNAC concentration (tmax,SNAC,SD and tmax,SNAC,SS, respectively) was derived from the observed SNAC pharmacokinetic profiles.
2.6 Statistical Analyses
In the single-dose trial, no formal sample size calculation was performed for part 1. A total of 10 subjects receiving oral semaglutide and 2 subjects receiving placebo with SNAC per treatment group was considered adequate for the primary endpoint (safety assessment). In part 2 of the single-dose trial, the sample size of 14 subjects receiving oral semaglutide and 3 subjects receiving placebo with SNAC per treatment group was based on a comparison of AUC between the oral semaglutide treatment groups and the treatment group receiving intravenous semaglutide (data not shown).
In the multiple-dose trial, no formal sample size calculation was performed for the primary endpoint (number of AEs). The sample size was based on the precision of the ratio of AUC0–24 h,semaglutide,SS between the oral semaglutide treatment groups. Based on a previous trial, it was assumed that the standard deviation for the log-transformed AUC0–24 h,semaglutide,SS was 0.51. A total of 13 subjects with evaluable pharmacokinetic profiles would then result in a probability of at least 80% to obtain a 95% confidence interval (CI) for any treatment group ratio within the range of 0.67 to 1.59 times the estimated mean treatment group ratio. This range was assessed to be sufficiently narrow. In order to account for withdrawn subjects and non-evaluable pharmacokinetic profiles, it was planned to randomise 16 subjects in each oral semaglutide treatment group.
In both trials, safety endpoints were summarised by descriptive statistics using the safety analysis set comprising all subjects receiving at least one dose of trial product.
Statistical analyses were performed using SAS versions 9.3 or 9.4 (SAS Institute, Cary, NC, USA). All statistical tests were two-sided with 5% significance level and were performed on the full analysis set comprising all randomised subjects receiving at least one dose of trial product. Statistical analyses were not controlled for multiplicity.
In the single-dose trial, AUC0–5 h,SNAC,SD, Cmax,SNAC,SD and tmax,SNAC,SD were summarised by descriptive statistics. SNAC pharmacokinetics in subjects receiving placebo with SNAC were not included since the main purpose of administering placebo with SNAC was to investigate safety.
In the multiple-dose trial, AUC0–24 h,semaglutide,SS and Cmax,semaglutide,SS on days 68, 69 and 70 were log-transformed and analysed in linear mixed models with treatment (oral semaglutide 20 and 40 mg in healthy subjects and oral semaglutide 40 mg in subjects with T2D) as a fixed effect, subject as a random effect, and allowing for different within-subject and between-subject variations in each cohort. The t½,semaglutide,SS, AUC0–24 h,SNAC,SS, Cmax,SNAC,SS and tmax,SNAC,SS were all summarised by descriptive statistics. Changes from baseline in body weight and HbA1c were analysed using mixed models for repeated measures (MMRM) with time, cohort (healthy, T2D), and treatment (oral semaglutide 20 and 40 mg, placebo, placebo with SNAC) as fixed effects, and baseline value as a covariate. The effect of the baseline parameter was allowed to vary by cohort and treatment, while the treatment effect was allowed to vary by cohort and visit. Within-subject repeated measurements were incorporated with an unstructured covariance structure.
3 Results
3.1 Subject Disposition and Characteristics
In the single-dose trial, 389 male subjects were screened, 135 were randomised and exposed to oral administration, and 134 completed the trial (electronic supplementary Fig. S1). An additional 20 subjects were randomised to subcutaneous or intravenous administration of semaglutide (not shown). All 135 exposed subjects were included in the safety analysis set and the full analysis set. In the multiple-dose trial, 245 male subjects were screened, 107 were enrolled, randomised and exposed (84 healthy males and 23 males with T2D), and 92 completed the trial (electronic supplementary Fig. S2). All 107 exposed subjects were included in the safety analysis set and the full analysis set.
Subject characteristics overall for each of the two trials are shown in Table 1, while subject characteristics for each treatment group are shown in electronic supplementary Tables S3 and S4.
