
Abstract
Pyoderma gangrenosum is a rare inflammatory skin disease classified within the group of neutrophilic dermatoses and clinically characterized by painful, rapidly evolving cutaneous ulcers with undermined, irregular, erythematous-violaceous edges. Pyoderma gangrenosum pathogenesis is complex and involves a profound dysregulation of components of both innate and adaptive immunity in genetically predisposed individuals, with the follicular unit increasingly recognized as the putative initial target. T helper 17/T helper 1-skewed inflammation and exaggerated inflammasome activation lead to a dysregulated neutrophil-dominant milieu with high levels of tumor necrosis factor-α, interleukin (IL)-1β, IL-1α, IL-8, IL-12, IL-15, IL-17, IL-23, and IL-36. Low-evidence studies and a lack of validated diagnostic and response criteria have hindered the discovery and validation of new effective treatments for pyoderma gangrenosum. We review established and emerging treatments for pyoderma gangrenosum. A therapeutic algorithm based on available evidence is also provided. For emerging treatments, we review target molecules and their role in the pathogenesis of pyoderma gangrenosum.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Treatment of pyoderma gangrenosum is mostly based on clinical experience. |
Treatment of pyoderma gangrenosum is often challenging and continues to rely on immunosuppressive therapy as the main cornerstone. |
Increased understanding of the molecular basis for pyoderma gangrenosum will lead to the discovery of new effective targeted therapies. |
1 Introduction
Pyoderma gangrenosum (PG) is a rare debilitating inflammatory skin disease clinically characterized by painful, rapidly evolving cutaneous ulcers with undermined, irregular, erythematous-violaceous edges [1,2,3]. First described by Brocq and Simon in 1908 as “phagédénisme géométrique” [4] and subsequently renamed by Brunsting et al. in 1930 [5], PG represents the prototype of neutrophilic dermatoses and is currently classified within deep/hypodermal neutrophilic dermatoses [6], with a worldwide estimated incidence of 3–10 cases/million people/year [1, 7]. Several clinical variants have been described, including classic ulcerative, bullous, pustular, vegetative, peristomal, and postoperative [1]. Bullous PG is hallmarked by blisters that are present at onset and later evolve into ulcerative lesions [8]. Vegetating lesions usually develop on the surface of a previously classic ulcerative PG, often during treatment [9], while pustular lesions usually precede or are concomitant to PG ulcers. Pyoderma gangrenosum lesions mostly affect the lower limbs [1]. Pyoderma gangrenosum may be idiopathic, associated with systemic conditions such as inflammatory bowel diseases (IBD), rheumatological disorders, and hematological malignancies, or present in the setting of autoinflammatory syndromes such as PAPA (Pyogenic Arthritis, PG and Acne), PASH (PG, Acne, and Suppurative Hidradenitis), PAPASH (Pyogenic Arthritis, PG, Acne, and Suppurative Hidradenitis), and in a small proportion of SAPHO (Synovitis, Acne, Pustulosis, Hyperostosis, and Osteitis) cases [10, 11].
Diagnostic criteria for classic ulcerative PG have recently been validated by means of a Delphi consensus of international experts [2]. Another set of criteria, the PARACELSUS score, has been proposed by a German group, particularly for the differential diagnosis with venous leg ulcers [3].
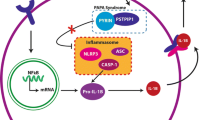
The pathogenesis of PG is complex and involves a profound dysregulation of components of both innate and adaptive immunity, with the follicular unit increasingly recognized as the putative initial target [12]. Following antigenic priming in predisposed individuals, T helper (Th)17/Th1 skewing [13] leads to the establishment of a neutrophil-dominant, self-maintaining, autoinflammatory milieu, with elevated levels of tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1β, IL-1α, IL-8, IL-12, IL-15, IL-17, IL-23, and IL-36 [14, 15]. Indeed, pathogenic variants of genes involved in inflammasome formation, including PSTPIP1, MEFV, NLRP3, NLRP12, and NOD2, leading to an exaggerated release of IL-1β, have been documented in both syndromic (e.g., PAPA) and sporadic cases of PG [16, 17]. Unsurprisingly, trauma (i.e., pathergy) is among the best documented trigger factors of PG, as it entails the release of PG-driving cytokines such as IL-36 and IL-8 from keratinocytes, an event that may be sufficient in the setting of concurrent genetic predisposition [1]. Complement system, and particularly neutrophil-attractant anaphylatoxin C5a [18, 19], NETosis, regulatory T-cell unbalance [20], B cells as well as fibroblasts and monocytes/macrophages all add to the multi-layered pathophysiology of PG [21].
Although PG pathogenesis remains incompletely elucidated, our understanding of its molecular underpinnings will pave the way for targeted treatments. To date, the majority of published studies on the treatment of PG are of low clinical evidence (level 3–5), i.e., retrospective case-series and single case reports, with only a few controlled clinical trials (Table 1). Further, there remains an unmet need for studies that assess treatments in refractory or recurrent PG, as well as the optimal duration of therapy once healing has been reached. Moreover, the lack of standardized outcomes has hampered the comparability of PG clinical trials. The main therapeutic options for classic PG include those listed by Garcovich et al. [22]. Moreover, evidence from a large, multicenter, retrospective cohort study as well as an expert survey study shows that patients with PG receive an average of two different systemic agents, underlining the importance of combination treatment regimens in real-life clinical practice [23, 24]. In a prospective study, additional use of systemic immunomodulators has been associated with healing [25]. The main treatment options discussed in the following paragraphs are summarized in Table 2.
2 Classical Immunosuppressive and Immunomodulating Drugs
2.1 Systemic Corticosteroids (Level of Evidence 1B) [First-Line Treatment]
The anti-inflammatory action of corticosteroids (CS) is attributed to their transcription-altering effects, and to NF-κB inhibition particularly, with subsequent downregulation of many proinflammatory cytokines, chemokines, and cell adhesion molecules [26]. The rapid onset of action has made systemic CS a first-line option in PG [27].
Treatment with systemic CS (dose 0.5–1 mg/kg/day) induces a clinical response in about 40–50% of cases [28], with widely variable complete response rates depending on associated systemic diseases and PG severity [29]. Once healing has been reached, the CS dose can be tapered over a variable time period (4–6 weeks to 12–24 weeks), according to clinical course, comorbidities, and the risk of relapse [30]. Pulse therapy with 1000 mg of intravenous methylprednisolone for 3–5 consecutive days, followed by oral CS, may have a faster onset of action and may also help taper oral CS [27, 31]. As complete remission is achieved only in 40% and relapse prevention only in 20% of those with multi-lesional PG, it is recommended to combine systemic CS with immunosuppressive/immunomodulatory adjuvants in severe cases, with the most common agent being cyclosporine [30].
2.2 Cyclosporine (Level of Evidence 1B) [First-Line Treatment]
Cyclosporine is an immunosuppressive drug used as a first-line choice for PG treatment. Cyclosporine is a calcineurin inhibitor that hampers the synthesis of ILs, in particular IL-2, which is crucial in blocking T-lymphocyte activation [32].
A multicenter randomized controlled trial, the Study of Treatments fOr Pyoderma GAngrenosum Patients (STOP GAP), was conducted to compare prednisolone and cyclosporine, i.e., the two first-line treatment options for PG. In the STOP GAP trial, patients either received oral prednisolone 0.75 mg/kg/day or cyclosporine 4 mg/kg/day. Limitations of this study included the possibility of PG misdiagnosis as no diagnostic framework for PG was used as well as the inclusion of mostly mild PG cases. There was no difference between the two treatments in terms of speed of healing over 6 weeks, time to healing, treatment response, inflammation resolution, pain, quality of life, treatment failures, and time to recurrence. Of note, almost half of the enrolled patients receiving systemic CS or cyclosporine did not obtain healing of PG ulcers at 6 months, and almost a third of patients in both treatment groups developed a disease recurrence, with a median time to recurrence of 582 days in both groups. Additionally, about two thirds of patients experienced adverse effects in each group. The choice of prednisolone vs cyclosporine depends on patients’ comorbidities. Pre-existing conditions favoring prednisolone over cyclosporine include renal insufficiency or malignancy. Whereas patients with obesity, diabetes mellitus, osteoporosis, peptic ulcer disease, or a history of mental illness may benefit from using cyclosporine over prednisolone. Pre-existing hypertension also favors prednisolone usage, although with caution [33]. A subsequent cost-effectiveness analysis recommended cyclosporine for the treatment of patients with large PG ulcers (size > 20 cm2) [34].
Cyclosporine also proved effective in a case series of 26 patients with classical ulcerative PG. The study utilized the term episodes, defined from the appearance of lesion(s) until complete skin healing. Twenty-two patients had 51 episodes of PG and they received a mean dose of 4.9 mg/kg/day of cyclosporine with a mean treatment duration of 14 weeks. Forty-nine episodes achieved complete healing and the remaining two episodes achieved partial response. Monotherapy was utilized in 22 of the 51 episodes and the cyclosporine with prednisone combination was used in 13 episodes. Although most episodes resulted in completed healing, 14 patients experienced relapse when cyclosporine was tapered or discontinued [35].
A study of 21 patients highlighted the benefits of combining cyclosporine and prednisone in treating multi-lesional PG. Four patients with multi-lesional PG, refractory to systemic corticosteroid monotherapy, healed and achieved remission after the addition of cyclosporine. Three patients with disseminated PG achieved complete response, with two remaining in remission at the time of follow-up [30].
Overall, and also in our experience, cyclosporine is highly effective in treating classical ulcerative PG and should also be considered a first-line option for PG [30, 35].
2.3 Methotrexate (Level of Evidence 4)
Methotrexate (MTX) is an immunomodulating drug widely used for the treatment of chronic inflammatory skin diseases. Its mechanisms of action include: (i) increased adenosine release, which inhibits many inflammatory and immune responses; (ii) nitric oxide synthase uncoupling, which translates into an enhanced sensitivity of T cells to apoptosis; and (iii) increased expression of long intergenic non-coding RNA p21, which in turn modulates a variety of immune and inflammatory signaling pathways [36].
Evidence concerning MTX for the treatment of PG is mostly limited to isolated reports where it has been employed as a steroid-sparing agent, either alone [37, 38] or in combination with other immunosuppressant/immunomodulating drugs [39] and/or biologics [40, 41].
Combination therapy with MTX, dapsone, systemic CS, and minocycline was shown to be effective and well tolerated in a case of classic, genetically proven PAPA syndrome. Although maintenance was with MTX and dapsone alone, it may be difficult to dissect the relative contribution of MTX [39]. Maintenance MTX therapy was also successful in a patient with PG and comorbid psoriasis/psoriatic arthritis, after disease control was achieved by means of cyclosporine and systemic CS [42].
Interestingly, whereas systemic oral MTX plus systemic CS failed to determine a response, a switch to intralesional MTX injected weekly along the ulcer border led to a dramatic improvement in a patient with classic ulcerative PG, with almost complete healing by the seventh week of therapy [43].
Furthermore, MTX may serve as a useful rescue therapy for patients with severe PG developing human anti-chimeric antibodies to infliximab. Indeed, Wang et al. showed improvement and even remission after the introduction of MTX in three patients (two with PG and hidradenitis suppurativa and one with PG alone) treated with infliximab, who had developed antichimeric antibodies [40]. In our experience, MTX is best used as an adjuvant option in patients with PG with a concomitant neoplasm in which cyclosporine and other immunosuppressants as well as biologics are contraindicated.
