
Abstract
Graft-versus-host disease (GVHD) is an immunological reaction and a frequent complication following allogeneic hematopoietic stem cell transplantation. It is associated with high mortality rates and may have a significant negative impact on the patient’s quality of life, particularly in the chronic-stage setting. Many different organs can be involved, which leads to a wide range of clinical manifestations. In this context, dermatologists play a key role by diagnosing and treating GVHD from the outset since cutaneous features are not just the most common but are also usually the presenting sign. Several skin-direct therapies are available and may be indicated as monotherapy or adjuvant treatment in order to allow faster tapering and withdrawal of systemic immunosuppression. Treatment of steroid-refractory patients remains a challenge and, to date, no consensus has been reached for one single agent in second-line therapy. This article aims to review skin involvement as well as provide and update discussion on therapeutic options for both acute and chronic cutaneous GVHD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Cutaneous manifestations are the most common and are often the presenting sign of graft-versus-host disease (GVHD). |
Cutaneous GVHD may mimic well-known inflammatory and autoimmune disorders. |
The frequent overlapping of both clinical and histopathological findings of cutaneous GVHD with other entities can make it difficult to establish a definite diagnosis. |
1 Background
Graft-versus-host disease (GVHD) is an immune-mediated reaction and a major complication following allogeneic hematopoietic stem cell transplantation (HSCT). It can affect between 40 and 60% of patients, depending on host and donor factors, and accounts for 15% of mortality after HSCT [1, 2]. Although extremely rare, GVHD may also occur after transfusion of blood products, after solid organ transplantation, and even after autologous HSCT [3,4,5].
In the allogeneic HSCT setting, human leucocyte antigen (HLA) mismatch is the strongest determinant of GVHD occurrence, but minor histocompatibility antigens are also thought to play a role in its pathophysiology [6]. Additional risk factors include advanced age of the recipient, myeloablative conditioning regimens, gender disparity between host and donor, donor multiparity, nonconventional GVHD prophylaxis, and the use of peripheral blood stem cells as the graft source [2, 7,8,9,10].
In spite of all its negative consequences, GVHD is associated with a beneficial effect known as graft-versus-leukemia (GVL) and lower relapse rates of hematologic malignancies. This is of particular relevance in reduced-intensity conditioning regimens that have limited cytotoxicity, and may not be able to promote complete destruction of the malignant cells themselves. From this perspective, depending on the patient’s underlying disease and conditioning regimen applied, a mild presentation of GVHD is considered beneficial in order to ensure an immunological antitumor effect [11].
Dermatologists play a critical role in the context of allogeneic HSCT, not only by attending patients who can benefit from this kind of therapy, such as those with cutaneous T-cell lymphomas, but also by diagnosing and treating GVHD from the outset since cutaneous manifestations are the most common and are often the presenting sign.
2 Classification
GVHD was originally classified as acute or chronic depending on the time of onset after HSCT. GVHD signs and symptoms appearing within the first 100 days after transplantation were considered acute, whereas those occurring beyond 100 days were assumed as chronic, independent of clinical presentation. However, expanding transplant practices affecting the recipient’s immune status, such as reduced-intensity conditioning regimens, infusion of donor lymphocytes (DLI), and second allogeneic HSCT, have changed the classical onset of acute and chronic manifestations [12,13,14]. In addition, tapering and withdrawal of systemic immunosuppression are frequently related to relapse of acute GVHD after 100 days of HSCT [13, 14]. Since these two forms of GVHD can differ in terms of prognosis and treatment, a new classification was considered necessary.
In 2005, the National Institutes of Health (NIH) Working Group redefined both acute and chronic GVHD, primarily according to its clinical and histopathological features, and divided them into two subcategories (classic acute GVHD, and persistent, recurrent or late-onset acute GVHD; classic chronic GVHD and overlap syndrome), which were reviewed and ratified by the 2014 NIH Chronic GVHD Diagnosis and Staging Consensus (Table 1) [13, 15].
