
Abstract
Background
Antibiotics for the treatment of complicated, multidrug-resistant Gram-positive infections are limited, especially when prolonged treatment is necessary. Oritavancin is approved for the treatment of serious skin infections as a 1200 mg single-dose regimen, but case reports describe supplemental doses given at weekly intervals ranging from 800 mg to 1200 mg.
Objective
This study determined population pharmacokinetic estimates for a 1200 mg single dose with and without an 800 mg dose 1 week apart.
Methods
A simulated oritavancin 1200 mg dose was infused over 3 h followed 7 days later by a simulated 800 mg dose infused over 3 h for pharmacokinetic estimation.
Results
The oritavancin dosing displayed predictable linear pharmacokinetics and therapeutic concentrations. The total and free oritavancin concentrations remained above the susceptibility breakpoint (0.12 mg/L) for 8 weeks and 4.6 weeks, respectively, with the two-dose regimen. This was significantly greater than the single-dose regimen. This regimen also results in a greater area under the drug concentration–time curve (AUC) above the susceptibility breakpoint compared to the single-dose regimen (p < 0.001), and it maintains a high AUC:minimum inhibitory concentration (MIC) ratio against organisms with MICs up to 0.25 mg/L.
Conclusion
These results along with the observational clinical reports of success and safety with this dosing scheme of 1200 mg followed by 800 mg 7 days later provide evidence for further evaluation of this approach when prolonged oritavancin treatment may be indicated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Therapeutic concentrations of a novel oritavancin dosing regimen of 1200 mg and 800 mg administered 7 days apart were assessed in a pharmacokinetic model. |
This dosing regimen achieved oritavancin concentrations above the susceptibility breakpoint (0.12 mg/L) for 8 weeks, and maintained a high AUC:MIC ratio for efficacy against organisms with MICs up to 0.25 mg/L. |
Along with the observational clinical reports of success and safety with this dosing scheme, this study provides evidence for further evaluation of this approach when prolonged oritavancin treatment may be indicated. |
1 Introduction
Despite the available antibiotic options for invasive, multidrug-resistant Gram-positive infections, the treatment remains challenging for clinicians. New antibiotics with activity against these organisms, such as methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus spp. (VRE), are being used, but limitations exist that prevent broad applications. A major issue is the continued difficulties involved in treating specific infection types such as bone and joint infections, bacteremia and endocarditis, and pneumonia, among others [1,2,3]. Additionally, prolonged duration of antibiotic treatment associated with these infection types introduces toxicity concerns with some treatment options [4,5,6,7]. Further, the pharmacokinetics of antibiotics in difficult-to-penetrate compartments, such as bone, have not been well studied in patients during long-term treatment [8].
The prolonged half-life of the new lipoglycopeptide antibiotics, such as oritavancin, make them attractive options for patients requiring intravenous management of complicated infections [9, 10]. These agents possess broad-spectrum Gram-positive activity, particularly for oritavancin, with activity against both MRSA and VRE. Multidrug-resistant organisms continue to account for a significant percentage of complicated infection types, therefore reliable activity against these strains is an important characteristic.
Oritavancin is frequently used for acute bacterial skin and skin-structure infections (ABSSSI) in susceptible Gram-positive organisms. Although oritavancin demonstrated activity against vancomycin-susceptible Entercococcus spp. in the SOLO I and II clinical trials [11, 12], recent clinical data suggest that it has high effectiveness in complicated infections due to VRE [10, 13]. This is despite oritavancin not having a defined pharmacokinetic–pharmacodynamic target against VRE. Case reports of successful treatment with oritavancin in patients with complicated infections also suggest that this antibiotic has a place in therapy for prolonged use beyond the initial 1200 mg single dose.