2.4 Mycophenolate Mofetil (Level of Evidence 2B)
Mycophenolate mofetil (MMF) is an immunosuppressive agent that was originally approved for preventing acute renal allograft rejection but then became known as an option for immune-mediated skin diseases. It acts by inhibiting inosine monophosphate dehydrogenase in the de novo purine synthesis pathway to interfere with the production of guanosine triphosphate. The lack of guanosine nucleotides subsequently impairs RNA, DNA, and protein synthesis. Lymphocytes lack the purine salvage pathway, which is an alternative way to replenish adenosine and guanosine. Therefore, MMF selectively decreases lymphocyte proliferation without interfering with other cells. By preventing lymphocyte and monocyte glycoprotein glycosylation, MMF may also alter adhesion dynamics to endothelial cells, thereby inhibiting the recruitment of leukocytes to sites of inflammation [44, 45].
Mycophenolate mofetil has been evaluated as a first-line or second-line steroid-sparing agent in patients with PG. In one retrospective chart review of 26 patients, MMF was started at 1 or 2 g daily with a maintenance dose of 2 or 3 g daily. The average treatment length was around 1 year. Almost 85% of patients demonstrated clinical improvement, with 13 patients achieving complete ulcer healing. All patients were taking concomitant prednisolone, 15 were taking another immunomodulatory medication, several others were taking another immunosuppressant for their comorbid conditions, and many took a tetracycline antibiotic. The starting dose of prednisolone was 40 mg daily and was tapered to 18 mg daily by the end of the study or when MMF was ceased. While a little over half of the patients experienced side effects, most were minor gastrointestinal upsets. Mycophenolate mofetil was the main form used, but mycophenolate sodium was used if MMF was not tolerated [46]. Similar findings regarding the efficacy, safety, and tolerability of MMF have been documented also in smaller case series and case reports, even in the setting of severe, tendon-exposing, refractory PG [47, 48] or if patients declined biologic treatment [49].
2.5 Azathioprine (Level of Evidence 4)
Azathioprine has been used in dermatology for several decades to treat immunobullous diseases. It acts by disrupting purine synthesis, which can selectively decrease lymphocyte counts as lymphocytes lack the purine salvage pathway. Moreover, azathioprine impairs T-cell activation and decreases circulating monocytes in a dose-dependent manner [50, 51]. Azathioprine may be helpful in treating refractory/severe PG, as a corticosteroid-sparing agent or as an alternative to first-line treatments [52]. It is also a good choice in patients with underlying IBD [27]. Complete ulcer healing was noted in a patient with diffuse prednisolone-refractory PG 3 weeks after initiation of azathioprine [53]. Azathioprine is generally well tolerated, and it has a more favorable therapeutic index compared with traditional immunosuppressants [54, 55]. The most common side effects are gastrointestinal upsets, such as nausea, vomiting, and diarrhea. Rarer adverse effects include hepatotoxicity and myelosuppression, which is why it is important to monitor transaminase levels and blood count [27, 50]. Thiopurine methyltransferase deficiency will lead to the accumulation of thioguanine nucleotides, which is related to hematopoietic toxicity [50]. Individual levels of thiopurine methyltransferase activity should ideally dictate the dosage requirements to help improve therapeutic benefits while decreasing risks for adverse effects. In our experience, azathioprine is used mainly as an adjuvant treatment in patients who have another indication, such as IBD.
2.6 Systemic Tacrolimus (Level of Evidence 4)
Tacrolimus reduces the transcription of proinflammatory cytokines by binding to the FK-binding proteins in the cytoplasm, which associate with calcium-dependent calcineurin/calmodulin complexes, to interfere with lymphocyte signal transduction. It has also been found to be more potent than dexamethasone and cyclosporine A, common first-line options, in inhibiting T-cell activation-induced TNF-α and IL-1β production by human peripheral blood mononuclear cells [54].
In a study of four patients, the efficacy of systemic tacrolimus was evaluated for classic ulcerative PG resistant to conventional therapy. All patients had a dramatic reduction in pain, erythema, and drainage within 1 week of treatment initiation. Three of four patients were able to achieve complete healing within 4–8 weeks [55].
In another report, two patients diagnosed with refractory PG were successfully treated with systemic tacrolimus. Both patients were started on a treatment regimen consisting of 30 mg/day of prednisolone combined with 2 mg/day of tacrolimus. The first patient healed in 3 months and the second patient’s ulcer decreased to a negligible size in 6 months [56].
Potential adverse effects of tacrolimus include hyperkalemia, hypomagnesemia, elevation in serum creatinine and blood urea nitrogen, and arterial hypertension. It is crucial to monitor its blood concentrations and blood pressure and adjust the dosage as needed.
2.7 Dapsone (Level of Evidence 2B)
Dapsone is a sulfone that has established itself as a first choice in many inflammatory skin diseases with predominant neutrophil and/or eosinophil accumulation [57]. It causes disruption of neutrophil adhesion and chemotaxis, inhibition of myeloperoxidase-induced oxidative burst, and production of damaging reactive oxygen species. The use of dapsone to treat PG has been sparsely reported in some case reports, and small reviews [22]. Indeed, in a retrospective series of 27 patients with PG, oral dapsone was used as an adjuvant in conjunction with systemic, topical, or intralesional therapies. Systemic CS were the most common concurrent therapy, followed by antibiotics, cyclosporine, and TNF-α inhibitors. Despite the 96.9% response rate, only 15.6% of patients achieved a complete response. The average time to initial response was 5.3 weeks. Unfortunately, around a third of patients developed adverse effects, the most frequent being hemolytic anemia [58].
In our experience, dapsone works well in superficial/epidermal NDs, such as Sneddon–Wilkinson disease or amicrobial pustulosis of the folds, but is usually less effective in deep/hypodermal NDs, such as PG [6, 59, 60]. However, we favor the use of dapsone as adjuvant treatment.
2.8 Colchicine (Level of Evidence 4)
Colchicine has been reported to be successful in the management of neutrophilic dermatoses, such as Sweet syndrome or Behҫet’s disease, making it appealing also for PG. Colchicine disrupts microtubule polymerization, thereby perturbing intracellular trafficking, chemokine secretion, cell migration, and cell division among many others. Its main appeal in PG treatment relies on the inhibition of innate immunity, NALP3 inflammasome assembly, and caspase 1 activation. At low concentrations, its effect on microtubules inhibits neutrophil recruitment and prevents neutrophil adhesion, whereas, at slightly higher concentrations, it can also inhibit neutrophil activation and the release of proinflammatory cytokines, such as IL-1, IL-8, and superoxide [61].
There are limited cases reporting the use of colchicine in PG. Low-dose colchicine monotherapy resulted in rapid regression of multiple 2–3 cm ulcers in two patients with refractory PG. Both patients tapered their colchicine without recurrence after 2 years and 6 months, respectively [62].
2.9 Thalidomide (Level of Evidence 4)
Thalidomide has been shown to be effective in treating refractory PG in many case reports. It may modulate NF-κB-related proinflammatory cytokines and chemokines, and reduce CD8+ T cells. Thalidomide can also selectively inhibit TNF-α production from monocytes [63].
Case reports show that it can be used in the case of primary or idiopathic PG when traditional treatments fail. It has been proposed as monotherapy [64] or in combination with CS or immunomodulatory agents, such as dapsone [65, 66]. Reported dosages of thalidomide range from 100 to 400 mg/day [66]. It is important to keep in mind the teratogenicity of thalidomide and the rare side effect of peripheral neuropathy [66].
2.10 Intravenous Immunoglobulin (Level of Evidence 3A)
Intravenous immunoglobulin (IVIG) has been utilized as an adjunct steroid-sparing agent and has shown success in treating refractory PG. In particular, IVIG has been used as a second-line systemic therapy for disseminated or moderate-to-severe PG [22]. Intravenous immunoglobulin is an appealing option because PG comorbidities may underlie contraindications to other, more commonly used systemic therapies. The anti-inflammatory actions of IVIG include: hampering of immune complex-mediated activation of FcγRs; half-life reduction of circulating antibodies through neonatal Fc receptor binding; blockade of complement activation; disruption of auto-reactive T-cell/antigen-presenting cell interactions; restoring balance of proinflammatory and anti-inflammatory cytokines; and downregulation of antibody production [67].
In a retrospective case series, 88% of patients responded to IVIG as adjunct therapy, with 53% achieving complete response after an average treatment length of 5.9 months. Mean time to initial response was 3.5 weeks. The most common concurrent therapy was systemic CS. Of note, the time to initial response was reduced with increasing IVIG dose (> 2 g/kg), although the dosage did not have the same beneficial effect on overall treatment length [68]. A retrospective study sought to determine the effects of IVIG incorporation in patients with treatment-resistant PG. A statistically significant improvement in complete healing with the addition of IVIG to treatment regimens was found in patients who had one resistant ulcer and no comorbidities compared with those with multiple refractory ulcers. The most common concurrent medication leading to complete healing was cyclosporine [69].
In addition to its use as an adjuvant option, IVIG has also been documented to be an alternative steroid-sparing agent [70]. The range of dosages reported include 0.4 g/kg/day up to 2 g/kg/day and 2–5 consecutive day monthly infusions. The treatment length ranged from 3 to 11 months [70]. In our experience, IVIG is an adequate alternative in patients with PG with repetitive and concomitant severe infections, particularly skin/soft-tissue infections and sepsis.
2.11 Granulocyte and Monocyte Adsorption Apheresis (Level of Evidence 4)
Granulocyte and monocyte adsorption apheresis (GMCAP) was first reviewed for use in ulcerative colitis. It uses an extracorporeal column filled with cellulose acetate beads that can adsorb granulocytes and monocytes [71]. The ability to reduce the amount of circulating leukocytes can help decrease inflammation and tissue damage that can be caused by the recruitment of neutrophils in PG.
Some cases have shown the effectiveness of GMCAP in treating refractory PG as monotherapy or in combination with systemic CS. The successful regimens included ten sessions of GMCAP at either 5-day or 7-day intervals [72]. No adverse events were reported but thromboembolism was noted as a potential adverse effect of GMCAP [73].
3 Biologics
3.1 Tumor Necrosis Factor-Alpha Inhibitors (Level of Evidence 1B)
Tumor necrosis factor-α is a key proinflammatory pleiotropic cytokine that regulates IL1-β, IL-6, and IL-8. The latter is a potent chemokine that serves as a chemoattractant for neutrophils and works synergistically with TNF-α to promote and maintain a proinflammatory state [17]. Expression of TNF-α and its receptors is increased in lesional PG skin [1, 74].
Infliximab, adalimumab, etanercept, certolizumab pegol, and golimumab have been used in the treatment of PG. Whereas etanercept binds only the soluble form of TNF-α, infliximab, adalimumab, and golimumab bind the soluble-bound and membrane-bound forms of TNF-α and induce apoptosis of TNF-α-expressing cells. Certolizumab lacks an Fc domain and therefore cannot induce apoptosis, complement-dependent, or antibody-dependent cell-mediated cytotoxicity. There is growing evidence supporting the use of TNF-α inhibitors as first-line treatments, particularly infliximab and adalimumab [75]. Indeed, a semi-systematic review found a 67% (238/356 patients) complete response rate and an 87% response rate to TNF-α inhibitors overall. There was a trend towards a higher response rate for adalimumab and infliximab compared with etanercept, although this was not statistically significant [76]. Additionally, response rates were not significantly different for PG type or associated diseases (i.e., inflammatory bowel disease, hematological diseases, or other inflammatory disorders) [76,77,78].
To date, infliximab remains the sole anti-TNF-α agent of demonstrated efficacy in classic PG, as shown in a randomized double-blind controlled trial [79]. Recently, a phase III, open-label multicenter study was conducted on 22 Japanese patients to evaluate the efficacy and safety of adalimumab for refractory PG. Complete healing was achieved in 55% of participants at the end of the 26-week treatment period [80]. Although no head-to-head studies have been performed to compare efficacy, in a recent review, 75% (38/51) of patients taking adalimumab showed complete PG resolution, whereas only 61% (17/28) of those taking etanercept had complete healing [77]. Certolizumab has been demonstrated to be effective in three cases [81,82,83]. Golimumab has had mixed treatment responses, with two reports of complete response, one partial response, one treatment failure, and a case of new-onset PG [84,85,86,87,88].