3 Pathophysiology
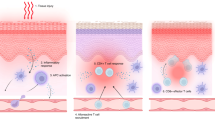
3.1 Acute Graft-Versus-Host Disease (GVHD) Pathophysiology
Acute GVHD is mediated by donor T cells that migrate to lymphoid tissues immediately following graft infusion. Chemotherapy and radiotherapy used during the conditioning regimens induce tissue damage and consequently release exogenous (lipopolysaccharides) and endogenous (e.g. interleukin [IL]-1, tumor necrosis factor [TNF]-α, IL-6, and interferon [IFN]-γ) molecules that activate the innate immune response via toll-like receptors [16]. Host antigen-presenting cells (APCs) in the early post-HSCT phase, and emerging donor APCs, detect antigen histocompatibility disparity and provide co-stimulatory molecules for the activation of alloreactive T lymphocytes, which expand and differentiate into various subtypes, preferentially T helper (Th) 1/T cytotoxic (Tc) 1 and Th17/Tc17 [17, 18]. Cytotoxic effector T cells exit lymphoid tissues and traffic to the target organs (mainly the skin, gut, liver, and thymus) through chemokine receptor, selectin-ligand and integrin-ligand interaction, causing tissue damage with direct cytotoxic activity and recruitment of other inflammatory cells [19]. The loss of normal thymic repertoire selection in addition to an imbalance between the effector and regulatory T cells are also thought to play a role in its pathophysiology [20,21,22,23].
3.2 Chronic GVHD Pathophysiology
Chronic GVHD pathophysiology involves both alloimmune and autoimmune reactions. The post-fetal thymus is not effective in eliminating autoreactive T cells and may be further compromised by the conditioning regimens and alloreactive T cells during acute GVHD [24]. It has been proposed that impaired negative selection in the thymus may be implicated in production of autoreactive and alloreactive T CD4+ cells [24, 25]. In the past, the Th2 pathway was correlated with fibrosis due to IL-4 and IL-13 binding and activation of fibroblasts [26, 27]. However, emerging evidence supports the role of the Th17 pathway by showing increased IL-17 messenger RNA transcripts, and infiltration of Tc17 in the skin [28]. Other studies suggest that reduced levels of FOXP3+ CD4+CD25+ regulatory T cells may also play a role by revealing decreased frequency and reduced gene expression of T-regulatory transcription factor FOXP3 in chronic GVHD patients when compared with individuals without GVHD or healthy donors [29]. In addition, increased activity of B lymphocytes with both autoantibody and alloantibody secretion have been shown to be implicated [30, 31]. The interaction of these antibodies with tissue macrophages may explain its aberrant differentiation, resulting in transforming growth factor (TGF)-β production, myofibroblast activation, and fibrosis [32, 33].
4 Biomarkers
Several studies have sought to identify the role of biomarkers in GVHD in order to improve clinical and histopathological diagnosis, prediction of disease occurrence, and response to therapy. Although some trials have demonstrated the value of biomarkers in the GVHD setting, limited evidence exists to date to support their use in clinical routine.
Initial studies have failed to demonstrate correlation between some cytokines (e.g. IL-2, TNFα) and acute GVHD development and severity due to their lack of specificity in the context of HSCT [34, 35]. More recent trials have focused on additional cytokines, cell surface molecules, and soluble biomarkers that may foretell end-organ injury. Paczesny et al. showed direct association of plasmatic levels of elafin, a soluble protein produced by keratinocytes, and disease severity in cutaneous GVHD [36], while Ahmed et al., in a cohort study, identified B-cell-activating factor (BAFF) and the chemokine receptors CXCL10 and CXCL11 as predictors for both acute and chronic GVHD diagnosis [37]. In a prospective, randomized trial, Levine et al. concluded that a biomarker algorithm composed of TFNR1, ST2, and REG3α was able to better predict 6-month nonrelapse mortality at acute GVHD diagnosis than Glucksberg grade [38]. Hartwell et al. reported that an algorithm based on the concentrations of two biomarkers (ST2 and REG3α) 7 days after HSCT, predicted patients at high risk for lethal GVHD and nonrelapse mortality [39]. In addition, Pidala et al. demonstrated that a 3 RNA marker panel (IRS2, PLEKHF1, and IL1R2) in combination with clinical variables could distinguish chronic GVHD cases from non-GVHD controls with a high degree of accuracy [40]. Newer markers have been suggested but need broader clinical confirmation.