For prolonged oritavancin treatment, clinicians have used a variety of dosing strategies. Doses are most reportedly given at weekly intervals and have ranged from 800 mg to 1200 mg. In this study, we used population pharmacokinetic estimates to simulate oritavancin concentrations following a 1200 mg single dose with and without subsequent 800 mg dosing administered 1 week later. This study validates that 1200 mg followed by an 800 mg dose 7 days later prolongs targeted pharmacokinetic parameters for efficacy.
2 Methods
A simulation was performed to assess the predicted oritavancin concentrations that would result from doses of 1200 and 800 mg administered intravenously over 3 h 7 days apart. The population pharmacokinetic model used was based upon relatively sparse sampling (0, 3, 12, 24, 72, and 576 h) after a single 1200 mg intravenous dose of oritavancin infused over 3 h in 297 patients [14]. The covariate relationships included in the final simulation were median age and height based on the previously published pharmacokinetic model [14]. The model yielded similar parameters for a three-compartment model that was described by the same group in 2009 [15].
NONMEM (v7.4) was used to simulate total plasma concentrations for 500 hypothetical patients after a single 1200 mg intravenous dose over 3 h, as well as after doses of 1200 mg and 800 mg administered 1 week apart. The interindividual variances for the six pharmacokinetic parameters described for the three-compartment model by Rubino et al. were used, as were the additive and proportional residual errors. The NONMEM simulation also calculated both the cumulative time and the area under the drug concentration–time curve from zero to infinity [AUC0-∞ (mg · days/L)], during which the oritavancin concentration was above 0.12 mg/L. This threshold was used as it is the lowest breakpoint for susceptibility for oritavancin among Gram-positive organisms. The results of the NONMEM simulations were processed using R (version 3.4.4) in RStudio (1.1.1335). A two-sided t-test was used to analyze the pharmacokinetic results between the single-dose and two-dose regimens with p ≤ 0.05 for significance.
3 Results
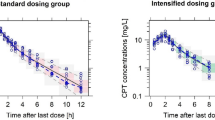
An oritavancin 1200 mg dose infused over 3 h followed 7 days later by an 800 mg dose infused over 3 h displayed predictable and therapeutic concentrations. Figure 1 compares the simulated plasma oritavancin concentrations following one versus two doses. The blue and red lines show the median concentrations expected after one 1200 mg dose or two doses of 1200 mg and 800 mg, respectively. Similarly, the blue and red ribbons show the respective 90% confidence intervals for the two dosing regimens, with purple representing the overlap intervals between the regimens.

Comparison of simulated oritavancin concentrations. a Total oritavancin concentrations, b unbound free oritavancin concentrations. The blue line displays the median plasma concentrations expected after one 1200 mg dose followed by an 800 mg dose 1 week apart, and the red line represents the single 1200 mg dose. The ribbons represent the 90% confidence intervals, with purple ribbons representing overlap between the dosing schemes
Table 1 displays the pharmacokinetic parameters achieved with the single- and two-dose regimens. The simulated AUC0-72 h for total drug exposure after the single 1200 mg dose were 778.7, 1399.1, and 2481.4 (mg · h/L) for the fifth, 50th, and 95th percentiles, respectively. These percentiles were within 2.2–14.4% difference versus those determined by Rubino et al. with a similar 1200 mg dose simulation, corroborating the model consistency [14]. As expected, the second 800 mg dose provides slightly lower but overall similar maximum concentrations (Cmax) versus the initial 1200 mg dose, and the minimum concentration (Cmin) total and unbound free drug concentrations were significantly higher at day 29 with the two-dose regimen. Also, the AUC0-∞ was predictably 67% higher with the two-dose regimen over the estimation duration. The oritavancin concentrations remained above the susceptibility breakpoint (0.12 mg/L) with the two-dose regimen for the entire 8-week simulation for total drug and 4.6 weeks for unbound free drug. Compared to the single 1200 mg dose, the two-dose regimen provided oritavancin concentrations above the susceptibility breakpoint for 11.3 more days (p < 0.0001). The two-dose regimen simulated in our study provides a total drug AUC/minimum inhibitory concentration (MIC) ≥ 17,568 (free unbound > 2,635.2) against organisms with an MIC value up to 0.25 mg/L.