In our experience, anti-TNF-α agents, especially infliximab and adalimumab, represent the best options in cases resistant to systemic CS, cyclosporine, or combination therapy with both [9]. Anti-TNF-α agents can also be used as sparing agents to avoid long-term side effects from CS and/or cyclosporine.
3.2 Interleukin-1β Inhibitors (Level of Evidence 2B)
Interleukin-1β is upregulated in lesions from both syndromic and sporadic PG cases [14, 15]. In PAPA syndrome, genetic mutations in Proline-Serine-Threonine Phosphatase Interacting Protein 1 (PSTPIP1) result in increased binding affinity to pyrin, enhancing the assembly of inflammasomes. These activate caspase 1, which subsequently cleaves inactive pro-IL-1β to its active form, IL-1β. In non-genetically determined cases of PG, IL-1β contributes to the release of proinflammatory cytokines and chemokines, which then contribute to neutrophil recruitment and activation. Interleukin-1β can be produced by macrophages, T lymphocytes, endothelial cells, fibroblasts, and activated keratinocytes [74].
Interleukin-1 inhibitors used for the treatment of PG include anakinra (IL-1 receptor antagonist that blocks IL1α and IL-1β), canakinumab (IL-1β inhibitor), and gevokizumab (IL-1β inhibitor). Use of IL-1 inhibitors remains limited to case reports and phase II open-label trials. In those reports, IL-1 inhibitors have shown some benefit [77]. Specifically, anakinra showed significant clinical improvement or complete remission in 10/13 patients and in nine case reports [77]. Canakinumab has also shown complete remission of PG in 6/11 patients, with either clinical improvement (1/11) or no response (4/11) in the remainder [77, 89, 90].
3.3 Interleukin-1α Blockade (Level of Evidence 5)
Interleukin-1α is also suggested to play a role in autoinflammatory diseases, independently of IL-1β [1]. Interleukin-1α is constitutively expressed by keratinocytes, stored intracellularly and released upon cellular injury [91]. Extracellular IL-1α regulates the production of granulocyte colony-stimulating factor, IL-6, CXCL-1, and CXCL-8, recruiting and activating neutrophils. Evidence from a murine model of neutrophilic dermatoses illustrated that Protein Tyrosine Phosphatase Non-Receptor Type 6 (PTPN6) gene variants lead to an excessive IL-1α-mediated inflammatory response, which appears to rely on receptor-interacting serine/threonine-protein kinase 1 and caspase recruitment domain-containing protein 9 signaling [92,93,94,95]. Indeed, PTPN6 gene variants have been documented in patients with PG and Sweet syndrome [96,97,98]. Bermekimab (RA-18C3) is an IL1α inhibitor currently under investigation for the treatment of PG [77].
3.4 Interleukin-17 Inhibitors (Level of Evidence 2B)
The IL-17 family of cytokines encompass six structurally related cytokines, IL-17A through IL17F. The prototypical member of the family, IL-17A, has a proinflammatory role in autoimmune disease [99]. Interleukin-17 and its receptor have been seen to be significantly expressed in lesional skin biopsies of PG [12, 15, 74]. Interleukin-17 signals to induce chemokines that recruit neutrophils and stimulates release of neutrophil-activating cytokines including IL-6, granulocyte colony-stimulating factor, and IL-8 [74, 99]. T helper 17 cells are also augmented in PG [14, 15, 20].
Sparse reports have described successful PG treatment with IL-17 inhibitors, including secukinumab (anti-IL-17A), brodalumab (anti-IL-17 receptor), and ixekizumab (anti-IL-17A/F) [100,101,102,103,104]. A very recent phase I–II pilot study was conducted to further evaluate the efficacy and safety of secukinumab monotherapy in treating PG. Seven patients received secukinumab 300 mg weekly at weeks 0–4, followed by every 4 weeks until week 16 for two patients, and every 2 weeks until week 32 for five patients, while remaining on the same dosage. Two patients continued treatment until week 32 and had marked improvement in ulcer size, inflammatory markers, and Dermatological Life Quality Index. All patients involved reported pain reduction [105]. Brodalumab has been used at doses of 210 mg weekly or biweekly, with improvement in three patients [103, 104]. Finally, a recent open-label trial has been completed on ixekizumab (NCT03137160) [106]. Interestingly, IL-17 inhibition may also induce PG [107,108,109,110,111,112], possibly due to paradoxical IL-23 upregulation [113]. Pyoderma gangrenosum induction has also been documented upon switching between anti-IL-17 agents [107].
3.5 Interleukin-23 Inhibitors (Level of Evidence 3A)
Interleukin-23 is overexpressed in PG lesions, and has also been demonstrated to be upregulated via immunofluorescence [15, 114]. Interleukin-23 is important for expanding and maintaining Th17 cells, which produce IL-17 and subsequently increase neutrophil recruitment.
Interleukin-23 inhibitors include ustekinumab (which blocks common p40 subunit of IL-12 and IL-23), tildrakizumab, guselkumab, and risankizumab (which target the p19 subunit of IL-23 without IL-12 blockade). In a recent semi-systematic review, complete response rates were seen in 71% of patients treated with ustekinumab [90]. Additionally, a recent summary of case reports of ustekinumab for PG showed that 68% of patients (19/28) had complete healing of their ulcers and 32% (9/28) had partial improvement [115]. Use of ustekinumab 90 mg every 2–3 months for refractory PG has been seen to improve all subtypes of PG including ulcerative, peristomal, pustular, and vegetant subtypes [115]. One patient with refractory, tendon-exposing PG on the lower extremities showed improvement with 3 months of treatment with guselkumab [116]. Additionally, a single case report showed complete healing after four doses of guselkumab with a modified regimen [117]. Finally, another case report showed improvement of recurrent PG after four doses of risankizumab [118].
3.6 C5a Inhibitors (Level of Evidence 2B)
C5a is a potent chemoattractant for neutrophils. It also enhances phagocytosis, release of granule enzymes, and oxidative burst, exerting its effects through its receptors C5aR and C5L2/C5aR2 [18]. C5aR1 and C5aR2 are both upregulated in PG lesional skin [15]. In a murine model, C5a has also been shown to have a role in wound healing, with C5a receptor deficiency resulting in more effective wound closure [119]. Collectively, the known roles of C5a in neutrophil recruitment, wound healing, and complement dysregulation present in PG lesional skin suggest C5a as a potential therapeutic target in PG [19]. Importantly, the membrane attack complex C5b-9 retains its ability to form and promote bacterial defence in the absence of C5a [15, 120].
IFX-1 (vilobelimab) is a chimeric monoclonal IgG4 antibody that inhibits C5a [120, 121]. In an open-label single-arm trial, 12 patients with HS were treated with IFX-1 800 mg delivered at 30-min intravenous infusion on days 1, 4, 8, 15, 22, 29, 36, 43, and 50 with the number of adverse events as the primary endpoint. Four of the five serious adverse events were exacerbation of HS that required hospitalization [121]. A phase IIa open-label study is currently investigating the efficacy and safety of vilobelimab in patients with PG (NCT03971643). A total of 19 patients were enrolled in the study. Seven patients were in the dosing cohort at 2400 mg biweekly, and all had elevated C5a levels at baseline. Six of these seven patients achieved clinical remission with closure of the target ulcer and showed suppression of C5a levels. Two of 19 patients had serious adverse events including erysipelas leading to hospitalization (not thought to be medication related), while another developed a delayed hypersensitivity reaction [122]. Final outcomes from the study are expected to be reported in 2022.
3.7 Interleukin-6 Inhibitors (Level of Evidence 4)
Interleukin-6 is a strong inducer of the acute phase response. Interleukin-6 binds to the IL-6 receptor/gp130 complex, which activates intracellular signaling cascades through Janus kinase 1 (JAK1) and signal transducer and activator of transcription 3 (STAT3). T helper cells differentiate towards a Th17 phenotype in the presence of IL-6, transforming growth factor-beta, and IL-23 [123]. Both IL-6 and its receptor have been seen to be elevated in the serum and skin of patients with PG [15, 124]. Elevated IL-6 levels have been shown to decrease after successful PG treatment [124, 125]. Along with other cytokines, IL-6 is involved in the activation and accumulation of neutrophils in tissue.
The humanized anti-IL-6 receptor monoclonal antibody tocilizumab has activity against soluble-bound and membrane-bound receptors. A single case report showed improvement of PG in a patient with rheumatoid arthritis [126]. Given its use for rheumatoid arthritis, tocilizumab may be considered as a treatment for refractory PG associated with rheumatoid arthritis. Another case showed improvement of PG in a patient with Takayasu arteritis [127]. However, a paradoxical case of PG has also been reported with the use of tocilizumab for Takayasu arteritis [128]. Ongoing investigations are required to determine the utility of tocilizumab for PG [129].
3.8 CD3 Inhibitors (Level of Evidence 4)
Early PG lesions show perivascular and peri-pilosebaceous T-cell infiltrates and are dominated by T cells with a Th1 phenotype [12]. Accordingly, targeting T-cell surface receptors may be a potential therapeutic strategy for this disease. CD3 is a T-cell co-receptor involved in activating Th cells and cytotoxic T cells. CD3 comprises the CD3 gamma chain, the CD3 delta chain, and two CD3 epsilon chains. These form a TCR complex with TCR and CD3 zeta and mediate T-cell activation. Inhibition of CD3 leads to apoptosis of activated T cells [127]. Visilizumab is a humanized IgG2 monoclonal antibody that binds to the CD3 epsilon chain [130]. Visilizumab induced improvement of steroid-refractory PG in a patient with concomitant ulcerative colitis [131]. Unfortunately, in a pilot study, visilizumab was not effective for severe corticosteroid-refractory ulcerative colitis and had safety issues [132]. The safety and effectiveness of CD3 inhibitors for idiopathic PG are not known.
3.9 CD20 Inhibitors (Level of Evidence 4)
The role of B cells in PG, if any, remains unknown. A broad autoantibody response, also against neutrophil extracellular traps, has been documented in serum and skin from patients with hidradenitis suppurativa, associating with immune complex generation and macrophage activation [133]. Similar mechanisms may occur in PG [21]. Rituximab is a monoclonal antibody that targets CD20, an antigen expressed at most stages of B-cell development. Pyoderma gangrenosum has been associated with conditions with aberrancies in B-cell populations including monoclonal gammopathies, leukemias, lymphomas, and myelodysplastic syndromes. Rituximab has been seen to improve PG-like ulcers in patients with associated granulomatous polyangiitis and refractory PG ulcers in those without an underlying comorbidity [134,135,136]. Conversely, a recent systematic review demonstrated that rituximab may be associated with the onset of vulvovaginal PG [137]. In fact, rituximab appears to be responsible for the majority of new-onset PG cases reported during biologic therapies [138].
3.10 Integrin Inhibitors (Level of Evidence 4)
Integrins are important for leukocyte migration. Integrin Subunit Beta 4 (ITGB4) and Integrin Subunit Beta 7 (ITGB7) genes are upregulated in PG lesions. These genes encode the proteins that form the heterodimeric integrin receptor α4β7 [15].
Vedolizumab is a humanized monoclonal antibody that has been approved by the US Food and Drug Administration for the treatment of IBD since 2014. It binds to α4β7 integrin and blocks interaction with a subset of gastrointestinal-homing T cells [139]. Four patients with IBD experienced PG improvement while taking vedolizumab [140, 141]. Conversely, vedolizumab-induced PG has also been observed in patients with IBD [142, 143], including a case improving with granulocyte and monocyte apheresis and prednisolone [144].