5 Clinical Manifestations
5.1 Acute GVHD Clinical Manifestations
Skin, gut, and liver are the major target organs in acute GVHD, and therefore the classic triad of exanthema, diarrhea and elevated bilirubin levels strongly suggest the diagnosis [41]. On the other hand, each site can also be individually involved, which may represent a challenge for physicians dealing with allogeneic bone marrow-transplanted patients.
Acute GVHD signs and symptoms are usually present after neutrophil engraftment, but a relatively rare and severe hyperacute form of GVHD has been reported within the first 2 weeks of HSCT [42]. Saliba et al. carried out a prospective clinical trial with 265 grade II–IV acute GVHD patients and evidenced that skin involvement was significantly more common (88 vs. 44%) and extensive (stages III–IV, 88 vs. 66%) in hyperacute forms compared with acute GVHD. In addition, these authors reported higher mortality and lower response rates to first-line therapy in the hyperacute group of patients [42].
Cutaneous manifestations are the most common and are often the presenting sign of the disease. They are classically described as erythematous maculopapular morbilliform eruptions starting on the face, ears, palms, and soles. Follicular erythema is a frequent acute GVHD early manifestation, and both erythematous macular and papular rashes can occur [13, 43]. Skin lesions often spread to the trunk and may evolve to erythroderma, affecting an extensive body surface area (BSA) (Fig. 1a, b), while epidermolysis can be present in severe cases resembling toxic epidermal necrolysis (Figs. 2, 3a, b) [13]. Itching and dysesthesia are commonly reported, but some patients remain asymptomatic. Atypical presentations mimicking pityriasis rubra pilaris, acquired ichthyosis, and psoriasis vulgaris-like eruption have been previously reported in the literature [44,45,46].

Acute cutaneous graft-versus-host disease. a Maculopapular rash; b erythroderma

Acute cutaneous graft-versus-host disease affecting the palms

Acute cutaneous GVHD. a Blisters; b complete detachment of the epidermis in acute GVHD mimicking toxic epidermal necrolysis. GVHD graft-versus-host disease
When the skin is the only organ involved, the frequent overlapping of both clinical and histopathological features of acute GVHD with other entities such as drug hypersensitivity reactions, viral exanthems, and lymphocyte recovery eruptions makes it difficult to establish a definite diagnosis [47, 48]. However, the involvement of particular sites, such as face, palms, and soles, may favor acute GVHD in some cases [41]. In a retrospective study on allogeneic bone marrow-transplanted patients who had undergone bone marrow transplant within the first 100 days, Byun et al. concluded that facial involvement was more common among acute GVHD patients than those with drug hypersensitivity reactions (59 vs. 24%). In addition, involvement of the face, palms, and soles was even more frequent in acute GVHD individuals when compared with patients with drug eruptions (36% vs. no occurrence) [41]. Recently, Kaminska-Winciorek et al. carried out a prospective study using dermoscopy on cutaneous acute GVHD and demonstrated a pinkish or reddish background and well-visible, multiple telangiectasias [49].
Although rarely related to conditioning regimens applied for allogeneic stem-cell transplants, toxic erythema induced by chemotherapy may resemble cutaneous acute GVHD and, likewise, involve palms and soles [48, 50]. However, particular histological findings, such as eccrine squamous syringometaplasia, when present, can be a clue for this diagnosis [50].
Involvement of the oral mucosa is unusual in the acute GVHD setting but may predict severe outcome. In a 15-year study of 2578 recipients of allogeneic HSCT, Ion et al. documented 82% of oral involvement in patients with grade III or IV acute GVHD [51]. Lesions were characterized by erythema and ulcerations resembling the mucositis induced by conditioning regimens and recrudescent herpes simplex virus [51]. Other mucous membranes such as ocular, genital, and nasal can also be affected [52].
Photo-induced rashes have been reported as acute GVHD isomorphic phenomenon triggered by phototherapy and sun exposure [53]. However, photosensitivity reactions are not part of the cutaneous GVHD spectrum of manifestations, and photosensitizing drugs should be extensively investigated in these cases. Voriconazole deserves added attention for being a frequent cause of phototoxicity reactions in patients undergoing HSCT.