4 Discussion
Oritavancin is increasingly used for a broad spectrum of Gram-positive pathogens and more complicated infections other than ABSSSI [13, 16]. The established safety profile of oritavancin despite the prolonged half-life has stimulated clinical experimentation with use and dosing strategies. Along with initial case reports of use in complicated infection types, the studies in this supplement demonstrate more evidence of the favorable safety, effectiveness, and pharmacoeconomics of oritavancin treatment. In a retrospective study by Brownell et al., most of the 75 patients received an initial 1200 mg dose followed by 1200 mg or 800 mg weekly thereafter, resulting in cure or improvement in 93% of these patients [17]. Our pharmacokinetic simulation of 1200 mg and 800 mg doses administered 7 days apart support this novel dosing approach when more than a single dose is used for infection treatment. It should be noted that some patients in the Brownell et al. study along with patients in a previous study by this group [13] received a full 1200 mg dose, often at day 10, if the second dose was not administered within 7 days. This dosing scheme was not analyzed in our study but should be considered for future work.
SIMPLIFI was a multicenter, randomized, double-blind, phase II clinical trial evaluating the safety and effectiveness of oritavancin in complicated skin and skin structure infections. It compared three different oritavancin dosing regimens: daily—200 mg for 3–7 days; infrequent—800 mg on day 1 with the option for 400 mg on day 5; single 1,200 mg dose. In the intent-to-treat groups clinical cure/improvement was similar; 72.4% in the daily-dose group, 78.2% in the infrequent group, and 81.1% in single-dose group. Oritavancin was well tolerated—most adverse events were mild or moderate, and more than half were unrelated to study treatment [18, 19]. This study did not evaluate pharmacokinetics; however, it does support the safety of oritavancin at different doses and frequency, as well as efficacy.
The previous oritavancin pharmacokinetic studies by Rubino et al. used data from two separate populations: (1) healthy subjects and patients with complicated skin and skin structure infections and bacteremia receiving fixed and weight-based dosing ranges [14], and (2) SOLO I and II patients with ABSSSI receiving a single 1200 mg dose [11, 12]. With our study of the novel 800 mg second dose, the Cmax was similar to the “loading dose” of 1200 mg, indicating that this regimen will achieve similar pharmacokinetic parameters. Furthermore, when comparing drug exposures, the median AUC while above 0.12 mg/L in our study was similar to AUC0-∞ for the single 1200 mg dose (110 mg · days/L vs. ~ 117 mg · days/L). However, the 1200 mg and 800 mg doses 1 week apart in our study resulted in a total drug AUC while above 0.12 mg/L of 183.5 mg · days/L and unbound free drug of 26.2 mg · days/L. Based on animal models of infection, the antimicrobial activity of oritavancin appears to correlate with the ratio of AUC0-72 h/MIC [20]. This is consistent with lipoglycopeptide antibiotics such as vancomycin and daptomycin [21, 22]. In preliminary analysis of clinical studies assessing pharmacokinetic target attainment, AUC/MIC > 17,568 appears to be associated with clinical response in patients [23]. In our study, this AUC was achieved in organisms above the susceptibility breakpoint with MICs up to 0.25 mg/L. Future pharmacokinetic studies may indicate the target attainment and pharmacokinetic parameters with additional 800 mg doses.
Schulz et al. described the 800 mg dosage using estimated pharmacokinetic linear assumptions, and overall the clinical outcomes were promising [13]. However, the two-thirds (800 mg) supplemental weekly-dosing approach has not been validated by pharmacokinetic modeling, which could help inform clinicians’ use of this approach.