4 Small Molecules
4.1 Phosphodiesterase 4 Inhibitors (Level of Evidence 4)
Phosphodiesterase 4 is an enzyme that hydrolyzes cyclic adenosine monophosphate (cAMP), which is a key secondary messenger molecule found in all cells. Cyclic adenosine monophosphate is activated by G-protein coupled receptor ligands and mediates its action through protein kinase A. Protein kinase A activates cyclic adenosine monophosphate response element-binding protein, which then modulates that activity of numerous genes including those that encode for IL-2, IL-6, and TNFα [145]. Tumor necrosis factor-α and IL-6 that are downstream of the cyclic adenosine monophosphate signaling cascade are dysregulated in PG lesions.
Apremilast is an oral selective phosphodiesterase 4 inhibitor that has been approved since 2014 for the treatment of psoriasis and psoriatic arthritis. Two cases of PG treated with apremilast have been reported. One patient with PG, palisaded neutrophilic granulomatous dermatitis, and rheumatoid arthritis healed after apremilast was added to infliximab and glucocorticoids [146]. In another patient with recalcitrant vegetant PG, apremilast 30 mg twice daily was added to prednisone 7.5 mg daily, and methotrexate 18 mg subcutaneously once weekly and complete healing was achieved at 4 months [147].
4.2 Janus Kinase Inhibitors (Level of Evidence 4)
The JAK/STAT signaling pathways are involved in many inflammatory skin disorders. Cytokines upstream of JAK/STAT signaling include ILs 1–31, TNF-α, and colony-stimulating factors, among others. Janus kinase activation results in phosphorylation of specific STAT proteins (e.g., STAT1–4, 5a, 5b, 6). When activated, STAT proteins translocate to the nucleus and stimulate or inhibit the transcription of genes involved in hematopoiesis and immune function [148, 149].
The JAK/STAT pathway has been shown to be dysregulated in PG lesions with JAK-1, JAK-2, and JAK-3 and STAT1 through 6 being overexpressed in the skin of patients with PG compared with healthy controls [15, 150]. Mutations in the JAK/STAT pathway are also implicated in PG progression [150,151,152,153].
While there are many topical and oral JAK inhibitors in development or currently used to treat other inflammatory skin disorders, only a few JAK inhibitors have been used for the treatment of PG, including tofacitinib (oral inhibitor of JAK-1 and JAK-3), ruxolitinib (oral inhibitor of JAK-1 and JAK-2), and baricitinib (JAK-1 and JAK-2 inhibitor).
Tofacitinib is approved by the Food and Drug Administration to treat ulcerative colitis and rheumatoid arthritis, which can be associated with PG. To date, ten cases of PG treated with tofacitinib have been reported, with four achieving complete healing. Most patients had not responded to other systemic treatments prior to initiation of tofacitinib at doses of 10 or 11 mg/day [154].
Ruxolitinib has been used to treat polycythemia vera and other hematologic disorders associated with PG [155]. Two patients with polycythemia vera experienced PG resolution after treatment with ruxolitinib [156, 157]. Baricitinib 4 mg daily has led to complete ulcer healing in isolated reports [158] and is currently under investigation in a phase II clinical trial (NCT04901325) in combination with prednisone [159].
5 Topical Treatment and Wound Care
5.1 Topical Corticosteroids (Level of Evidence 2B)
Topical CS can be effective for the treatment of mild or localized/unilesional PG. Typically, high-potency CS, such as clobetasol propionate, are used, although others were also reported to be beneficial. In a prospective cohort study, 49 patients were treated with clobetasol propionate 0.05% cream monotherapy, resulting in a median healing time of 136 days. Overall, 42.6% of patients healed completely at 6 months; however, 21.1% of the former had subsequent recurrences. The size of the lesion at presentation was noted to be a significant predictor of time to healing [160].
5.2 Topical Calcineurin Inhibitors (Level of Evidence 2B)
Topical calcineurin inhibitors include tacrolimus, pimecrolimus, and cyclosporine. Topical calcineurin inhibitors inhibit lymphocyte proliferation and activation, and decrease the production of inflammatory cytokines such as IL-2, IL-3, and interferon-γ [161].
In a retrospective review, five patients with recent-onset localized idiopathic PG were successfully treated with topical tacrolimus alone. Topical tacrolimus was applied twice a day, then tapered to once a day for 2 months if no new lesions appeared. A further reduction to twice a week for 6–12 months was recommended before discontinuing. All five patients achieved a complete response with an average time to healing of 6 weeks. Patients tolerated the treatment well with only one complaining of a burning sensation during the first week of application. Three patients who had already discontinued the drug did not experience relapses at the mean follow-up period of 13.5 months. Finally, the other two patients also remained disease free but were still applying topical tacrolimus at the time of writing [162]. In a prospective cohort study, ten patients were treated with topical tacrolimus ointment (0.03% and 0.01%) alone with a median healing time of 161 days [160]. Although reported posology varies, topical tacrolimus ointment (0.03% and 0.1%) and topical tacrolimus solution (0.5%) appear to be beneficial in treating PG [160, 163]. It should be noted that temporary renal failure has been recorded following direct application of topical tacrolimus on open wounds [164].
Topical pimecrolimus also reduces T-cell proliferation, activation, and secretion of proinflammatory cytokines. In addition, pimecrolimus may also inhibit degranulation of mast cells, neutrophils, and basophils. Use of topical pimecrolimus may be appealing over topical tacrolimus as the former has lower systemic absorption. Evidence is mostly limited to isolated case reports, documenting complete healing in 6–12 weeks. Topical pimecrolimus may be regarded as an option for mild localized PG or as an adjunct to systemic therapies for severe or unresponsive PG [165, 166].
Topical cyclosporine has been used in four patients with refractory PG. Topical cyclosporine was administered via a piece of lint cloth saturated with an intravenous preparation of cyclosporine (1 ampoule of 50 mg/mL) diluted 1:1 with distilled water. Treatment was applied daily and then tapered to every other day depending on patient response. Three patients healed completely within 4 months, with the fourth showing a considerable response. Indeed, topical cyclosporine use could be appealing, as it seems to be exempt from the tolerability issues associated with systemic administration [167].
5.3 Wound Care (Level of Evidence 3A)
Wound care is crucial for the appropriate management of PG cases [168]. Pyoderma gangrenosum ulcers evolve through inflammatory and healing phases, requiring a differentiated approach depending on depth and exudation [169]. Recently, an algorithm based on the Tissue, Infection or inflammation, Moisture and Edge (TIME) model has been adapted for PG [170]. Adequate control of wound bed and perilesional inflammation with topical CS or topical calcineurin inhibitors, as well as lidocaine for pain management, is key during the inflammatory phase. In this setting, to avoid pathergy only gentle cleansing without sharp debridement should be performed. Similarly, enzymatic (collagenases) or autolytic debridement (hydrogel) can aid reducing fibrin and necrotic tissues, and may be followed be the application of either absorbent (alginate, hydrofiber) or nonadherent (silicone, foam) dressings to control the exudate. Alginates are best employed for bleeding PG ulcers because of their hemostatic properties, while polyurethane foam with a silicon layer could be useful on inflamed PG lesions. Antimicrobial dressings to decrease microbial burden and compressive bandages to reduce the exudate, edema, and overgranulation of PG ulcers may also have a role during the inflammatory phase. Indeed, according to a systematic review of the literature, antimicrobial and hyperabsorbent dressings appear to be the most commonly used dressings for the management of PG ulcers, requiring less frequent changes and manipulation [168]. Bioactive dressings such as collagen sheets, dermal and/or epidermal substitutes, and grafts may be considered if the wound bed is not excessively inflamed or devitalized [170].
Classically, surgical intervention has been considered controversial in PG. However, the paradigm may be changing with split-thickness skin grafts giving good results in terms of healing if combined with negative pressure wound therapy, assuming that adequate immunosuppression is obtained to prevent pathergy beforehand [171]. Negative pressure wound therapy (NPWT) is a well-known option for various types of wounds, as it decreases intracellular edema, stimulates granulation tissue formation, promotes micro-vascularization of the wound bed, and preserves appropriate wound humidity. Pichler et al. described 15 patients with large and deep ulcers who underwent NPWT 1 week prior to STSG. Ten patients healed completely. Among these, nine had no recurrence after the first treatment cycle and one required a second treatment cycle. The others had a marked improvement with only minor recurrences. Failure to respond was observed only in one case [172]. Indeed, a recent systematic review highlighted that NPWT can be considered a safe adjuvant option, with 85.1% of treated cases showing improvement in wound healing [173]. Further elaborating on the available literature, Eisendle et al. underscored that despite halting the inflammatory process, NPWT alone does not significantly accelerate the healing time, whereas the best approach appears to be the combination of NPWT with skin grafting, also with porcine xenografts, while the patient is medically treated for PG [171]. Negative pressure wound therapy is an appropriate alternative also in patients with tendon-exposing PG ulcers [171].
Finally, hyperbaric oxygen therapy may represent an option for PG ulcers, owing to its effects on edema reduction, inflammation control, collagen formation, and bacterial burden mitigation [174]. Rescue therapy with hyperbaric oxygen resulted in complete healing and/or improvement in treatment-refractory PG cases, with a good safety profile [175,176,177,178]. However, more studies are needed.
5.4 Other Topical Treatment Alternatives
5.4.1 Topical Timolol (Level of Evidence 4)
Topical timolol has been used to treat ulcerated infantile hemangiomas, chronic venous leg ulcers, and other wounds. By antagonizing β2-adrenergic receptors, timolol may enhance keratinocyte migration, promoting wound healing. Antagonism of β2-adrenergic receptors on neutrophils may also decrease neutrophil recruitment. Two reports documented the effectiveness of topical timolol maleate in PG, either alone [179] or in combination with collagenase ointment [180]. Topical timolol, either gel or ophthalmic solution, may be considered as an adjunct for localized/persistent PG especially when the inflammatory component of PG is already under control.
5.4.2 Topical Phenytoin (Level of Evidence 4)
Topical phenytoin has been used in decubitus, diabetic foot, as well as venous stasis ulcers. Phenytoin may modulate fibroblast proliferation, enhance granulation tissue formation, promote collagen deposition, and decrease bacterial load on wounds. A series of six patients showed a dramatic improvement of PG lesions, although five were receiving concurrent systemic therapy. Topical phenytoin may be considered as an adjunct to systemic therapy in severe cases, or as monotherapy in milder cases [181].
6 Conclusions and Future Directions
The treatment of PG can be challenging. In addition to medical therapy, wound care, with appropriate dressings depending on inflammatory vs non-inflammatory phase [168], and pain control are crucial in the management of PG cases. Based on current knowledge and a previously published severity classification [30], the authors are proposing an algorithm (Fig. 1), which could be useful in the management of patients with classic ulcerative PG. There is insufficient evidence to clearly define the preferred therapeutic management for each PG variant. However, based on our experience as well as the literature, few considerations may be presented. Management of peristomal and postoperative PG is usually similar to that of unilesional/localized PG. For peristomal PG particularly, management is generally that of the underlying condition, for example, IBD. For bullous and vegetative PG, the therapeutic ladder is substantially the same as in classic ulcerative PG, although wound care approaches may need to be adapted to these settings, favoring hyperabsorbing and nonadherent dressings.

Proposed algorithm for the treatment of classic ulcerative pyoderma gangrenosum. Created with BioRender.com. BSA body surface area, IL-1 interleukin-1, IL-12/23 interleukin-12/23, IL-17 interleukin-17, IL-23 interleukin-23, TNFα tumor necrosis factor-alpha
In addition to the well-known diagnostic challenges in PG, its clinical heterogeneity as well as a lack of validated severity and response criteria has complicated the planning of clinical trials to assess the efficacy and safety of therapeutic agents. Recently, the UPGRADE (Understanding Pyoderma Gangrenosum: Review of Adverse Disease Effects) initiative has developed a platform for clinicians, researchers, industry, and patients to collaborate in the creation of a core outcome set for PG that could be used for future clinical trials (http://cs-cousin.org/understanding-pyoderma-gangrenosum-review-and-analysis-of-disease-effects-upgrade/). Research for PG and other neutrophilic dermatoses seems to be promising as more specific and targeted therapies are currently being developed.
