
Abstract
Background and Objectives
Dalbavancin is a novel, once-weekly glycopeptide antibiotic approved for treatment of acute bacterial skin infections. Given the importance of understanding any pharmacokinetic variability across different patient populations, a double-blind, placebo-controlled study was conducted to evaluate the pharmacokinetics, safety, and tolerability of a single 500-mg and a single 1000-mg intravenous dose of dalbavancin in healthy Japanese subjects.
Methods
Ten subjects received intravenous dalbavancin 1000 mg, five subjects received intravenous dalbavancin 500 mg, and three subjects received intravenous placebo.
Results
After a single infusion of dalbavancin, the maximal plasma concentration (C max) and area under the plasma concentration–time curve (AUC) increased in a proportional manner from 500 mg to 1000 mg (C max: 157 μg/ml and 299 μg/ml; AUClast: 10,850 μg·h/ml and 22,679 μg·h/ml, on the 500-mg and 1000-mg regimens, respectively) with low inter-subject variability. The mean terminal phase half-life (t 1/2) was 204 and 193 h after the 500-mg and 1000-mg dose, respectively. Clearance and volume of distribution were similar for the two dose concentrations. Treatment-emergent adverse events reported were considered to be of mild intensity. There were no relevant changes in laboratory values or vital signs over time in subjects in either treatment group.
Conclusions
Overall, dalbavancin 500 mg and dalbavancin 1000 mg, administered as a single 30-min infusion, was well tolerated in this population and resulted in plasma exposures similar to those in non-Asians.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Dalbavancin demonstrated dose-proportional pharmacokinetics in Japanese patients and is well tolerated. |
The pharmacokinetic profile in Japanese patients is similar to that of non-Asian subjects. |
1 Introduction
Dalbavancin is a novel, semisynthetic, lipoglycopeptide antibiotic with in vitro and in vivo activity against Gram-positive pathogens, including a minimal inhibitory concentration (MIC) required to inhibit the growth of 90 % of the isolates (MIC90) for Staphylococcus aureus of 0.06 μg/ml [1]. Dalbavancin has a terminal half-life (t 1/2) of 2 weeks, related in part to 93 % protein binding, and reduces the bacterial inoculum in animal models when administered as larger, less frequent doses, as compared with smaller, more frequent doses [2]. Pharmacokinetic studies have demonstrated that plasma concentrations obtained after once-weekly dosing of dalbavancin provide mean total and calculated free plasma concentrations above the S. aureus bactericidal concentration for 7–14 days [3]. Efficacy was shown in a development program that included four trials involving 1086 patients with skin infection [4–7].
Dalbavancin, like other glycopeptides, binds to the d-ala-d-ala terminus of pentapeptides in nascent peptidoglycan located within the bacterial cell wall [8], thus preventing cross-linking. Dalbavancin does not undergo cytochrome P450 (CYP) oxidative metabolism and is not a substrate, inhibitor or inducer of any known CYP-isozyme. Elimination occurs by means of non-oxidative metabolism through the hepato-gastrointestinal routes as well as renally as active intact dalbavancin. The pharmacokinetics of intravenous dalbavancin have been previously studied in subjects given single-dose regimens ranging from 70 to 1500 mg, multiple-dose regimens ranging from 480 to 1600 mg administered over 7 days, and once-weekly doses up to a cumulative total of 4500 mg given over 8 weeks [3, 9, 10]. Plasma concentrations were determined in healthy volunteers and clinically infected patients. Special populations have also been evaluated including those with renal and hepatic impairment, and adolescent children [3, 11–14]. Dalbavancin pharmacokinetics are linear, exhibit low inter-individual variability, and are similar in patients and healthy subjects. The area under the plasma concentration–time curve (AUC)/MIC ratio appears to be the best predictor of bacterial eradication and clinical efficacy [2]. There is no evidence of QTc prolongation [17]. The long elimination t 1/2 of dalbavancin of ~8.5 days (~200 h) and its terminal t 1/2 of more than 346 h (14.4 days) support the approved dosing regimen of 1000 mg intravenously followed 1 week later by 500 mg, delivered over 30 min [18], and provide an alternative to current therapy dosing of patients with acute bacterial skin and skin structure infection (ABSSSI) [3, 7]. The primary objective of this study was to evaluate the safety, tolerability, and pharmacokinetics of both single 500-mg and single 1000-mg intravenous doses of dalbavancin in healthy Japanese subjects in order to understand if any dose adjustment in this patient population, relative to non-Asians, is required.