3.2 Single-Dose Trial
3.2.1 Safety After a Single Dose of Oral Semaglutide
An overview of reported AEs in the single-dose trial is shown in electronic supplementary Table S5. A total of 104 AEs were reported in 55 subjects (41%). No systematic differences in the overall proportion of subjects reporting AEs were observed between treatment groups (where the pooled placebo group consisted of all subjects receiving placebo with different amounts of SNAC). The most common AEs were headache (15% vs. 4% of subjects receiving oral semaglutide vs. placebo with SNAC) and gastrointestinal disorders (14% vs. 13%). The majority of AEs were mild (99 AEs), while 5 AEs were moderate. All subjects recovered from the AEs. There were no severe or serious AEs and no subjects were withdrawn due to AEs. A total of 13 confirmed hypoglycaemic episodes were reported in 9 subjects, with no systematic distribution between treatment groups. No severe hypoglycaemic episodes were reported. No clinically relevant changes were observed in vital signs and laboratory safety parameters, and in the overall interpretation of physical examination and electrocardiogram.
3.2.2 Semaglutide Pharmacokinetics After a Single Dose
In the single-dose trial, a high proportion of subjects distributed among all oral semaglutide treatment arms had no measurable semaglutide in plasma (64 of 112 subjects).
The effect of varying the amount of SNAC in the oral semaglutide tablet from 150 to 600 mg on the plasma exposure of semaglutide is illustrated in Fig. 2. Semaglutide exposure levels were highest with 300 mg SNAC, suggesting that 300 mg, compared with 150 or 600 mg, is the optimal amount of SNAC to enhance absorption of oral semaglutide.

Modified from Buckley et al. [5]
Arithmetic mean semaglutide plasma concentration-time profiles after a single dose of oral semaglutide with varying amounts of SNAC in healthy male subjects (single-dose trial). Semaglutide plasma concentrations are expressed in percentage of the maximum mean concentration for a 5 mg oral semaglutide with 300 mg SNAC, or b 10 mg oral semaglutide with 300 mg SNAC (see Sect. 2.4 for further details). For subjects with no measurable semaglutide plasma concentration, concentrations were set to 0. SNAC sodium N-(8-[2-hydroxybenzoyl] amino) caprylate
At a fixed amount of 300 mg SNAC, both the proportion of subjects with measurable semaglutide in plasma, and the semaglutide exposure, appeared to increase with increasing dose of oral semaglutide from 2 to 10 mg (Table 2; Fig. 3).

Arithmetic mean semaglutide plasma concentration-time profiles after ascending single doses of oral semaglutide with 300 mg SNAC in healthy male subjects (single-dose trial). Semaglutide plasma concentrations are expressed in percentage of the maximum mean concentration for 10 mg oral semaglutide with 300 mg SNAC (see Sect. 2.4 for further details). For subjects with no measurable semaglutide plasma concentration, concentrations were set to 0. SNAC sodium N-(8-[2-hydroxybenzoyl] amino) caprylate
3.2.3 SNAC Pharmacokinetics After a Single Dose of Oral Semaglutide
Geometric mean AUC0–5 h,SNAC,SD ranged from 383 ng·h/mL for 5 mg semaglutide/150 mg SNAC to 1821 ng·h/mL for 20 mg semaglutide/600 mg SNAC (electronic supplementary Table S6). Geometric mean Cmax,SNAC,SD ranged from 432 ng/mL for 5 mg semaglutide/150 mg SNAC to 1673 ng/mL for 20 mg semaglutide/600 mg SNAC. Both AUC0–5 h,SNAC,SD and Cmax,SNAC,SD appeared to increase with increasing amount of SNAC. Median tmax,SNAC,SD appeared similar in all treatment groups, ranging between 0.38 and 0.75 h.