References
Maverakis E, Marzano AV, Le ST, et al. Pyoderma gangrenosum. Nat Rev Dis Primers. 2020;6(1):81. https://doi.org/10.1038/s41572-020-0213-x.
Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol. 2018;154(4):461–6. https://doi.org/10.1001/jamadermatol.2017.5980.
Jockenhöfer F, Wollina U, Salva KA, Benson S, Dissemond J. The PARACELSUS score: a novel diagnostic tool for pyoderma gangrenosum. Br J Dermatol. 2019;180(3):615–20. https://doi.org/10.1111/bjd.16401.
Brocq L, Simon C. Contribution à l’étude du phagédénisme. Bull Soc Méd Hop Paris. 1908;290–307.
Brunsting LA, Goeckerman WH, O’Leary PA. Pyoderma (ecthyma) gangrenosum. Arch Derm Syphilol. 1930;22(4):655–80.
Marzano AV, Borghi A, Wallach D, Cugno M. A comprehensive review of neutrophilic diseases. Clin Rev Allergy Immunol. 2018;54(1):114–30. https://doi.org/10.1007/s12016-017-8621-8.
Ruocco E, Sangiuliano S, Gravina AG, Miranda A, Nicoletti G. Pyoderma gangrenosum: an updated review. J Eur Acad Dermatol Venereol. 2009;23(9):1008–17. https://doi.org/10.1111/j.1468-3083.2009.03199.x.
Marzano AV, Trevisan V, Galloni C, Alessi E. Fatal bullous pyoderma gangrenosum in a patient with Klinefelter’s syndrome. Acta Derm Venereol. 2008;88(2):158–9. https://doi.org/10.2340/00015555-0346.
Marzano AV, Tourlaki A, Alessi E, Caputo R. Widespread idiopathic pyoderma gangrenosum evolved from ulcerative to vegetative type: a 10-year history with a recent response to infliximab. Clin Exp Dermatol. 2008;33(2):156–9. https://doi.org/10.1111/j.1365-2230.2007.02607.x.
Cugno M, Borghi A, Marzano AV. PAPA, PASH and PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. 2017;18(4):555–62. https://doi.org/10.1007/s40257-017-0265-1.
Marzano AV, Borghi A, Meroni PL, Cugno M. Pyoderma gangrenosum and its syndromic forms: evidence for a link with autoinflammation. Br J Dermatol. 2016;175(5):882–91. https://doi.org/10.1111/bjd.14691.
Wang EA, Steel A, Luxardi G, et al. Classic ulcerative pyoderma gangrenosum is a T cell-mediated disease targeting follicular adnexal structures: a hypothesis based on molecular and clinicopathologic studies. Front Immunol. 2018;8:1980. https://doi.org/10.3389/fimmu.2017.01980.
Antiga E, Maglie R, Volpi W, et al. T helper type 1-related molecules as well as interleukin-15 are hyperexpressed in the skin lesions of patients with pyoderma gangrenosum. Clin Exp Immunol. 2017;189(3):383–91. https://doi.org/10.1111/cei.12989.
Ortega-Loayza AG, Nugent WH, Lucero OM, Washington SL, Nunley JR, Walsh SW. Dysregulation of inflammatory gene expression in lesional and nonlesional skin of patients with pyoderma gangrenosum. Br J Dermatol. 2018;178(1):e35–6. https://doi.org/10.1111/bjd.15837.
Ortega-Loayza AG, Friedman MA, Reese AM, et al. Molecular and cellular characterization of pyoderma gangrenosum: implications for the use of gene expression. J Investig Dermatol. 2021. https://doi.org/10.1016/j.jid.2021.08.431.
Marzano AV, Damiani G, Ceccherini I, Berti E, Gattorno M, Cugno M. Autoinflammation in pyoderma gangrenosum and its syndromic form (pyoderma gangrenosum, acne and suppurative hidradenitis). Br J Dermatol. 2017;176(6):1588–98. https://doi.org/10.1111/bjd.15226.
Marzano AV, Ortega-Loayza AG, Heath M, Morse D, Genovese G, Cugno M. Mechanisms of inflammation in neutrophil-mediated skin diseases. Front Immunol. 2019;10:1059. https://doi.org/10.3389/fimmu.2019.01059.
Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–52. https://doi.org/10.1146/annurev.immunol.23.021704.115835.
Lu JD, Milakovic M, Ortega-Loayza AG, Marzano AV, Alavi A. Pyoderma gangrenosum: proposed pathogenesis and current use of biologics with an emphasis on complement C5a inhibitor IFX-1. Expert Opin Investig Drugs. 2020;29(11):1179–85. https://doi.org/10.1080/13543784.2020.1819981.
Caproni M, Antiga E, Volpi W, et al. The Treg/Th17 cell ratio is reduced in the skin lesions of patients with pyoderma gangrenosum. Br J Dermatol. 2015;173(1):275–8. https://doi.org/10.1111/bjd.13670.
Flora A, Kozera E, Frew JW. Pyoderma gangrenosum: a systematic review of the molecular characteristics of disease. Exp Dermatol. 2022. https://doi.org/10.1111/exd.14534.10.1111/exd.14534.
Garcovich S, De Simone C, Berti E, Marzano AV. Drug management of neutrophilic dermatoses. Expert Rev Clin Pharmacol. 2017;10(10):1119–28. https://doi.org/10.1080/17512433.2017.1356719.
Herberger K, Dissemond J, Hohaus K, Schaller J, Anastasiadou Z, Augustin M. Treatment of pyoderma gangrenosum: retrospective multicentre analysis of 121 patients. Br J Dermatol. 2016;175(5):1070–2. https://doi.org/10.1111/bjd.14619.
Afifi L, Ortega-Loayza AG, Shinkai K. Management of classic ulcerative pyoderma gangrenosum. Cutis. 2020;106(3):119-E3. https://doi.org/10.12788/cutis.0076.
Orfaly VE, Reese AM, Friedman M, Latour E, Ortega-Loayza AG. Pyoderma gangrenosum study pilot registry: the first step to a better understanding. Wound Repair Regen. 2022. https://doi.org/10.1111/wrr.13005 (Epub ahead of print, 1 Apr 2022).
Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011;335(1):2–13. https://doi.org/10.1016/j.mce.2010.04.005.
Wollina U. Clinical management of pyoderma gangrenosum. Am J Clin Dermatol. 2002;3(3):149–58. https://doi.org/10.2165/00128071-200203030-00002.
Kolios AGA, Gübeli A, Meier B, et al. Clinical disease patterns in a regional Swiss cohort of 34 pyoderma gangrenosum patients. Dermatology. 2017;233(4):268–76. https://doi.org/10.1159/000481432.
Vacas AS, Torre AC, Bollea-Garlatti ML, Warley F, Galimberti RL. Pyoderma gangrenosum: clinical characteristics, associated diseases, and responses to treatment in a retrospective cohort study of 31 patients. Int J Dermatol. 2017;56(4):386–91. https://doi.org/10.1111/ijd.13591.
Marzano AV, Trevisan V, Lazzari R, Crosti C. Pyoderma gangrenosum: study of 21 patients and proposal of a ‘clinicotherapeutic’ classification. J Dermatol Treat. 2011;22(5):254–60. https://doi.org/10.3109/09546631003686069.
Ehling A, Karrer S, Klebl F, Schäffler A, Müller-Ladner U. Therapeutic management of pyoderma gangrenosum. Arthritis Rheum. 2004;50(10):3076–84. https://doi.org/10.1002/art.20559.
Tapia C, Nessel TA, Zito PM. Cyclosporine [updated 2021 Nov 15]. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2022 Jan. https://www.ncbi.nlm.nih.gov/books/NBK482450/. Accessed 16 May 2022.
Ormerod AD, Thomas KS, Craig FE, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial [published correction appears in BMJ. 2017 Mar 21;356:j1462]. BMJ. 2015;350: h2958. https://doi.org/10.1136/bmj.h2958.
Mason JM, Thomas KS, Ormerod AD, et al. Ciclosporin compared with prednisolone therapy for patients with pyoderma gangrenosum: cost-effectiveness analysis of the STOP GAP trial. Br J Dermatol. 2017;177(6):1527–36. https://doi.org/10.1111/bjd.15561.
Vidal D, Puig L, Gilaberte M, Alomar A. Review of 26 cases of classical pyoderma gangrenosum: clinical and therapeutic features. J Dermatol Treat. 2004;15(3):146–52. https://doi.org/10.1080/09546630410031909.
Cronstein BN, Aune TM. Methotrexate and its mechanisms of action in inflammatory arthritis. Nat Rev Rheumatol. 2020;16(3):145–54. https://doi.org/10.1038/s41584-020-0373-9.
Teitel AD. Treatment of pyoderma gangrenosum with methotrexate. Cutis. 1996;57(5):326–8.
Loloi J, MacDonald SM. Pyoderma gangrenosum of the penis. Can J Urol. 2021;28(1):10560–4.
Sardana K, Bajaj S, Bose SK. Successful treatment of PAPA syndrome with minocycline, dapsone, deflazacort and methotrexate: a cost-effective therapy with a 2-year follow-up. Clin Exp Dermatol. 2019;44(5):577–9. https://doi.org/10.1111/ced.13792.
Wang LL, Micheletti RG. Low-dose methotrexate as rescue therapy in patients with hidradenitis suppurativa and pyoderma gangrenosum developing human antichimeric antibodies to infliximab: a retrospective chart review. J Am Acad Dermatol. 2020;82(2):507–10. https://doi.org/10.1016/j.jaad.2019.09.012.
Vekic DA, Woods J, Lin P, Cains GD. SAPHO syndrome associated with hidradenitis suppurativa and pyoderma gangrenosum successfully treated with adalimumab and methotrexate: a case report and review of the literature. Int J Dermatol. 2018;57(1):10–8. https://doi.org/10.1111/ijd.13740.
Spangler JG. Pyoderma gangrenosum in a patient with psoriatic arthritis. J Am Board Fam Pract. 2001;14(6):466–9.
Del Puerto C, Navarrete-Dechent CP, Carrasco-Zuber JE, Vera-Kellet C. Intralesional methotrexate as an adjuvant treatment for pyoderma gangrenosum: a case report. Indian J Dermatol Venereol Leprol. 2017;83(2):277. https://doi.org/10.4103/0378-6323.186497.
Oğuz O, Keskin S. Use of mycophenolate mofetil in dermatology. J Turk Acad Dermatol. 2007;1(4):71401r.
Mydlarski PR. Mycophenolate mofetil: a dermatologic perspective. Skin Ther Lett. 2005;10(3):1–6.
Li J, Kelly R. Treatment of pyoderma gangrenosum with mycophenolate mofetil as a steroid-sparing agent. J Am Acad Dermatol. 2013;69(4):565–9. https://doi.org/10.1016/j.jaad.2013.05.028.
Lee MR, Cooper AJ. Mycophenolate mofetil in pyoderma gangrenosum. J Dermatol Treat. 2004;15(5):303–7. https://doi.org/10.1080/09546630410020154.
Husein-ElAhmed H, Callejas-Rubio JL, Ríos Fernandez R, Ortego CN. Effectiveness of mycophenolic acid in refractory pyoderma gangrenosum. J Clin Rheumatol. 2010;16(7):346–7. https://doi.org/10.1097/RHU.0b013e3181f6059f.
Young JN, Elgash M, Ortega-Loayza AG. Hidden in plain sight: considerations for an ulcer of the scalp. Head Neck. 2022;44(4):E11-5. https://doi.org/10.1002/hed.26988.