5 Conclusion
This population pharmacokinetic estimate analysis provides evidence that 1200 mg and 800 mg doses 1 week apart achieve appropriate oritavancin concentrations with serum levels above the susceptibility breakpoint of 0.12 mg/L for over 4 weeks, which is approximately 11 days longer than the 1200 mg single-dose simulation. When prolonged treatment is required, our analysis suggests pharmacokinetic advantages over single-dose treatment with this regimen. Combined with previous reports of the clinical success and safety of this dosing scheme, our analysis further validates use of this dosing when prolonged oritavancin treatment is desired [13]. Additional modeling is needed for multiple supplemental doses as well as in tertiary compartments such as bone and deep tissue to further understand novel oritavancin treatment approaches.
References
Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis. 2011;52(3):285–92.
Tong SYC, Lye DC, Yahav D, Sud A, Robinson JO, Nelson J, et al. Effect of vancomycin or daptomycin with vs without an antistaphylococcal beta-lactam on mortality, bacteremia, relapse, or treatment failure in patients with MRSA bacteremia: a randomized clinical trial. JAMA. 2020;323(6):527–37.
McCreary EK, Kullar R, Geriak M, Zasowski EJ, Rizvi K, Schulz LT, et al. Multicenter cohort of patients with methicillin-resistant Staphylococcus aureus bacteremia receiving daptomycin plus ceftaroline compared with other MRSA treatments. Open Forum Infect Dis. 2020;7(1):538.
Kullar R, Sakoulas G, Deresinski S, van Hal SJ. When sepsis persists: a review of MRSA bacteraemia salvage therapy. J Antimicrob Chemother. 2016;71(3):576–86.
Lodise TP, Lomaestro B, Graves J, Drusano GL. Larger vancomycin doses (at least four grams per day) are associated with an increased incidence of nephrotoxicity. Antimicrob Agents Chemother. 2008;52(4):1330–6.
Narita M, Tsuji BT, Yu VL. Linezolid-associated peripheral and optic neuropathy, lactic acidosis, and serotonin syndrome. Pharmacotherapy. 2007;27(8):1189–97.
LaVie KW, Anderson SW, O'Neal HR Jr, Rice TW, Saavedra TC, O'Neal CS. Neutropenia associated with long-term ceftaroline use. Antimicrob Agents Chemother. 2016;60(1):264–9.
Osmon DR, Berbari EF, Berendt AR, Lew D, Zimmerli W, Steckelberg JM, et al. Diagnosis and management of prosthetic joint infection: clinical practice guidelines by the infectious diseases society of America. Clin Infect Dis. 2013;56(1):e1–e25.
Chambers HF. Pharmacology and the treatment of complicated skin and skin-structure infections. N Engl J Med. 2014;370(23):2238–9.
Redell M, Moeck G, Lucasti C, Durso S, Kennedy C, Fusaro K, et al. A real-world patient registry for oritavancin demonstrates efficacy and safety consistent With the phase 3 SOLO program. Open Forum Infect Dis. 2018;5(6):051.
Corey GR, Good S, Jiang H, Moeck G, Wikler M, Green S, et al. Single-dose oritavancin versus 7–10 days of vancomycin in the treatment of gram-positive acute bacterial skin and skin structure infections: the SOLO II noninferiority study. Clin Infect Dis. 2015;60(2):254–62.
Corey GR, Kabler H, Mehra P, Gupta S, Overcash JS, Porwal A, et al. Single-dose oritavancin in the treatment of acute bacterial skin infections. N Engl J Med. 2014;370(23):2180–90.
Schulz LT, Dworkin E, Dela-Pena J, Rose WE. Multiple-dose oritavancin evaluation in a retrospective cohort of patients with complicated infections. Pharmacotherapy. 2018;38(1):152–9.
Rubino CM, Bhavnani SM, Moeck G, Bellibas SE, Ambrose PG. Population pharmacokinetic analysis for a single 1,200-milligram dose of oritavancin using data from two pivotal phase 3 clinical trials. Antimicrob Agents Chemother. 2015;59(6):3365–72.