3.3 Multiple-Dose Trial
3.3.1 Safety After Multiple Doses of Oral Semaglutide
An overview of reported AEs in the multiple-dose trial is shown in Table 3, while the AEs reported in the highest proportion of subjects are summarised per system organ class and preferred term in electronic supplementary Table S7. A total of 662 AEs were reported in 99 subjects (93%). A similar proportion of subjects reported AEs across treatment groups. The most commonly reported AEs were gastrointestinal disorders. Among the healthy subjects, 50%, 84%, 28% and 72% of subjects reported gastrointestinal disorders for oral semaglutide 20 mg, oral semaglutide 40 mg, placebo and placebo with SNAC, respectively. Among subjects with T2D, 73%, 50% and 33% of subjects reported gastrointestinal disorders for oral semaglutide 40 mg, placebo and placebo with SNAC, respectively. The majority of AEs were mild; however, the severity of AEs increased with increasing dose of semaglutide. The majority of subjects recovered from the AEs at the end of the trial. Four serious AEs were reported in 3 subjects: vomiting (healthy placebo), atrial fibrillation (T2D placebo) and upper abdominal pain and aortic aneurysm (T2D 40 mg). All 3 subjects recovered from the serious AEs. A total of 11 healthy subjects and 4 subjects with T2D were withdrawn from the trial due to AEs. The AE withdrawals in healthy subjects were in the oral semaglutide 20 mg (1 subject; decreased appetite and nausea), oral semaglutide 40 mg [7 subjects; increased lipase (4), decreased appetite and nausea (2), vomiting], placebo (2 subjects; increased lipase and a serious AE of vomiting) and placebo with SNAC (1 subject; medication error) treatment groups, and those AE withdrawals in subjects with T2D were in the oral semaglutide 40 mg (3 subjects; increased lipase, vomiting and a serious AE of aortic aneurysm) and placebo (1 subject; serious AE of atrial fibrillation) treatment groups (increased lipase > 3 times the upper normal limit was a withdrawal criterion). A total of four confirmed hypoglycaemic episodes were reported in 4 healthy subjects. No severe hypoglycaemic episodes were reported. In the oral semaglutide treatment groups, mean levels of lipase increased by approximately 30–75% during the course of the trial. Compared with placebo, a mean increase in pulse rate of 7, 8 and 5 beats/min was observed for healthy subjects receiving 20 and 40 mg and subjects with T2D receiving 40 mg, respectively. Mean systolic blood pressure did not change systematically in healthy subjects, but decreased 13 mmHg (from 142 mmHg at baseline) with oral semaglutide 40 mg versus placebo in subjects with T2D. No systematic changes were observed in diastolic blood pressure and in the overall interpretation of physical examination and electrocardiogram.
3.3.2 Semaglutide Pharmacokinetics After Multiple Doses
At steady state, in healthy subjects, semaglutide plasma exposure was approximately twofold higher with oral semaglutide 40 mg vs. 20 mg. Furthermore, semaglutide plasma exposure did not differ between healthy subjects receiving 40 mg and subjects with T2D receiving 40 mg (Figs. 4, 5).

Geometric mean semaglutide plasma concentration-time profiles at steady state (multiple-dose trial). Profiles represent geometric means of the last 3 days of once-daily oral semaglutide treatment for 10 weeks. T2D type 2 diabetes

Comparison of a AUC0–24 h,semaglutide,SS and bCmax,semaglutide,SS between 20 and 40 mg doses of oral semaglutide in healthy males and between healthy males and males with T2D receiving 40 mg oral semaglutide (multiple-dose trial). Bars are estimated means and 95% CIs. Treatment comparisons show estimated treatment ratios [95% CI] and p value. AUC0–24 h,semaglutide,SS area under the semaglutide plasma concentration-time curve from 0 to 24 h at steady state, Cmax,semaglutide,SS maximum plasma semaglutide concentration at steady state, T2D type 2 diabetes, CI confidence interval
The t½,semaglutide,SS was comparable between treatment groups, with geometric means of 153, 161 and 158 h in healthy subjects receiving 20 and 40 mg and subjects with T2D receiving 40 mg, respectively, i.e. approximately 1 week for all groups.
The estimated mean within-subject day-to-day variability in AUC0–24 h,semaglutide,SS and Cmax,semaglutide,SS ranged between 19.7% and 34.9% across the three treatment groups, while the total variability (which consists of both the within-subject day-to-day variability and the between-subject variability) ranged between 63.6% and 84.4% (electronic supplementary Table S8).