Patel AA, Swerlick RA, McCall CO. Azathioprine in dermatology: the past, the present, and the future. J Am Acad Dermatol. 2006;55(3):369–89. https://doi.org/10.1016/j.jaad.2005.07.059.
Broen JCA, van Laar JM. Mycophenolate mofetil, azathioprine and tacrolimus: mechanisms in rheumatology. Nat Rev Rheumatol. 2020;16(3):167–78. https://doi.org/10.1038/s41584-020-0374-8.
Bennett ML, Jackson JM, Jorizzo JL, Fleischer AB Jr, White WL, Callen JP. Pyoderma gangrenosum: a comparison of typical and atypical forms with an emphasis on time to remission. Case review of 86 patients from 2 institutions. Medicine (Baltimore). 2000;79(1):37–46. https://doi.org/10.1097/00005792-200001000-00004.
August PJ, Wells GC. Pyoderma gangrenosum treated with azathioprine and prednisolone. Br J Dermatol. 1974;91:80–2.
Yoon KH. Efficacy and cytokine modulating effects of tacrolimus in systemic lupus erythematosus: a review. J Biomed Biotechnol. 2010;2010: 686480. https://doi.org/10.1155/2010/686480.
Abu-Elmagd K, Jegasothy BV, Ackerman CD, et al. Efficacy of FK 506 in the treatment of recalcitrant pyoderma gangrenosum. Transplant Proc. 1991;23(6):3328–9.
Sadati MS, Dastgheib L, Aflaki E. Recalcitrant cases of pyoderma gangrenosum, responding dramatically to systemic tacrolimus. G Ital Dermatol Venereol. 2017;152(3):308–10. https://doi.org/10.23736/S0392-0488.17.04876-3.
Wozel G, Blasum C. Dapsone in dermatology and beyond. Arch Dermatol Res. 2014;306(2):103–24. https://doi.org/10.1007/s00403-013-1409-7.
Din RS, Tsiaras WG, Li DG, Mostaghimi A. Efficacy of systemic dapsone treatment for pyoderma gangrenosum: a retrospective review. J Drugs Dermatol. 2018;17(10):1058–60.
Marzano AV, Ramoni S, Caputo R. Amicrobial pustulosis of the folds: report of 6 cases and a literature review. Dermatology. 2008;216(4):305–11. https://doi.org/10.1159/000113942.
Marzano AV, Tavecchio S, Berti E, Gelmetti C, Cugno M. Cytokine and Chemokine profile in amicrobial pustulosis of the folds: evidence for autoinflammation. Medicine (Baltimore). 2015;94(50): e2301. https://doi.org/10.1097/MD.0000000000002301.
Leung YY, Yao Hui LL, Kraus VB. Colchicine: update on mechanisms of action and therapeutic uses. Semin Arthritis Rheum. 2015;45(3):341–50. https://doi.org/10.1016/j.semarthrit.2015.06.013.
Kontochristopoulos GJ, Stavropoulos PG, Gregoriou S, Zakopoulou N. Treatment of pyoderma gangrenosum with low-dose colchicine. Dermatology. 2004;209(3):233–6. https://doi.org/10.1159/000079897.
Domingo S, Solé C, Moliné T, Ferrer B, Cortés-Hernández J. Thalidomide exerts anti-inflammatory effects in cutaneous lupus by inhibiting the IRF4/NF-ҡB and AMPK1/mTOR pathways. Biomedicines. 2021;9(12):1857. https://doi.org/10.3390/biomedicines9121857.
Hecker MS, Lebwohl MG. Recalcitrant pyoderma gangrenosum: treatment with thalidomide. J Am Acad Dermatol. 1998;38(3):490–1. https://doi.org/10.1016/s0190-9622(98)70513-4.
Farrell AM, Black MM, Bracka A, Bunker CB. Pyoderma gangrenosum of the penis. Br J Dermatol. 1998;138(2):337–40. https://doi.org/10.1046/j.1365-2133.1998.02087.x.
Federman GL, Federman DG. Recalcitrant pyoderma gangrenosum treated with thalidomide. Mayo Clin Proc. 2000;75(8):842–4. https://doi.org/10.4065/75.8.842.
Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol. 2013;13(3):176–89. https://doi.org/10.1038/nri3401.
Song H, Lahood N, Mostaghimi A. Intravenous immunoglobulin as adjunct therapy for refractory pyoderma gangrenosum: systematic review of cases and case series. Br J Dermatol. 2018;178(2):363–8. https://doi.org/10.1111/bjd.15850.
Haag CK, Ortega-Loayza AG, Latour E, Keller JJ, Fett NM. Clinical factors influencing the response to intravenous immunoglobulin treatment in cases of treatment-resistant pyoderma gangrenosum. J Dermatol Treat. 2020;31(7):723–6. https://doi.org/10.1080/09546634.2019.1606888.
de Zwaan SE, Iland HJ, Damian DL. Treatment of refractory pyoderma gangrenosum with intravenous immunoglobulin. Australas J Dermatol. 2009;50(1):56–9. https://doi.org/10.1111/j.1440-0960.2008.00506.x.
Yamamoto T, Umegae S, Matsumoto K. Safety and clinical efficacy of granulocyte and monocyte adsorptive apheresis therapy for ulcerative colitis. World J Gastroenterol. 2006;12(4):520–5. https://doi.org/10.3748/wjg.v12.i4.520.
Russo I, Miotto S, Colpo A, et al. Successful treatment of pyoderma gangrenosum with granulocyte and monocyte adsorption apheresis. Int Wound J. 2017;14(1):282–4. https://doi.org/10.1111/iwj.12684.
Kanekura T, Kawahara K, Maruyama I, Kanzaki T. Treatment of pyoderma gangrenosum with granulocyte and monocyte adsorption apheresis. Ther Apher Dial. 2005;9(4):292–6. https://doi.org/10.1111/j.1744-9987.2005.00284.x.
Marzano AV, Fanoni D, Antiga E, et al. Expression of cytokines, chemokines and other effector molecules in two prototypic autoinflammatory skin diseases, pyoderma gangrenosum and Sweet’s syndrome [published correction appears in Clin Exp Immunol. 2015 Jul;181(1):188]. Clin Exp Immunol. 2014;178(1):48–56. https://doi.org/10.1111/cei.12394.
Agarwal A, Andrews JM. Systematic review: IBD-associated pyoderma gangrenosum in the biologic era, the response to therapy. Aliment Pharmacol Ther. 2013;38(6):563–72. https://doi.org/10.1111/apt.12431.
Ben Abdallah H, Fogh K, Bech R. Pyoderma gangrenosum and tumour necrosis factor alpha inhibitors: a semi-systematic review. Int Wound J. 2019;16(2):511–21. https://doi.org/10.1111/iwj.13067.
McKenzie F, Cash D, Gupta A, Cummings LW, Ortega-Loayza AG. Biologic and small-molecule medications in the management of pyoderma gangrenosum. J Dermatol Treat. 2019;30(3):264–76. https://doi.org/10.1080/09546634.2018.1506083.
Ahronowitz I, Harp J, Shinkai K. Etiology and management of pyoderma gangrenosum: a comprehensive review. Am J Clin Dermatol. 2012;13(3):191–211. https://doi.org/10.2165/11595240-000000000-00000.
Brooklyn TN, Dunnill MG, Shetty A, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55(4):505–9. https://doi.org/10.1136/gut.2005.074815.
Yamamoto T. An update on adalimumab for pyoderma gangrenosum. Drugs Today (Barc). 2021;57(9):535–42. https://doi.org/10.1358/dot.2021.57.9.3293619.
Cinotti E, Labeille B, Perrot JL, Pallot-Prades B, Cambazard F. Certolizumab for the treatment of refractory disseminated pyoderma gangrenosum associated with rheumatoid arthritis. Clin Exp Dermatol. 2014;39(6):750–1. https://doi.org/10.1111/ced.12393.
Hurabielle C, Schneider P, Baudry C, Bagot M, Allez M, Viguier M. Certolizumab pegol: a new therapeutic option for refractory disseminated pyoderma gangrenosum associated with Crohn’s disease. J Dermatol Treat. 2016;27(1):67–9. https://doi.org/10.3109/09546634.2015.1034075.
Pender TM, Ayandibu G, Van Voorhees AS. Certolizumab for the treatment of localized pyoderma gangrenosum associated with Crohn’s disease: a case report. Dermatol Ther. 2020;33(6): e14352. https://doi.org/10.1111/dth.14352.
Goldminz AM, Botto NC, Gottlieb AB. Severely recalcitrant pyoderma gangrenosum successfully treated with ustekinumab. J Am Acad Dermatol. 2012;67(5):e237-8. https://doi.org/10.1016/j.jaad.2012.04.045.
Skalkou A, Manoli SM, Sachinidis A, et al. Pyoderma gangrenosum and pyogenic arthritis presenting as severe sepsis in a rheumatoid arthritis patient treated with golimumab. Rheumatol Int. 2018;38(1):161–7. https://doi.org/10.1007/s00296-017-3861-8.
Diotallevi F, Campanati A, Radi G, et al. Pyoderma gangrenosum successfully treated with golimumab: case report and review of the literature. Dermatol Ther. 2019;32(4): e12928. https://doi.org/10.1111/dth.12928.
Liu FC, Liu NT, Huang TY. Ulcerative colitis with refractory pyoderma gangrenosum. QJM Int J Med. 2020;113(8):567–8. https://doi.org/10.1093/qjmed/hcaa078.
Russi L, Scharl M, Rogler G, Biedermann L. The efficacy and safety of golimumab as third- or fourth-line anti-TNF therapy in patients with refractory Crohn’s disease: a case series. Inflamm Intest Dis. 2017;2(2):131–8. https://doi.org/10.1159/000481400.
Kolios AG, Maul JT, Meier B, et al. Canakinumab in adults with steroid-refractory pyoderma gangrenosum. Br J Dermatol. 2015;173(5):1216–23. https://doi.org/10.1111/bjd.14037.
Ben Abdallah H, Fogh K, Vestergaard C, Bech R. Pyoderma gangrenosum and interleukin inhibitors: a semi-systematic review. Dermatology. 2021. https://doi.org/10.1159/000519320.
Macleod T, Berekmeri A, Bridgewood C, Stacey M, McGonagle D, Wittmann M. The immunological impact of IL-1 family cytokines on the epidermal barrier. Front Immunol. 2021;12: 808012. https://doi.org/10.3389/fimmu.2021.808012.
Lukens JR, Vogel P, Johnson GR, et al. RIP1-driven autoinflammation targets IL-1α independently of inflammasomes and RIP3. Nature. 2013;498(7453):224–7. https://doi.org/10.1038/nature12174.
Lukens JR, Kanneganti TD. SHP-1 and IL-1α conspire to provoke neutrophilic dermatoses. Rare Dis. 2014;2: e27742. https://doi.org/10.4161/rdis.27742.
Tartey S, Gurung P, Dasari TK, Burton A, Kanneganti TD. ASK1/2 signaling promotes inflammation in a mouse model of neutrophilic dermatosis. J Clin Investig. 2018;128(5):2042–7. https://doi.org/10.1172/JCI98446.
Tartey S, Gurung P, Samir P, Burton A, Kanneganti TD. Cutting edge: dysregulated CARD9 signaling in neutrophils drives inflammation in a mouse model of neutrophilic dermatoses. J Immunol. 2018;201(6):1639–44. https://doi.org/10.4049/jimmunol.1800760.
Nesterovitch AB, Gyorfy Z, Hoffman MD, et al. Alteration in the gene encoding protein tyrosine phosphatase nonreceptor type 6 (PTPN6/SHP1) may contribute to neutrophilic dermatoses. Am J Pathol. 2011;178(4):1434–41. https://doi.org/10.1016/j.ajpath.2010.12.035.