2 Subjects and Methods
2.1 Study Design
The study was conducted between June 2011 and February 2012 as a double-blind, placebo-controlled design in healthy male and female Japanese volunteers. Eligible subjects were randomized to receive either intravenous dalbavancin or intravenous placebo in a 5:1 ratio. The study consisted of a screening visit (day −28 to day −2), a check-in visit (Day −1), a treatment phase (days 1–3), and outpatient visits on day 7, day 14, and day 21 (±1 day). Subjects returned for an end-of-study visit on day 28 (±1 day). Individual subject participation lasted up to 57 days.
The doses of 1000 and 500 mg of dalbavancin were chosen based on the dosage regimen for ABSSSI. Subjects received a single intravenous infusion over 30 min on day 1 of either placebo (250 ml 5 % dextrose for injection, 50 ml sterile water for injection), dalbavancin 500 mg (250 ml 5 % dextrose for injection, with 25 ml sterile water for injection, 500 mg dalbavancin with 25 ml sterile water), or dalbavancin 1000 mg (250 ml 5 % dextrose for injection, with 50 ml of sterile water for injection, containing 1000 mg dalbavancin). Safety was assessed through physical examinations, clinical laboratory tests (chemistry, hematology, and urinalysis), vital sign measurements (heart rate, blood pressure, respiratory rate, and oral body temperature), and adverse event and concomitant medication monitoring.
Subjects were queried regularly on all study days for any adverse events using non-leading questions. All adverse events reported spontaneously during the course of the study were also recorded, as were any clinically significant clinical laboratory, vital sign, or ECG findings. Serious adverse events are those that result in death, are life-threatening, require inpatient hospitalization or prolongation of existing hospitalization, result in persistent or significant disability or incapacity, result in congenital anomaly or birth defect, or are assessed as being a medically important event based on medical and scientific judgment. Blood sampling for plasma dalbavancin concentration determination occurred at scheduled time points.
2.2 Study Subjects
Subjects between the ages of 18 and 55 years who had a body mass index (BMI) between 18.5 and 30.0 kg/m2 were considered eligible to participate in the study if they voluntarily agreed to participate and signed an internal review board-approved informed consent form and a supplemental Japanese consent form prior to performing any of the screening procedures. Patients enrolled in the study were born in Japan, with both parents and both sets of grandparents also Japanese, had a valid Japanese passport, had not lived outside Japan for more than 5 years and presently lived in a Japanese community, generally eating a typical Japanese diet with lifestyle not notably different from that experienced in Japan.
Female subjects were surgically sterile (hysterectomy or bilateral tubal ligation), postmenopausal for at least 2 years and ≥50 years of age or confirmed by follicle stimulating hormone >40 U/L. If of childbearing potential, females were not pregnant or lactating and agreed to remain sexually abstinent from entry through 60 days post-dosing or have any male partner simultaneously use two proven effective contraceptive methods. Male patients, without a vasectomy or with a vasectomy within 6 months prior to enrollment into the study, were to use a condom and be instructed that their female partners were to use another effective form of contraception.