Skin biopsies should be performed as routine, as per European consensus recommendations, but they should never delay management since early treatment is associated with improved prognosis [54]. No direct relationship exists between clinical and histological grading (except in late stages when epidermal detachment occurs), therefore a clinical-histopathological correlation is essential. Skin histology reveals interface dermatitis, vacuolar degeneration of the basal layers, dyskeratosis, and superficial infiltrate, which are characteristic of, but not specific to, acute GVHD. The combination of histopathological findings of GVHD and other entities can be a clue for diagnosis in atypical presentations mimicking well-known dermatological disorders [44,45,46]. Histopathological grading was defined by Lerner et al. in 1974 and is currently used (Table 2) [55].
Such recommendations withstanding, some European transplant centers advocate that skin biopsies should only be performed for atypical cases or depending on the clinical course of the disease due to the low sensitivity and specificity of the histopathological findings [54]. Weaver and Bergfeld revealed that the presence of eosinophils in histopathological skin samples does not reliably distinguish drug hypersensitivity reaction from GVHD. In their concept, a very high number of eosinophils per high-power fields (>16) is necessary to definitely rule out GVHD diagnosis [56]. Furthermore, Vassallo et al. reported that normal-looking skin is not necessarily normal in histology after allogeneic HSCT, and may resemble GVHD features in approximately 30% of patients [57].
Once diagnosis is confirmed, rash type and extent, bilirubin levels, and volume of diarrhea must be evaluated in order to define overall acute GVHD grade, which has prognostic significance [58]. The modified Seattle Glucksberg criteria are recommended for grading acute GVHD (Table 3) [58].
5.2 Chronic GVHD Clinical Manifestations
Previous acute GVHD is a major risk factor for chronic GVHD, with the skin being the most commonly affected organ [1]. Involvement of other sites, such as oral, ocular and genital mucous, liver, gastrointestinal tract, joints, fascia, and lungs, can occur alone or concurrently, which leads to a wide variety of clinical manifestations and a possible significant negative impact on the patient’s quality of life [13, 15].
Skin involvement presents with many different nonsclerotic and sclerotic phenotypes and may simulate well-known chronic inflammatory and autoimmune diseases [13, 15]. Nonsclerotic GVHD tends to have earlier onset and may precede sclerotic forms, although it is not a mandatory prerequisite. Previous studies have associated detectable autoantibodies and antinuclear antibody (ANA) patterns with an increased risk for chronic GVHD, particularly its severe forms [59]; however, more recent trials do not corroborate these findings. A cross-sectional study with 206 chronic GVHD patients demonstrated no relationship between autoantibodies and sclerotic manifestations [60]. No correlation was found between platelet-derived growth factor receptor autoantibodies and severity of chronic GVHD in 39 patients [61]. Kuzmina et al. carried out a cross-sectional prospective study with 280 chronic GVHD patients and evidenced no association between a panel of 21 autoantibodies and GVHD activity or severity [62]. Only oral chronic GVHD was significantly associated with the detection of autoantibodies in this study [62].
A lichen planus-like eruption and poikiloderma (skin atrophy, pigmentary changes, telangiectasia) are the most typical eruptions in the nonsclerotic chronic GVHD setting. Lichenoid manifestation may be clinically undistinguishable from the idiopathic lichen planus and, likewise, present with erythematous or violaceous flat-topped papules with surface reticulations and shiny appearance (Fig. 4a–c) [13]. However, GVHD rash tends to involve sites usually spared by the idiopathic disease, such as the ears, face, palms, and soles [63]. Dyshidrosis-like lesions on the palms are reported (Fig. 5) as well as ocular hyperpigmentation, which may be a clinical predicting factor for extensive sclerotic forms [64]. Involvement of the oral mucosa can also resemble idiopathic lichen planus and present with white arboriform lines and erosive manifestations (Fig. 6) [13, 15]. Other oral features include painful ulcers, mucoceles, mucosal atrophy, pseudomembranes, and symptoms of sicca syndrome that can interfere with patient food intake [13, 15].

Chronic cutaneous graft-versus-host disease. a–c Lichen planus-like lesions

Dyshidrosis-like lesions in chronic cutaneous graft-versus-host disease

White arboriform lines resembling oral lichen planus in chronic cutaneous graft-versus-host disease
Less specific papulosquamous lesions are reported and may mimic well-known dermatological entities, including psoriasis, pityriasis rosea (Fig. 7) and eczema [65,66,67]. Jang et al. described a generalized psoriasiform chronic GVHD rash resulting in secondary vitiligo [67].