Nesterovitch AB, Szanto S, Gonda A, et al. Spontaneous insertion of a b2 element in the ptpn6 gene drives a systemic autoinflammatory disease in mice resembling neutrophilic dermatosis in humans. Am J Pathol. 2011;178(4):1701–14. https://doi.org/10.1016/j.ajpath.2010.12.053.
Nesterovitch AB, Arbieva Z, Toth DM, Tharp MD, Glant TT. A differential gene expression study: Ptpn6 (SHP-1)-insufficiency leads to neutrophilic dermatosis-like disease (NDLD) in mice. J Dermatol Sci. 2016;83(1):17–25. https://doi.org/10.1016/j.jdermsci.2016.03.005.
McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity. 2019;50(4):892–906. https://doi.org/10.1016/j.immuni.2019.03.021.
Herberger K, Dissemond J, Brüggestrat S, Sorbe C, Augustin M. Biologics and immunoglobulins in the treatment of pyoderma gangrenosum: analysis of 52 patients. J Dtsch Dermatol Ges. 2019;17(1):32–41. https://doi.org/10.1111/ddg.13741.
Moreno García M, Madrid González M, Prada Lobato JM. Secukinumab for pyoderma gangrenosum: a case report. Med Clin (Barc). 2019;152(6):246. https://doi.org/10.1016/j.medcli.2018.06.011.
McPhie ML, Kirchhof MG. Pyoderma gangrenosum treated with secukinumab: a case report. SAGE Open Med Case Rep. 2020. https://doi.org/10.1177/2050313X20940430.
Huang CM, Tsai TF. Use of brodalumab for the treatment of pyoderma gangrenosum: a case report. Dermatol Sin. 2021;39(1):57. https://doi.org/10.4103/ds.ds_40_20.
Tee MW, Avarbock AB, Ungar J, Frew JW. Rapid resolution of pyoderma gangrenosum with brodalumab therapy. JAAD Case Rep. 2020;6(11):1167–9. https://doi.org/10.1016/j.jdcr.2020.08.033.
Lauffer F, Seiringer P, Böhmer D, Oesterlin C, Eyerich K. 044 Safety and efficacy of anti-IL-17 (secukinumab) for the treatment of pyoderma gangrenosum. J Investig Dermatol. 2021;141(10):S156. https://doi.org/10.1016/j.jid.2021.08.046.
Kaffenberger BH. An open-label, proof-of-concept study of ixekizumab in the treatment of pyoderma gangrenosum. Ohio State University. https://www.clinicaltrials.gov/ct2/show/NCT03137160. Accessed 16 May 2022.
Sadik CD, Thieme M, Zillikens D, Terheyden P. First emergence of pyoderma gangraenosum, palmoplantar pustulosis and sacroiliitis in a psoriasis patient associated with switching from secukinumab to brodalumab. J Eur Acad Dermatol Venereol. 2019;33(11):e406-7. https://doi.org/10.1111/jdv.15714.
Munera-Campos M, Ballesca F, Carrascosa JM. Paradoxical reactions to biologic therapy in psoriasis: a review of the literature. Actas Dermosifiliogr (Engl Ed). 2018;109(9):791–800. https://doi.org/10.1016/j.ad.2018.04.003.
Toussirot É, Aubin F. Paradoxical reactions under TNF-α blocking agents and other biological agents given for chronic immune-mediated diseases: an analytical and comprehensive overview. RMD Open. 2016;2(2): e000239. https://doi.org/10.1136/rmdopen-2015-000239.
Wollina U, Schönlebe J, Fürll C. Pyoderma gangrenosum induced by secukinumab: a late paradoxical drug reaction. Dermatol Ther. 2020;33(1): e13161. https://doi.org/10.1111/dth.13161.
Jin K, Matsuzaki Y, Akasaka E, Nakano H, Sawamura D. Pyoderma gangrenosum triggered by switching from adalimumab to secukinumab. J Dermatol. 2019;46(3):e108-9. https://doi.org/10.1111/1346-8138.14611.
Pollack IR, Wolner ZJ, Hammett J, Swerlick RA. Pyoderma gangrenosum in a patient on ixekizumab. JAAD Case Rep. 2021;16:152–4. https://doi.org/10.1016/j.jdcr.2021.08.021.
Petty AJ, Whitley MJ, Balaban A, Ellington K, Marano AL. Pyoderma gangrenosum induced by secukinumab in a patient with psoriasis successfully treated with ustekinumab. JAAD Case Rep. 2020;6(8):731–3. https://doi.org/10.1016/j.jdcr.2020.06.011.
Guenova E, Teske A, Fehrenbacher B, et al. Interleukin 23 expression in pyoderma gangrenosum and targeted therapy with ustekinumab. Arch Dermatol. 2011;147(10):1203–5. https://doi.org/10.1001/archdermatol.2011.168.
Westerdahl JS, Nusbaum KB, Chung CG, Kaffenberger BH, Ortega-Loayza AG. Ustekinumab as adjuvant treatment for all pyoderma gangrenosum subtypes. J Dermatol Treat. 2021. https://doi.org/10.1080/09546634.2021.1937475.
Baier C, Barak O. Guselkumab as a treatment option for recalcitrant pyoderma gangrenosum. JAAD Case Rep. 2020;8:43–6. https://doi.org/10.1016/j.jdcr.2020.12.005.
Reese AM, Erickson K, Reed KB, Ortega-Loayza AG. Modified dose of guselkumab for treatment of pyoderma gangrenosum. JAAD Case Rep. 2022;21:38–42. https://doi.org/10.1016/j.jdcr.2021.11.030.
Burgdorf B, Schlott S, Ivanov IH, Dissemond J. Successful treatment of a refractory pyoderma gangrenosum with risankizumab. Int Wound J. 2020;17(4):1086–8. https://doi.org/10.1111/iwj.13359.
Rafail S, Kourtzelis I, Foukas PG, et al. Complement deficiency promotes cutaneous wound healing in mice. J Immunol. 2015;194(3):1285–91. https://doi.org/10.4049/jimmunol.1402354.
Sun S, Zhao G, Liu C, et al. Treatment with anti-C5a antibody improves the outcome of H7N9 virus infection in African green monkeys. Clin Infect Dis. 2015;60(4):586–95. https://doi.org/10.1093/cid/ciu887.
Giamarellos-Bourboulis EJ, Argyropoulou M, Kanni T, et al. Clinical efficacy of complement C5a inhibition by IFX-1 in hidradenitis suppurativa: an open-label single-arm trial in patients not eligible for adalimumab. Br J Dermatol. 2020;183(1):176–8. https://doi.org/10.1111/bjd.18877.
InflaRx N.V. 10-2021-InflaRx announces positive data from third cohort of phase IIa open-label study with vilobelimab in pyoderma gangrenosum. https://www.inflarx.de/Home/Investors/Press-Releases/10-2021-InflaRx-Announces-Positive-Data-from-Third-Cohort-of-Phase-IIa-Open-Label-Study-with-Vilobelimab-in-Pyoderma-Gangraenosum-0.html. Accessed 29 Dec 2021.
Schlapbach C, Navarini AA. The continuing evolution of targeted therapy for inflammatory skin disease. Semin Immunopathol. 2016;38(1):123–33. https://doi.org/10.1007/s00281-015-0524-2.
Kozono K, Nakahara T, Kikuchi S, Itoh E, Kido-Nakahara M, Furue M. Pyoderma gangrenosum with increased levels of serum cytokines. J Dermatol. 2015;42(12):1186–8. https://doi.org/10.1111/1346-8138.12970.
Kawakami T, Yamazaki M, Soma Y. Reduction of interleukin-6, interleukin-8, and anti-phosphatidylserine-prothrombin complex antibody by granulocyte and monocyte adsorption apheresis in a patient with pyoderma gangrenosum and ulcerative colitis. Am J Gastroenterol. 2009;104(9):2363–4. https://doi.org/10.1038/ajg.2009.271.
Lee WS, Choi YJ, Yoo WH. Use of tocilizumab in a patient with pyoderma gangrenosum and rheumatoid arthritis. J Eur Acad Dermatol Venereol. 2017;31(2):e75-7. https://doi.org/10.1111/jdv.13736.
Choong DJ, Ng JL, Vinciullo C. Pyoderma gangrenosum associated with Takayasu’s arteritis in a young Caucasian woman and response to biologic therapy with tocilizumab. JAAD Case Rep. 2021;9:4–6. https://doi.org/10.1016/j.jdcr.2020.12.034.
Borgia F, Sutera D, Spagnolo A, et al. Onset of pyoderma gangrenosum after tocilizumab therapy for Takayasu arteritis: a new undescribed paradoxical reaction. Br J Clin Pharmacol. 2021;87(8):3378–9. https://doi.org/10.1111/bcp.14756.
Choong DJ, Tan E. Does tocilizumab have a role in dermatology? A review of clinical applications, its adverse side effects and practical considerations. Dermatol Ther. 2021;34(4): e14990. https://doi.org/10.1111/dth.14990.
Hsu DH, Shi JD, Homola M, et al. A humanized anti-CD3 antibody, HuM291, with low mitogenic activity, mediates complete and reversible T-cell depletion in chimpanzees. Transplantation. 1999;68(4):545–54. https://doi.org/10.1097/00007890-199908270-00018.
Lorincz M, Kleszky M, Szalóki T Jr, Szalóki T. Refrakter pyoderma gangraenosum sikeres visilizumabkezelése colitis ulcerosás betegben. Orv Hetil. 2010;151(4):144–7. https://doi.org/10.1556/OH.2010.28786.
Sandborn WJ, Colombel JF, Frankel M, et al. Anti-CD3 antibody visilizumab is not effective in patients with intravenous corticosteroid-refractory ulcerative colitis. Gut. 2010;59(11):1485–92. https://doi.org/10.1136/gut.2009.205443.
Carmona-Rivera C, O’Neil LJ, Patino-Martinez E, et al. Autoantibodies present in hidradenitis suppurativa correlate with disease severity and promote the release of proinflammatory cytokines in macrophages. J Investig Dermatol. 2022;142(3 Pt B):924–35. https://doi.org/10.1016/j.jid.2021.07.187.
Sen M, Dogra S, Rathi M, Sharma A. Successful treatment of large refractory pyoderma gangrenosum-like presentation of granulomatosis with polyangiitis by rituximab. Int J Rheum Dis. 2017;20(12):2200–2. https://doi.org/10.1111/1756-185X.12882.
Murthy RK, Jackson J, Chatham WW, Sami N. Extensive pyoderma gangrenosum associated with granulomatosis with polyangiitis with both responsive to rituximab. J Clin Rheumatol. 2016;22(7):393–5. https://doi.org/10.1097/RHU.0000000000000447.
DaCunha M, Siscos S, Downing M, Tarantino I, Hall J. Pyoderma gangrenosum controlled with rituximab. JAAD Case Rep. 2019;5(7):593–5. https://doi.org/10.1016/j.jdcr.2019.04.019.
Croitoru D, Nathanielsz N, Seigel K, et al. Clinical manifestations and treatment outcomes of pyoderma gangrenosum following rituximab exposure: a systematic review. J Am Acad Dermatol. 2021. https://doi.org/10.1016/j.jaad.2021.12.028.
Lytvyn Y, Mufti A, Maliyar K, Sachdeva M, Yeung J. Onset of pyoderma gangrenosum in patients on biologic therapies: a systematic review. Adv Skin Wound Care. 2022. https://doi.org/10.1097/01.ASW.0000820252.96869.8e (Epub ahead of print).
Wyant T, Fedyk E, Abhyankar B. An overview of the mechanism of action of the monoclonal antibody vedolizumab. J Crohns Colitis. 2016;10(12):1437–44. https://doi.org/10.1093/ecco-jcc/jjw092.