Rubino CM, Van Wart SA, Bhavnani SM, Ambrose PG, McCollam JS, Forrest A. Oritavancin population pharmacokinetics in healthy subjects and patients with complicated skin and skin structure infections or bacteremia. Antimicrob Agents Chemother. 2009;53(10):4422–8.
Morrisette T, Miller MA, Montague BT, Barber GR, McQueen RB, Krsak M. On- and off-label utilization of dalbavancin and oritavancin for Gram-positive infections. J Antimicrob Chemother. 2019;74(8):2405–16.
Brownwell LE, Adamsick ML, McCreary EK, Vanderloo JP, Ernst EJ, Jackson ER, et al. Clinical outcomes and economic impact of oritavancin for gram-positive infections: a single academic medical center experience. Drugs Real World Outcomes. 2020.
Brade KD, Rybak JM, Rybak MJ. Oritavancin: a new lipoglycopeptide antibiotic in the treatment of gram-positive infections. Infect Dis Ther. 2016;5(1):1–15.
Dunbar LM, Milata J, McClure T, Wasilewski MM, Team SS. Comparison of the efficacy and safety of oritavancin front-loaded dosing regimens to daily dosing: an analysis of the SIMPLIFI trial. Antimicrob Agents Chemother. 2011;55(7):3476–84.
Boylan CJ, Campanale K, Iversen PW, Phillips DL, Zeckel ML, Parr TR Jr. Pharmacodynamics of oritavancin (LY333328) in a neutropenic-mouse thigh model of Staphylococcus aureus infection. Antimicrob Agents Chemother. 2003;47(5):1700–6.
Rybak MJ, Lomaestro BM, Rotschafer JC, Moellering RC Jr, Craig WA, Billeter M, et al. Therapeutic monitoring of vancomycin in adults summary of consensus recommendations from the American Society of health-system pharmacists, the infectious diseases society of America, and the society of infectious diseases pharmacists. Pharmacotherapy. 2009;29(11):1275–9.
Safdar N, Andes D, Craig WA. In vivo pharmacodynamic activity of daptomycin. Antimicrob Agents Chemother. 2004;48(1):63–8.
Bhavnani SM, Hammel JP, Rubino CM, Moeck G, Jiang H, Bellibas SE, et al. Oritavancin (ORI) Pharmacokinetic-Pharmacodynamic (PK-PD) Analyses for Efficacy Based on Data from Patients with Acute Bacterial Skin and Skin Structure Infections (ABSSSI) Enrolled in SOLO I and II. 54th Interscience Conference on Antimicrobial Agents and Chemotherapy; Washington, DC2014.
Arhin FF, Belley A, McKay G, Beaulieu S, Sarmiento I, Parr TR Jr, et al. Assessment of oritavancin serum protein binding across species. Antimicrob Agents Chemother. 2010;54(8):3481–3.
Author information
Authors and Affiliations
Contributions
All authors had a role in the study design and in conceiving and writing the manuscript. According to the guidelines of the International Committee of Medical Journal Editors (ICMJE, www.icmje.org), all authors met the criteria for authorship and no deserving authors have been omitted.
Corresponding author
Ethics declarations
Funding
No funding was received for the conduct of this study. This article is part of a supplement wholly funded by Melinta Therapeutics.
Conflict of interest
W. Rose receives speaker honoraria from Melinta Therapeutics, consulting fees from Visante, Inc., and grant funding from Theravance. PH has no conflicts to declare.
Additional information
Digital features To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12221690.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Rose, W.E., Hutson, P.R. A Two-Dose Oritavancin Regimen Using Pharmacokinetic Estimation Analysis. Drugs - Real World Outcomes 7 (Suppl 1), 36–40 (2020). https://doi.org/10.1007/s40801-020-00188-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40801-020-00188-6