Subjects were not considered eligible to participate in the clinical study if they had a clinically significant history or evidence of cardiovascular, respiratory, hepatic, renal, gastrointestinal, endocrine, neurological, immunological, hematologic, oncologic, or psychiatric disorders as determined by the investigator or designee or had an estimated creatinine clearance of ≤80 ml/min based on the Cockcroft–Gault formula [15]. In addition, subjects were excluded if any concurrent disease or condition was present that, in the opinion of the investigator, would have made the subject unsuitable for participation in the clinical study, the subject had an aspartate aminotransferase (AST) or alanine aminotransferase (ALT) >2× the upper limit of normal (ULN), total bilirubin >2× ULN, alkaline phosphatase (AP) > 2× ULN, hemoglobin <12.0 g/dl, or any significant clinical, laboratory, or ECG findings at baseline. Female subjects who were breastfeeding, planning a pregnancy during the course of the study, or who had a positive serum pregnancy test at screening or Day-1 were excluded, as were subjects having a history of alcohol or illicit drug abuse within 2 years of entry, those regular alcohol intake exceeded 24 ounces of beer or the equivalent per day, or who had a positive breath test for ethanol.
Other exclusion criteria at screening or Day-1 included: positive test for hepatitis B surface antigen, hepatitis C antibody, human immunodeficiency virus antibody, positive urine drug test, smoking more than 10 cigarettes a day, recent donation of >500 ml of blood or blood products, inability to abstain from the use of over-the-counter medications, prescription medications, or herbal remedies, or use of an investigational drug within the last 28 days. Subjects with known hypersensitivity or intolerance to a glycopeptide, lipoglycopeptide, or any constituent of dalbavancin were excluded. Overall general health was determined by pre-study medical evaluation and principal investigator discretion.
2.3 Administration of the Investigational Medicinal Product
Subjects received a 300-ml intravenous infusion of the active solution containing 500 or 1000 mg of dalbavancin or the placebo solution over 30 min (−2 to +10 min). The intravenous line was flushed with 5 % dextrose solution (20–30 ml) at the end of the infusion to ensure delivery of the entire dose. The intravenous line was inserted in the contra-lateral arm used to obtain blood draws for safety and pharmacokinetic sampling.
2.4 Blinding
The clinical study was performed in a double-blind manner. With the exception of unblinded pharmacy staff, neither the study subjects nor the clinical staff knew the treatment each subject received. Unblinded pharmacy staff covered the infusion bags with amber-colored bags to mask the slight color difference between dalbavancin and placebo solutions. The amber-colored bags were secured with staples or tamper-evident tape to prevent unblinding. The study blind was not to be broken except in a medical emergency where knowledge of the administration of dalbavancin would have affected the treatment of the emergency. The decision to break the blind was to be made on a case-by-case basis at the discretion of the principal investigator in collaboration with the Sponsor Medical Monitor. The applicable standard operating procedure was to be followed for blind-breaking procedures. After database lock, the overall randomization code was broken only for reporting purposes.
2.5 Pharmacokinetic Analyses
No efficacy or pharmacodynamic endpoints were evaluated in this clinical study.
Pharmacokinetic data were to be analyzed and presented according to the statistical analysis plan. The individual subject concentration–time data were listed and displayed graphically on linear and log scales. The non-compartmental pharmacokinetic parameters described later in this section were calculated using WinNonlin Professional Version 5.2 (Certara, L.P, Princeton, NJ, USA). All pharmacokinetic parameters were calculated from the concentration–time data as described. All individual parameter estimates were to be calculated based on the actual dose administered with the actual pharmacokinetic sampling times recorded in the case report form. For descriptive statistics, the nominal sampling time was to be used. Any subjects with missing concentration data were included in the pharmacokinetics analysis set provided that, in the opinion of the pharmacokinetic analyst, a complete set of pharmacokinetic parameters could be calculated. Concentrations below the lower limit of quantification (LLOQ) were substituted by 1/2 LLOQ value prior to the calculation of the summary statistics.