Chronic cutaneous graft-versus-host disease lesions resembling pityriasis rosea
Autoimmune diseases are also part of the spectrum of nonsclerotic chronic GVHD despite the lack of specificity of identified autoantibodies. In an observational prospective study of 50 chronic GVHD patients, Ĉeović et al. evidenced rates of vitiligo and alopecia areata of 8 and 12%, respectively [68]. Lupus erythematous-like, dermatomyositis-like and epidermolysis bullosa acquisita-like eruptions have also been reported [69,70,71].
Hyperpigmentation and hypopigmentation usually represent post-inflammatory signs instead of active disease and their occurrence has been associated with an increased risk of sclerosis [72, 73]. Other nonspecific features include keratosis pilaris, xerosis, acquired ichthyosis, erythema multiforme-like eruptions, and exfoliative dermatitis [74].
Although granulomatous diseases are not considered for chronic GVHD diagnosis according to the 2014 NIH consensus, there are reports in the literature of sarcoidosis with skin involvement following allogeneic HSCT. Manalo et al. described a case of cutaneous and pulmonary sarcoidosis occurring concurrently with lichen sclerosus-like chronic GVHD [75]. Recently, Kinsella et al. reported a case of folliculotropic mycosis fungoides in the post-HSCT setting, first diagnosed as cutaneous sarcoidosis based on clinical-histopathological correlation. In this study, skin eruptions presented as a pruritic lichenoid rash evolving to erythroderma in association with lymphadenopathy [76].
Sclerotic eruptions are characterized by fibrosis, which may be superficial and localized, resembling lichen sclerosus and morphea, or deep and disseminated, mimicking systemic scleroderma (Fig. 8a, b). Lichen sclerosus-like and morphea-like GVHD simulate the idiopathic forms, although the classic ‘lilac ring’ is not reported in morphea-like GVHD [13]. Diffuse sclerotic GVHD tends to involve deeper collagen bundles compared with the idiopathic scleroderma, and skin stiffening without any other evident cutaneous lesion may be the first symptom. However, hyperpigmentation or hypopigmentation of the overlying skin, as well as concurrent nonsclerotic lesions, are also reported [14]. Vascular proliferations within the areas of sclerosis resembling Kaposi’s sarcoma have been described, particularly on the extremities [77]. Recently, Kaffenberger et al. proposed the term GVHD-associated angiomatosis due to its specificity for sclerotic-type chronic GVHD when compared with other fibrosing entities [77].

Chronic cutaneous graft-versus-host disease. a Morphea-like eruption; b localized sclerotic lesion
Diffuse sclerotic GVHD affects both dermis and subcutaneous tissues, causing adnexal loss, alopecia, and ulcers (Fig. 9). Deep sclerosis may also lead to limitation of mouth opening, vaginal stenosis, restricted chest-wall expansion, and the inability to move or pinch the thickened skin. Involvement of joints and fascia may worsen the range of motion and induce contractures with severe disability [14]. Chronic GVHD manifestations mimicking eosinophilic fasciitis with cellulite-like appearance due to subcutaneous septal and fascial fibrosis are considered the deep variant of the disease by some authors [78]. Unlike the idiopathic systemic scleroderma, Raynaud phenomenon and sclerodactyly are unusual [14].

Diffuse sclerosis with ulcer in chronic cutaneous graft-versus-host disease
For diagnostic purposes, the NIH recommends that each clinical manifestation, and, likewise, skin involvement (as well as nails, hair, and mouth), should be further classified into four categories: diagnostic features, which establish the presence of chronic GVHD even without skin biopsies or additional tests and include poikiloderma, lichen planus-like, lichen sclerosus-like, morphea-like and deep sclerotic eruptions; distinctive manifestations, which, alone, are not sufficient for diagnosis and comprise depigmentation vitiligo-like and papulosquamous lesions; other features, referring to nonspecific, rare, or controversial eruptions, such as ichthyosis, keratosis pilaris, sweat impairment, hypopigmentation and hyperpigmentation; and common manifestations to both acute and chronic GVHD, including erythema, maculopapular rash and pruritus (Table 4) [15].