3.3.3 SNAC Pharmacokinetics After Multiple Doses of Oral Semaglutide
Arithmetic mean 24-h SNAC plasma concentration-time profiles at steady state showed rapid absorption of SNAC and close to complete elimination at 12 h after dosing (electronic supplementary Fig. S3). The geometric mean AUC0–24 h,SNAC,SS and Cmax,SNAC,SS appeared to be comparable between the oral semaglutide treatment groups, ranging from 1206 to 1636 ng·h/mL for AUC0–24 h,SNAC,SS and from 579 to 658 ng/mL for Cmax,SNAC,SS (electronic supplementary Table S9). Median tmax,SNAC,SS was 0.50 h in both treatment groups of healthy subjects and 1.43 h in subjects with T2D. No accumulation of SNAC plasma exposure was observed during the 10 weeks of once-daily dosing (data not shown).
3.3.4 Pharmacodynamics After Multiple Doses of Oral Semaglutide
In subjects with T2D, a statistically significant decrease in HbA1c from a mean baseline level of 7.5% was seen after 10 weeks of treatment with 40 mg oral semaglutide versus placebo (treatment difference in change from baseline − 1.5% [95% CI − 1.8, − 1.3]; p < 0.001).
Body weight loss was observed after 10 weeks of oral semaglutide treatment in the multiple-dose trial in both healthy subjects (mean baseline body weight approximately 84 kg) and subjects with T2D (mean baseline body weight approximately 95 kg). The body weight reduction was statistically significantly greater for oral semaglutide versus placebo in healthy subjects (treatment difference in change from baseline, oral semaglutide—placebo: − 4.3 kg [95% CI − 6.3, − 2.3; p < 0.001] for 20 mg, and − 7.2 kg [95% CI − 8.9, − 5.4; p < 0.001] for 40 mg), and in subjects with T2D (− 5.4 kg [95% CI − 8.5, − 2.3]; p < 0.001).
4 Discussion
The two current trials investigated the safety and pharmacokinetics of the novel human GLP-1 analogue oral semaglutide during ascending single and multiple dosing. No safety concerns were identified in the two trials and the overall safety profile of oral semaglutide was as expected for the GLP-1 receptor agonist drug class. The pharmacokinetic properties of oral semaglutide were comparable in healthy subjects and subjects with T2D, and support the use of oral semaglutide for once-daily dosing.
The current single-dose trial investigated the pharmacokinetic properties of different combinations of semaglutide and SNAC in the tablet in order to determine the amount of SNAC that provided the largest systemic absorption of semaglutide. At 5 mg semaglutide, co-formulation with 300 mg SNAC resulted in substantially greater semaglutide plasma exposure compared with co-formulation with 150 mg SNAC. At 10 mg semaglutide, co-formulation with 300 mg SNAC resulted in slightly greater semaglutide plasma exposure compared with co-formulation with 600 mg SNAC. Thus, it seems that 300 mg SNAC in the oral semaglutide tablet is optimal to ensure the largest possible systemic absorption of semaglutide compared with both smaller and larger amounts of SNAC. Interestingly, a greater amount of SNAC in the tablet did not always lead to increased semaglutide exposure. Similar results were previously demonstrated when ibandronate was co-formulated with different amounts of SNAC [7]. Based on the current findings, a fixed amount of 300 mg SNAC was chosen for all dose strengths of semaglutide for further clinical development.
The therapeutic potential of oral semaglutide in the treatment of T2D was demonstrated in the multiple-dose trial. In subjects with T2D, once-daily dosing of 40 mg oral semaglutide for 10 weeks led to an HbA1c reduction of 1.5% points and a body weight reduction of 5.4 kg relative to placebo treatment. Body weight was also reduced with oral semaglutide versus placebo in healthy subjects. The current findings have subsequently been supported by results from the phase II dose-finding trial with oral semaglutide in subjects with T2D [10]. During 26 weeks of oral semaglutide treatment, dose levels of 2.5–40 mg significantly reduced HbA1c by 0.4 to 1.6% points versus placebo, and dose levels of 10–40 mg led to a significant body weight loss of 3.6–5.7 kg versus placebo [10]. The ability of oral semaglutide to improve glycaemic control and reduce body weight, as well as its effect on other clinical outcomes, are currently being investigated in phase III clinical trials.