The following were calculated using the linear up/logarithmic down trapezoidal method: AUC, AUC from time zero until the last quantifiable concentration (AUClast), AUC from time zero through day 7 (AUC0–7 days), and AUC from time zero to day 14 (AUC0–14 days). AUC from time zero (predose) to infinite time (AUC0–∞) was calculated using the following equation: AUC0–∞ = AUClast + C t /λ z , where C t is the last quantifiable concentration and λ z is the terminal elimination rate constant. Percentage of AUC0–∞ obtained by extrapolation (AUC%extrap) was calculated as (1 − [AUClast/AUC0–∞]) × 100. The terminal elimination rate constant was estimated at terminal phase by linear regression of concentration versus time data presented in a log-linear scale. Only those data points that were judged to describe the terminal log-linear decline were used in the regression. A minimum number of three data points in the terminal phase were used in calculating λ z with the line of regression starting at any post-C max data point [maximal plasma concentration (C max) was not to be part of the regression slope]. Clearance (CL) was calculated as dose/AUC0–∞. The volume of distribution (V d) was calculated as CL/λ z . Corrections for free drug portion of dalbavancin were not made; rather, all values reported are total drug concentrations for dalbavancin.
2.6 Sampling Scheme
Blood was sampled pre-dose and at end of infusion, and 1, 2, 4, 12, 24, and 48 h post-dose, as well as on Days 7, 14, 21 (±1 day), and 28 (±1 day) at approximately the time of Day 1 dosing. Ten milliliters of blood was collected in sodium heparin-containing vacutainers from each subject at each time point. Samples were cooled by an ice bath or cooling device until processed, centrifuged within 30 min of collection at approximately 1500 g and 4 °C for 10 min, and then returned to an ice bath or cooling device. Plasma was evenly divided and transferred into duplicate polypropylene tubes. Approximately 2 ml of plasma per tube was required. Immediately after separation samples were stored and frozen at approximately −20 °C or below until shipment to the bioanalytical laboratory. Plasma was analyzed by liquid chromatography–tandem mass spectrometry (LC–MS/MS) validated for dalbavancin in the linear range from 0.5 μg/ml to 500 μg/ml using 50 μl of K2 EDTA plasma [16]. The pharmacokinetic parameters listed in this section were calculated from the plasma concentration–actual time profiles. The non-compartmental analysis was performed using WinNonlin Version 5.2. Formal sample size calculations were not performed. A total of 18 subjects were considered suitable to achieve the study objectives.
2.7 Protocol Amendments
Two protocol amendments were issued during the study. The first was issued due to an update of the wording of the serious adverse event definition of the United States Food and Drug Administration Safety Reporting Requirements, and the second was to add an additional six subjects of whom five subjects received 500 mg dalbavancin and one subject received placebo.
3 Results
3.1 Pharmacokinetics
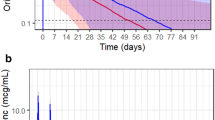
A single intravenous infusion of dalbavancin 500 mg was administered to five healthy Japanese subjects and 1000 mg to ten healthy Japanese subjects. Baseline demographic characteristics are provided in Table 1. Pharmacokinetic parameters following the administration of dalbavancin 1000-mg and 500-mg regimens are summarized in Tables 2 and 3, respectively. Peak plasma concentrations were reached following the end of the infusion. Mean concentration versus time plots in linear scales for those who received the 1000-mg or 500-mg dose regimens are illustrated in Figs. 1 and 2, respectively.