Fleisher M, Marsal J, Lee SD, et al. Effects of vedolizumab therapy on extraintestinal manifestations in inflammatory bowel disease. Dig Dis Sci. 2018;63(4):825–33. https://doi.org/10.1007/s10620-018-4971-1.
Groudan K, Gupta K, Singhania R. Vedolizumab (Entyvio®) for the treatment of pyoderma gangrenosum in a Crohn’s disease Patient. Cureus. 2021;13(1): e12582. https://doi.org/10.7759/cureus.12582.
Kim SB, Diaz L, Abreu M. Vedolizumab-induced pyoderma gangrenosum in a patient with ulcerative colitis: 2009. Am J Gastroenterol. 2017;112:S1106-7. https://doi.org/10.14309/00000434-201710001-02010.
Yeh JE, Tsiaras WG. Intractable pyoderma gangrenosum in a Crohn’s disease patient on vedolizumab. JAAD Case Rep. 2017;3(2):110–2. https://doi.org/10.1016/j.jdcr.2017.01.012.
Shibuya T, Haga K, Saeki M, et al. Pyoderma gangrenosum in an ulcerative colitis patient during treatment with vedolizumab responded favorably to adsorptive granulocyte and monocyte apheresis. J Clin Apher. 2020;35(5):488–92. https://doi.org/10.1002/jca.21821.
Li H, Zuo J, Tang W. Phosphodiesterase-4 inhibitors for the treatment of inflammatory diseases. Front Pharmacol. 2018;9:1048. https://doi.org/10.3389/fphar.2018.01048.
Zgraggen A, Maul LV, Hasler P, Mueller RB. Pyoderma gangrenosum and palisaded neutrophilic granulomatous dermatitis in a patient with rheumatoid arthritis treated with apremilast. Ann Clin Case Rep. 2020;5:1807.
Laird ME, Tong LX, Lo Sicco KI, Kim RH, Meehan SA, Franks AG Jr. Novel use of apremilast for adjunctive treatment of recalcitrant pyoderma gangrenosum. JAAD Case Rep. 2017;3(3):228–9. https://doi.org/10.1016/j.jdcr.2017.02.019.
Welsch K, Holstein J, Laurence A, Ghoreschi K. Targeting JAK/STAT signalling in inflammatory skin diseases with small molecule inhibitors. Eur J Immunol. 2017;47(7):1096–107. https://doi.org/10.1002/eji.201646680.
Solimani F, Meier K, Ghoreschi K. Emerging topical and systemic JAK inhibitors in dermatology. Front Immunol. 2019;10:2847. https://doi.org/10.3389/fimmu.2019.02847.
Alves de Medeiros AK, Speeckaert R, Desmet E, Van Gele M, De Schepper S, Lambert J. JAK3 as an emerging target for topical treatment of inflammatory skin diseases. PLoS One. 2016;11(10): e0164080. https://doi.org/10.1371/journal.pone.0164080.
DeFilippis EM, Feldman SR, Huang WW. The genetics of pyoderma gangrenosum and implications for treatment: a systematic review. Br J Dermatol. 2015;172(6):1487–97. https://doi.org/10.1111/bjd.13493.
Palanivel JA, Macbeth AE, Levell NJ. Pyoderma gangrenosum in association with Janus kinase 2 (JAK2V617F) mutation. Clin Exp Dermatol. 2013;38(1):44–6. https://doi.org/10.1111/j.1365-2230.2012.04375.x.
Hideki M, Tetsurou T, Sakae F, Kensei K, Makoto O, Toshinobu K. Idiopathic myelofibrosis and pyoderma gangrenosum involving a mutation of Janus kinase 2 (JAK2V617F), showing poor prognosis. Eur J Dermatol. 2013;23(2):256–7. https://doi.org/10.1684/ejd.2013.1956.
Orfaly VE, Kovalenko I, Tolkachjov SN, Ortega-Loayza AG, Nunley JR. Tofacitinib for the treatment of refractory pyoderma gangrenosum. Clin Exp Dermatol. 2021;46(6):1082–5. https://doi.org/10.1111/ced.14683.
Montagnon CM, Fracica EA, Patel AA, et al. Pyoderma gangrenosum in hematologic malignancies: a systematic review. J Am Acad Dermatol. 2020;82(6):1346–59. https://doi.org/10.1016/j.jaad.2019.09.032.
Shanmugam VK, McNish S, Shara N, et al. Chronic leg ulceration associated with polycythemia vera responding to ruxolitinib (Jakafi®). J Foot Ankle Surg. 2013;52(6):781–5. https://doi.org/10.1053/j.jfas.2013.07.003.
Nasifoglu S, Heinrich B, Welzel J. Successful therapy for pyoderma gangrenosum with a Janus kinase 2 inhibitor. Br J Dermatol. 2018;179(2):504–5. https://doi.org/10.1111/bjd.16468.
Scheinberg M, Machado LA, Castro M, LG, Ferreira SB, Michalany N. Successful treatment of ulcerated pyoderma gangrenosum with baricitinib, a novel JAK inhibitor. J Transl Autoimmun. 2021;4: 100099. https://doi.org/10.1016/j.jtauto.2021.100099.
ClinicalTrials.gov. Baricitinib in the treatment of adults with pyoderma gangrenosum (PG). https://clinicaltrials.gov/ct2/show/NCT04901325. Accessed 5 Mar 2022.
Thomas KS, Ormerod AD, Craig FE, et al. Clinical outcomes and response of patients applying topical therapy for pyoderma gangrenosum: a prospective cohort study. J Am Acad Dermatol. 2016;75(5):940–9. https://doi.org/10.1016/j.jaad.2016.06.016.
Kino T, Hatanaka H, Miyata S, et al. FK-506, a novel immunosuppressant isolated from a Streptomyces. II. Immunosuppressive effect of FK-506 in vitro. J Antibiot (Tokyo). 1987;40(9):1256–65. https://doi.org/10.7164/antibiotics.40.1256.
Marzano AV, Trevisan V, Lazzari R, Crosti C. Topical tacrolimus for the treatment of localized, idiopathic, newly diagnosed pyoderma gangrenosum. J Dermatol Treat. 2010;21(3):140–3. https://doi.org/10.3109/09546630903268239.
Schuppe HC, Homey B, Assmann T, Martens R, Ruzicka T. Topical tacrolimus for pyoderma gangrenosum. Lancet. 1998;351(9105):832. https://doi.org/10.1016/S0140-6736(05)78962-5.
Wollina U. Letter to the editor: Temporary renal insufficiency associated with topical tacrolimus treatment of multilocal pyoderma gangrenosum. J Dermatol Case Rep. 2013;7(3):106–7. https://doi.org/10.3315/jdcr.2013.1154.
Cecchi R, Pavesi M, Bartoli L, Brunetti L. Successful treatment of localized pyoderma gangrenosum with topical pimecrolimus. J Cutan Med Surg. 2012;16(5):295–7. https://doi.org/10.1177/120347541201600503.
Bellini V, Simonetti S, Lisi P. Successful treatment of severe pyoderma gangrenosum with pimecrolimus cream 1%. J Eur Acad Dermatol Venereol. 2008;22(1):113–5. https://doi.org/10.1111/j.1468-3083.2007.02280.x.
Azizan NZ, Gangaram HB, Hussein SH. A novel therapy for the treatment of pyoderma gangrenosum. Med J Malays. 2008;63(1):51–4.
Strunck JL, Cutler B, Latour E, Seminario-Vidal L, Ortega-Loayza AG. Wound care dressings for pyoderma gangrenosum. J Am Acad Dermatol. 2022;86(2):458–60. https://doi.org/10.1016/j.jaad.2021.09.053.
Croitoru D, Naderi-Azad S, Sachdeva M, Piguet V, Alavi A. A wound care specialist’s approach to pyoderma gangrenosum. Adv Wound Care (New Rochelle). 2020;9(12):686–94. https://doi.org/10.1089/wound.2020.1168.
Janowska A, Oranges T, Fissi A, Davini G, Romanelli M, Dini V. PG-TIME: a practical approach to the clinical management of pyoderma gangrenosum. Dermatol Ther. 2020;33(3): e13412. https://doi.org/10.1111/dth.13412.
Eisendle K, Thuile T, Deluca J, Pichler M. Surgical treatment of pyoderma gangrenosum with negative pressure wound therapy and skin grafting, including xenografts: personal experience and comprehensive review on 161 cases. Adv Wound Care (New Rochelle). 2020;9(7):405–25. https://doi.org/10.1089/wound.2020.1160.
Pichler M, Larcher L, Holzer M, et al. Surgical treatment of pyoderma gangrenosum with negative pressure wound therapy and split thickness skin grafting under adequate immunosuppression is a valuable treatment option: case series of 15 patients. J Am Acad Dermatol. 2016;74(4):760–5. https://doi.org/10.1016/j.jaad.2015.09.009.
Almeida IR, Coltro PS, Gonçalves HOC, et al. The role of negative pressure wound therapy (NPWT) on the treatment of pyoderma gangrenosum: a systematic review and personal experience. Wound Repair Regen. 2021;29(3):486–94. https://doi.org/10.1111/wrr.12910.
Teguh DN, Bol Raap R, Koole A, et al. Hyperbaric oxygen therapy for nonhealing wounds: treatment results of a single center. Wound Repair Regen. 2021;29(2):254–60. https://doi.org/10.1111/wrr.12884.
Altunay I, Kucukunal A, Sarikaya S, Tukenmez DG. A favourable response to surgical intervention and hyperbaric oxygen therapy in pyoderma gangrenosum. Int Wound J. 2014;11(4):350–3. https://doi.org/10.1111/j.1742-481X.2012.01102.x.
Vieira WA, Barbosa LR, Martin LMM. Hyperbaric oxygen therapy as an adjuvant treatment for pyoderma gangrenosum. An Bras Dermatol. 2011;86(6):1193–6.
de Sousa MR, Moreira MJ, Rosa B, Cotter J. Hyperbaric oxygen therapy for refractory pyoderma gangrenosum: a salvage treatment. BMJ Case Rep. 2021;14(2): e238638. https://doi.org/10.1136/bcr-2020-238638.
Tutrone WD, Green K, Weinberg JM, Caglar S, Clarke D. Pyoderma gangrenosum: dermatologic application of hyperbaric oxygen therapy. J Drugs Dermatol. 2007;6(12):1214–9.
Moreira C, Lopes S, Cruz MJ, Azevedo F. Topical timolol for the treatment of pyoderma gangrenosum. BMJ Case Rep. 2017;2017:bcr2016218589. https://doi.org/10.1136/bcr-2016-218589.
Liu DY, Fischer R, Fraga G, Aires DJ. Collagenase ointment and topical timolol gel for treating idiopathic pyoderma gangrenosum. J Am Acad Dermatol. 2014;71(5):e225-6. https://doi.org/10.1016/j.jaad.2014.07.060.
Fonseka HF, Ekanayake SM, Dissanayake M. Two percent topical phenytoin sodium solution in treating pyoderma gangrenosum: a cohort study. Int Wound J. 2010;7(6):519–23. https://doi.org/10.1111/j.1742-481X.2010.00725.x.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open access funding provided by Università degli Studi di Milano within the CRUI-CARE Agreement. No funding was received for the preparation of this article.
Conflicts of interest/competing interests
The authors have no conflicts of interest that are directly relevant to the content of this article.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Code availability
Not applicable.
Author contributions
All authors have made substantial contributions to the work and have approved the final version of this article. CAM, GG, MP, and ML reviewed the relevant literature. AVM, CAM, MP, and AOL edited and approved the final draft.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Maronese, C.A., Pimentel, M.A., Li, M.M. et al. Pyoderma Gangrenosum: An Updated Literature Review on Established and Emerging Pharmacological Treatments. Am J Clin Dermatol 23, 615–634 (2022). https://doi.org/10.1007/s40257-022-00699-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-022-00699-8