In the single-dose trial, a number of subjects had no measurable semaglutide in plasma following dosing of oral semaglutide. This finding may be explained by low bioavailability of oral semaglutide in combination with a relatively high degree of variability in exposure after a single dose. However, therapeutic plasma levels of semaglutide are obtained at steady state in all subjects treated with once-daily oral semaglutide. This is due to the long half-life of oral semaglutide of approximately 1 week, leading to overlap in exposure from consecutive doses during once-daily dosing. The consequence is a reduced impact if a single dose is absorbed to a limited extent and, furthermore, a decreased variability in day-to-day semaglutide exposure. Thus, in the multiple-dose trial, the within-subject day-to-day variability in oral semaglutide exposure was only 20–35% at steady state. However, a total variability in semaglutide exposure of 65–85% was observed, suggesting that some between-subject variability occurs in the absorption of oral semaglutide. Nevertheless, as discussed above, a clinically relevant effect of once-daily oral semaglutide was observed with respect to improvement of glycaemic control as well as body weight reduction, both in the current multiple-dose trial and in the phase II trial [10]. In fact, with the exception of the lowest treatment dose investigated (2.5 mg oral semaglutide), almost 100% of patients experienced a reduction in HbA1c in the phase II trial, indicating that the variability in semaglutide exposure is not likely to affect the proportion of subjects with a clinically relevant treatment response [10].
The safety and pharmacodynamic results in the multiple-dose trial must be interpreted in light of the high dose levels (20–40 mg oral semaglutide) and the relatively fast initial dose escalation in this trial compared with the phase III trials, where dose levels of 3–14 mg oral semaglutide are investigated and a slower initial dose escalation is used to minimise gastrointestinal adverse effects. Furthermore, subjects in the multiple-dose trial fasted for 2 h after each dosing of oral semaglutide, while the more recent dosing guidance used in the phase III trials requests subjects to fast for at least 30 min after dosing. Extended post-dose fasting leads to greater exposure of oral semaglutide [11] and may also increase the risk of headache per se. Therefore, the frequency of AEs seen in the current multiple-dose trial may not be reflective of the frequency of AEs for oral semaglutide at the doses expected to be used in the clinical setting and following the most recent recommendations on dosing conditions.
One strength of the present investigation was the comprehensive number of different treatment groups tested in the two trials, as well as the inclusion of placebo groups. In order to minimise the variability in hormone levels and other factors that might affect the pharmacokinetic response, only male subjects were included in the current trials. This may be seen as a potential limitation to the applicability of the present results. Following subcutaneous administration, there is no effect of gender on the pharmacokinetics of semaglutide [12]. However, additional trials will be needed in order to investigate if the pharmacokinetic properties of oral semaglutide are comparable between males and females. A limitation of the single-dose trial was the apparent underestimation of semaglutide plasma concentrations with the applied LOCI assay due to a matrix effect. However, the relative concentrations could still be used and provided a valid basis for choosing the optimal amount of SNAC in the tablet formulation for future clinical development. A limitation of the multiple-dose trial was the low number of subjects with T2D, implying that, in particular, the pharmacodynamic results from this trial should be interpreted carefully. Nonetheless, it is reassuring that the reductions in HbA1c and body weight from the multiple-dose trial are consistent with those results obtained in a substantially higher number of subjects in the phase II trial with oral semaglutide [10].
5 Conclusion
Following single and multiple ascending doses, oral semaglutide up to 40 mg was safe and tolerable, with a safety profile as expected for the GLP-1 receptor agonist drug class. The pharmacokinetic properties of oral semaglutide were comparable in healthy subjects and subjects with T2D, and supported that oral semaglutide is suitable for once-daily dosing. Based on the pharmacokinetic results, oral semaglutide co-formulated with a fixed amount of 300 mg SNAC was chosen for further clinical development. The current efficacy results suggest that oral semaglutide can improve glycaemic control and reduce body weight in patients with T2D.
References
Bruno BJ, Miller GD, Lim CS. Basics and recent advances in peptide and protein drug delivery. Ther Deliv. 2013;4:1443–67.
Morishita M, Peppas NA. Is the oral route possible for peptide and protein drug delivery? Drug Discov Today. 2006;11:905–10.
Ismail R, Csóka I. Novel strategies in the oral delivery of antidiabetic peptide drugs—insulin, GLP 1 and its analogs. Eur J Pharm Biopharm. 2017;115:257–67.