Mean (±SD) plasma concentrations of intravenous dalbavancin 1000 mg

Mean (±SD) plasma concentrations of intravenous dalbavancin 500 mg
After a single intravenous infusion of dalbavancin, C max and AUC increased in a proportional manner from the 500-mg dose to the 1000-mg dose. Plasma dalbavancin geometric mean (±SD) C max increased from 157 ± 18 μg/ml after the 500-mg dose to 299 ± 36 μg/ml for the 1000-mg dose level and occurred at a median time of 0.5 h for the 500-mg dose to 1 h for the 1000-mg dose post-start of infusion. Inter-subject variability was relatively low with a coefficient of variation (CV%) of 11.2 % for the 500-mg dose and 12.0 % for the 1000-mg dose level. Geometric mean AUClast increased from 10,850 ± 1781 μg·h/ml after the 500-mg dose to 22,679 ± 1982 μg·h/ml for the 1000-mg dose level. Similarly, geometric mean AUC0–7, AUC0–14, and AUC0–∞ increased from 5756 ± 671 μg·h/ml, 8554 ± 1148 μg·h/ml, and 12,042 ± 2460 μg·h/ml after the 500-mg dose to 12,155 ± 1466 μg·h/ml, 18,028 ± 1834 μg·h/ml, and 24,745 ± 2093 μg·h/ml for the 1000-mg dose level. Exposure measured in terms of the AUC also showed relatively low inter-subject variability, ranging from 11.6 to 20.1 % for the 500-mg dose and from 8.4 to 12.0 % for the 1000-mg dose level. The mean AUC%extrap ranged from 8.34 % for the 1000-mg dose to 9.83 % for the 500-mg dose, indicating reliable AUC0-∞ estimations for both dose concentrations. Based on mean partial AUCs, the percentage of total exposure (i.e., AUC0–∞) were similar for the two dose concentrations, with approximately 47 % after 7 days and 70 % after 14 days for the 500-mg dose and approximately 49 % after 7 days and 73 % after 14 days for the 1000-mg dose. The mean terminal phase t 1/2 was consistent across the dose concentrations: 204 h (range 182–240 h) after the 500-mg dose and 193 h (range 162–220 h) for the 1000-mg dose level. Clearance (CL) and volume of distribution were similar for the two dose concentrations. Mean CL was 42.2 ml/h (range 32.9–54.1 ml/h) for the 500 mg dose and 40.5 ml/h (range 34.8–45.8 ml/h) for the 1000-mg dose, and mean V d was 12,182 ml (range 11,412–14,205 ml) for the 500-mg dose and 11,301 ml (range 8999–13,691 ml) for the 1000-mg dose. Compartmental analysis was performed as an additional exercise to establish the probable error in determination of C max, T max, and AUC0–∞ due to the fact that in seven subjects receiving active drug (1000 mg), the end-of-infusion sample was taken just prior to the completion of the infusion. The percentage deviation between this predicted value and the observed value obtained by non-compartmental analysis (predicted/observed) did not exceed 10.7 % in any subject for C max (average C max deviation 7 % in seven subjects). Positive deviations were also recorded in two of the three remaining actively treated subjects in the cohort who had end-of infusion samples correctly taken. The AUC did not deviate significantly with a maximum recorded individual subject AUC0–∞ deviation of −3.3 % (average AUC0–∞ deviation was 2.0 % in seven subjects). Due to the small deviations recorded, it can be concluded that the pharmacokinetic samples taken during infusion did not significantly affect study conclusions.
3.2 Safety and Tolerability
Safety and tolerability were assessed and a summary of the demographics of the safety population is shown in Table 4. In the 500-mg dose group, five subjects received a single 500-mg infusion of dalbavancin and one subject received a single infusion of placebo. In the 1000-mg dose group, ten subjects received a single 1000-mg infusion of dalbavancin and two subjects received a single infusion of placebo. In the 500-mg dose group, two of the five subjects (40 %) receiving 500-mg dalbavancin experienced a single treatment-emergent adverse event (TEAE), and the one subject (100 %) receiving placebo experienced two TEAEs. In the dalbavancin group, these consisted of dyspepsia (mild) and dizziness (mild); dyspepsia was considered by the investigator to be not related and dizziness unlikely to be related to study medication. The subject receiving placebo reported eczema of mild intensity and cellulitis of moderate intensity, both of which were considered by the investigator to be not related to study medication. All TEAEs of the dalbavancin group were resolved at the end of the study.
For the 1000-mg dose group, a total of eight TEAEs were reported by three of the ten subjects (30 %) receiving dalbavancin; none of the two subjects receiving placebo reported a TEAE. The reported TEAEs consisted of palpitations, diarrhea (twice reported by one subject), dizziness, headache, oropharyngeal pain, blister, and rash. Of these, rash and diarrhea were considered by the investigator to be possibly related to study medication. All other TEAE reports were considered unlikely to be related. The palpitation TEAE was not resolved at the end of the study, whereas all other TEAEs had resolved. None of the subjects died, no serious adverse event occurred, and no subject discontinued study medication due to a TEAE.