The presence of at least one diagnostic manifestation or one distinctive feature confirmed by biopsy, laboratory tests, or radiology in the same or another organ is necessary for chronic GVHD diagnosis [13, 15]. Cutaneous histological findings are similar to the acute disease and reveal interface dermatitis with vacuolar degeneration and lymphocyte satellitosis. Lichenoid lesions demonstrate acanthosis and wedge-shaped hypergranulosis mimicking the idiopathic lichen planus. Hyperparakeratosis is frequently present and clinically corresponds to desquamation, which is not usually present in idiopathic disease. Sclerotic eruptions are histologically represented by collagen homogenization (sclerosis) of the dermis and/or subcutaneous tissues with little or no epidermal involvement. In lichen sclerosus-like GVHD, collagen alteration is confined to the papillary dermis, and associated atrophy, hyperkeratosis, and follicular plugging may occur [79].
Once diagnosis is confirmed, the severity of the affected organs should be properly scored according to a 4-point scale (0–3). The 2014 NIH group recommends that the rash type and its extent be separately evaluated, and the higher subscore must be considered for the overall skin score [15]. Skin subscores 1, 2, and 3 correspond to 1–18, 19–50 and >50% of BSA, respectively. Superficial sclerosis refers to subscore 2, and deep sclerotic eruptions associated with impaired mobility or ulcers refers to subscore 3. Since cutaneous pigmentary changes may represent post-inflammatory manifestations instead of active GVHD, they should not be punctuated [15]. In cases of poikiloderma, only the erythema should be taken into account in BSA skin score [15].
According to the NIH, the overall chronic GVHD grade must be defined as mild, moderate, or severe (Table 5) for prognostic and management purposes [15]. Other validated skin-scoring approaches have been reported, such as the Vienna Skin Score, but their use is more appropriate in the context of clinical studies [80].
6 Treatment
Therapeutic options for GVHD are likely to be determined by different relevant factors such as disease classification, overall grading, organs involved, and associated symptoms [81,82,83,84]. The importance of an immunological antitumor effect, depending on the patient’s underlying disease and the risk factors involved in GVHD severity, should also be carefully evaluated [11].
Although phototherapy and extracorporeal photopheresis (ECP) are established treatment options where available, direct sun exposure is thought to induce a GVHD flare effect and should be avoided by patients undergoing allogeneic HSCT [53, 83, 84]. In addition, transplanted recipients are also at increased risk for cutaneous malignancies, and appropriate clothing and high-protection, wide-spectrum sunscreen should be strongly recommended [83, 84].
6.1 Acute GVHD Treatment
6.1.1 First-Line Therapy
For grade I acute GVHD patients, management should include topical therapies in addition to optimizing systemic levels of calcineurin inhibitors (CNIs) [82]. Oral antihistamines and moisturizers to relieve itching and prevent skin breakdown may be necessary [82]. The recommended first-line therapy for isolated stages I and II cutaneous acute GVHD (overall grade I) is topical steroids of varying potencies [67]. Special concern should be taken in cases of long-term or high-potency topical steroids, particularly when applied on extensive BSAs or under occlusion, due to an increased risk of local and even systemic side effects. Topical CNIs are reserved for resistant cases and for sites where long-term use of topical steroids are contraindicated (e.g. intertriginous areas, face, and lips) [82, 85]. Skin-burning sensation is a common complaint with the use of CNIs and may initially be associated with topical steroids to improve tolerability. Systemic absorption with topical tacrolimus has been reported and special precaution is warranted in patients concurrently receiving the drug systemically [86].
In cases of grade II–IV acute GVHD, patients ought to have CNI levels optimized and are likely to benefit from other systemic immunosuppressive therapy [82]. Corticosteroids are the standard first-line agent, particularly methylprednisolone at an initial dose of 2 mg/kg/day intravenously [82]. Rashidi et al. carried out a systematic review and meta-analysis of randomized trials and concluded that standard treatment is better in terms of overall survival and response rates when compared with higher doses of systemic steroids or combined regimens of steroids and antithymocyte globulin (ATG), infliximab, an anti-IL-2 receptor antibody (daclizumab and BT563), a CD-5-specific immunotoxin, or mycophenolate mofetil [87]. An initial dose of 1 mg/kg/day for less severe forms (grade II GVHD) is associated with decreased toxicity without compromising therapy response or mortality rates, and should therefore be attempted [88]. Topical treatments follow grade I GVHD recommendations and may facilitate steroid dose tapering and withdrawal.