Tan X, Cao X, Zhou M, Zou P, Hu J. Efficacy and safety of once-weekly semaglutide for the treatment of type 2 diabetes. Expert Opin Investig Drugs. 2017;26:1083–9.
Buckley ST, Bækdal TA, Vegge A, et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci Transl Med. 2018;10:eaar7047.
Baughman RA, Kapoor SC, Agarwal RK, Kisicki J, Catella-Lawson F, FitzGerald GA. Oral delivery of anticoagulant doses of heparin. A randomized, double-blind, controlled study in humans. Circulation. 1998;98:1610–5.
Bittner B, McIntyre C, Tian H, et al. Phase I clinical study to select a novel oral formulation for ibandronate containing the excipient sodium N-[8-(2-hydroxybenzoyl) amino] caprylate (SNAC). Pharmazie. 2012;67:233–41.
Castelli MC, Wong DF, Friedman K, Riley MG. Pharmacokinetics of oral cyanocobalamin formulated with sodium N-[8-(2-hydroxybenzoyl)amino]caprylate (SNAC): an open-label, randomized, single-dose, parallel-group study in healthy male subjects. Clin Ther. 2011;33:934–45.
American Diabetes Association. Defining and reporting hypoglycaemia in diabetes: a report from the American Diabetes Association Workgroup on Hypoglycaemia. Diabetes Care. 2005;28:1245–9.
Davies M, Pieber TR, Hartoft-Nielsen ML, Hansen OKH, Jabbour S, Rosenstock J. Effect of oral semaglutide compared with placebo and subcutaneous semaglutide on glycemic control in patients with type 2 diabetes: a randomized clinical trial. JAMA. 2017;318:1460–70.
Bækdal TA, Borregaard J, Donsmark M, Breitschaft A, Søndergaard FL. Evaluation of the effects of water volume with dosing and post-dose fasting period on pharmacokinetics of oral semaglutide [abstract]. Diabetes. 2017;66(Suppl 1):A315.
Carlsson Petri KC, Ingwersen SH, Flint A, Zacho J, Overgaard RV. Semaglutide s.c. once-weekly in type 2 diabetes: a population pharmacokinetic analysis. Diabetes Ther. 2018;9:1533–47.
Acknowledgements
The authors would like to thank all the participants, investigators and trial site staff who were involved in the conduct of the trials. They would also like to thank Carsten Roepstorff, PhD, CR Pharma Consult, Copenhagen, Denmark, for providing medical writing support, funded by Novo Nordisk.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the studies.
Funding
This study was funded by Novo Nordisk.
Conflicts of Interest
Charlotte Granhall, Morten Donsmark, Thalia M. Blicher, Flemming L. Søndergaard and Tine A. Bækdal are employees and shareholders of Novo Nordisk A/S. Mette Thomsen is an employee of Novo Nordisk A/S. Georg Golor declares no conflicts of interest.
Data Availability
Will individual participant data be available (including data dictionaries)? Individual participant data will be shared in data sets in a de-identified/anonymised format. What data in particular will be shared? Data sets from Novo Nordisk sponsored clinical research completed after 2001 for product indications approved in both the EU and US. What other documents will be available? Study protocol and redacted Clinical Study Report (CSR) will be available according to Novo Nordisk data sharing commitments. When will data be available (start and end dates)? The data will be available permanently after research completion and approval of product and product use in both the EU and US. No end date. With whom will data be shared? With bona fide researchers submitting a research proposal requesting access to data. For what types of analyses? For use as approved by the Independent Review Board (IRB) according to the IRB Charter (see www.novonordisk-trials.com). By what mechanism will data be made available? Access request proposal form and the access criteria can be found at www.novonordisk-trials.com. The data will be made available on a specialised SAS data platform.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Granhall, C., Donsmark, M., Blicher, T.M. et al. Safety and Pharmacokinetics of Single and Multiple Ascending Doses of the Novel Oral Human GLP-1 Analogue, Oral Semaglutide, in Healthy Subjects and Subjects with Type 2 Diabetes. Clin Pharmacokinet 58, 781–791 (2019). https://doi.org/10.1007/s40262-018-0728-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-018-0728-4