Mean and median values of all parameters of clinical chemistry and hematology did not show any notable changes from baseline at any post-dose time point. None of the individual laboratory values of clinical chemistry, hematology, and urinalysis showed any relevant changes from baseline in any dose group or for placebo. A few isolated, abnormal values of some of the parameters were found in both dose groups and for placebo, but none were considered by the investigator to be of clinical significance. No clinically relevant, abnormal laboratory values were reported in any dose group or for placebo.
4 Discussion
The primary objectives of this study were to evaluate the safety, tolerability, and pharmacokinetics of single 500-mg and 1000-mg intravenous doses of dalbavancin in healthy male and female adult Japanese subjects. The results from this trial have confirmed that the pharmacokinetics of dalbavancin are similar in Japanese patients to those previously studied. The geometric mean dalbavancin C max and AUC increased in a proportional manner from the 500-mg to the 1000-mg dose level. The mean t 1/2 was consistent at 193–204 h across two dose concentrations, and the CL and V d were not dose dependent; mean CL ranged from 40.5 to 42.2 ml/h and mean V d ranged from 11,301 to 12,182 ml across two dose concentrations. The TEAEs in subjects receiving dalbavancin in both dose groups were considered to be of mild intensity and the sole adverse event of moderate intensity was reported in a subject receiving placebo and, while reported from a small number of subjects, is consistent with the experience in recent phase III trials [4, 7]. There were no relevant changes in laboratory values or vital signs over time in subjects in either treatment group. Overall, dalbavancin 500 mg and dalbavancin 1000 mg, administered as single 30-min infusions, were well tolerated in healthy male and female adult Japanese subjects.
The measured t 1/2 in the Asian patients enrolled in this study was 193–204 h (8.0–8.5 days) consistent with previous studies in non-Asians in which follow-up measurements were taken out for 28 days after dosing [3]. Other studies with follow-up periods as long as 70 days [19] have documented a terminal t 1/2 as long as 15.5 days, an observation consistent with the low clearance of dalbavancin.
Dalbavancin was the first long-acting lipoglycopeptide to be approved for use in the USA, in May 2014. Its long t 1/2 and relatively short infusion time allows for once-weekly dosing for treatment of acute bacterial skin and skin structure infections known or suspected to involve Gram-positive pathogens, while improving the experience of infected patients, allowing the avoidance of a need for a central line and the possibility of either preventing hospitalization in suitable patients or allowing an earlier discharge.
5 Conclusions
This study demonstrated in healthy Japanese volunteers that the C max and AUC of dalbavancin increased proportionally from the 500-mg to the 1000-mg dose level and were associated with a consistent t 1/2 of 193–204 h as has been described in other patient populations. This study supports the use of dalbavancin administered once weekly for the treatment of Gram-positive infections in the Japanese population.
References
Lopez S, Hackbarth C, Romano G, Trias J, Jabes D, Goldstein B. In vitro antistaphylococcal activity of dalbavancin, a novel glycopeptide. J Antimicrob Chemother. 2005;55(Suppl S2):ii21–4.
Andes D, Craig WA. In vivo pharmacodynamic activity of the glycopeptide dalbavancin. Antimicrob Agents Chemother. 2007;51:1633–42.
Dorr M, Jabes D, Cavaleri M, Dowell J, Mosconi G, Malabarba A, et al. Human pharmacokinetics and rationale for once-weekly dosing of dalbavancin, a semi-synthetic glycopeptide. J Antimicrob Chemother. 2005;55(Suppl 2):ii25–ii30.
Jauregui L, Babazadeh S, Seltzer E, Goldberg L, Krievins D, Frederick M, et al. Randomized, double-blind comparison of once-weekly dalbavancin versus twice-daily linezolid therapy for the treatment of complicated skin and skin structure infections. Clin Infect Dis. 2005;41:1407–15.
Raad I, Darouiche R, Vazquez J, Lentnek A, Hachem R, Hanna H, et al. Efficacy and safety of weekly dalbavancin therapy for catheter-related bloodstream infection caused by gram-positive pathogens. Clin Infect Dis. 2005;40:374–80.
Seltzer E, Dorr M, Goldstein B, Perry M, Dowell J, Henkel T. Once-weekly dalbavancin versus standard-of-care antimicrobial regimens for treatment of skin and soft-tissue infections. Clin Infect Dis. 2003;37:1298–303.
Boucher H, Wilcox M, Talbot G, Puttagunta S, Das A, Dunne M. Once-weekly dalbavancin versus daily conventional therapy for skin infection. N Engl J Med. 2014;370:2169–79.
Anderson J, Matsuhashi M, Haskin M, Strominger J. Lipid-phosphoacetylmuramylpentapeptide and lipid-phosphodisaccharide-pentapeptide: presumed membrane transport intermediates in cell wall synthesis. Proc Natl Acad Sci (US). 1965;53:881–9.
Dunne M, Puttagunta S, Sprenger C, Rubino C, Van Wart S, Baldassarre J. Extended-duration dosing and distribution of dalbavancin into bone and articular tissue. Antimicrob Agents Chemother. 2015;59:1849–55.
Leighton A, Gottlieb A, Dorr M, Jabes D, Mosconi G, VanSaders C, et al. Tolerability, pharmacokinetics, and serum bactericidal activity of intravenous dalbavancin in healthy volunteers. Antimicrob Agents Chemother. 2004;48:940–5.
Buckwalter M, Dowell J. Population pharmacokinetic analysis of dalbavancin, a novel lipoglycopeptide. J Clin Pharmacol. 2005;45:1279–87.
Dowell J, Goldstein B, Buckwalter M, Stogniew M, Damle B. Pharmacokinetic-pharmacodynamic modeling of dalbavancin, a novel glycopeptide antibiotic. J Clin Pharmacol. 2008;48:1063–8.
Marbury T, MD, Dowell JA, Seltzer E, Buckwalter M. Pharmacokinetics of dalbavancin in patients with renal or hepatic impairment. J Clin Pharmacol 2009;49:465–476.
Bradley JS, Puttagunta S, Rubino CM, Blumer JL, Dunne M, Sullivan JE. Pharmacokinetics, safety and tolerability of single dose dalbavancin in children 12–17 years of age. Pediatr Infect Dis J. 2015;34(7):748–52.
Cockcroft D, Gault MD. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41.
Alebic-Kolbah T, Demers R, Cojocaru L. Dalbavancin: quantification in human plasma and urine by a new improved high performance liquid chromatography–tandem mass spectrometry method. J Chromatogr B. 2011;879:2632–41.
Dunne M, Zhou M, Darpo B. A thorough QT study with dalbavancin—a novel lipoglycopeptide antibiotic for the treatment of acute bacterial skin and skin structure infections. Int J Antimicrob Agents. 2015;45:393–8.
Dalvance™ (dalbavancin for injection) [package insert]. Chicago, IL: Durata Therapeutics, Inc; 2014.
Nord CE, Rasmanis G, Wahlund E. Effect of dalbavancin on the normal intestinal microflora. J Antimicrob Chemother. 2006;58:627–31.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Durata Therapeutics, Inc., a subsidiary of Allergan plc.
Conflicts of interest
P.J.S, R.C.O., S.P., and M.W.D are employees of and stockholders in Durata Therapeutics, Inc.
Ethical approval
All procedures in this study were carried out in accordance with the 1964 Helsinki declaration and its amendments and the ethical committee or institutional review board which approved the study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Scoble, P.J., Owens, R.C., Puttagunta, S. et al. Pharmacokinetics, Safety, and Tolerability of a Single 500-mg or 1000-mg Intravenous Dose of Dalbavancin in Healthy Japanese Subjects. Clin Drug Investig 35, 785–793 (2015). https://doi.org/10.1007/s40261-015-0340-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-015-0340-4