Most centers consider steroid-refractory patients as those who have progressive symptoms after 3 days or do not respond to 5–7 days of intravenous methylprednisolone 2 mg/kg in conjunction with CNIs [82]. Despite being the standard therapy, the response rate to systemic steroids in acute GVHD is limited to 30–40%, and steroid-refractory patients are at an increased risk of transplantation related-mortality [89, 90].
6.1.2 Second-Line Therapy
Since no superiority of one second-line agent over another has been proven to date, the choice is often made according to their availability, costs, physician’s experience, and side-effect profile.
ECP is an attractive, well-tolerated immunomodulatory therapy that usually spares generalized immunosuppression and has not been associated with increased infections or immunosuppression [91]. Treatment consists of exposure of the peripheral blood mononuclear cells collected by apheresis to the photosensitizing compound 8-methoxypsoralen, and ultraviolet (UV) A radiation, which cause cross-linking of DNA in cell nuclei, inducing apoptosis. Apoptotic cells are re-infused to the patient and are thought to promote immune tolerance by modulating cytokine production and inducing T-regulatory cells. However, recently, Denney et al. did not show a correlation between T-regulatory cells and clinical improvement post-ECP treatment, suggesting that other mechanisms may also play a role in the responsiveness to ECP [92]. Greinix et al. carried out a prospective phase II study of steroid-refractory grade II–IV acute GVHD patients treated with ECP and demonstrated complete responses in 82% of those with skin involvement [93]. In a phase II randomized trial comparing steroids alone with steroids plus ECP as first-line therapy for grade II–IV acute GVHD patients, Alousi et al. showed higher response rates in the ECP arm, in addition to a faster reduction in steroid dosage. They also demonstrated ECP to be more beneficial for cutaneous GVHD (72 vs. 57% response rates) compared with the involvement of other organs (47 vs. 43%) [94].
Mycophenolate mofetil exerts selective antiproliferative effects on lymphocytes by acting as a reversible inhibitor of inosine monophosphate dehydrogenase. Inagaki et al. reported a 79% complete response rate for children with steroid-refractory acute GVHD treated with mycophenolate mofetil [95], while Hattori et al. evidenced better responses to mycophenolate mofetil in patients with skin steroid-refractory acute GVHD compared with those with only liver, only gut, skin and liver, or skin and gut involvement [96].
TNF antagonists (infliximab, etanercept) are mainly used for acute steroid-refractory GVHD with gastrointestinal tract involvement [97]. However, in a small retrospective study, Nogueira et al. evidenced all cutaneous steroid-refractory acute GVHD patients responding to infliximab [98]. High rates of severe infections were demonstrated and were confirmed by further studies [97,98,99].
The use of antibodies against IL-2 receptors has been reported for steroid-refractory acute GVHD patients, with variable response rates in the literature. In a recent single-center study with 64 patients, Tao et al. demonstrated complete and partial responses in 58 and 25%, respectively, of steroid-refractory acute GVHD individuals treated with daclizumab [100].
ATG has been used as a second-line agent in acute GVHD, mainly due to its T-lymphocyte depletion activity. MacMillan et al. carried out a retrospective study of 79 patients treated with equine ATG for steroid-refractory acute GVHD and revealed better response rates for skin involvement, in addition to 20 and 34% complete and partial response rates, respectively [101]. Recently, Nishimoto et al. reported efficacy of very-low-dose rabbit ATG therapy for acute steroid-refractory GVHD in a single-center analysis [102].
When only the skin is involved, phototherapy modalities have been shown to be beneficial for steroid-refractory and steroid-dependent acute GVHD patients [103,104,105,106]. The immunomodulating effects of UV irradiation may allow for reduction of systemic immunosuppressive agents, consequently minimizing its side effects [103,104,105,106]. On the other hand, the increased risk for skin malignancies due to cyclosporin therapy, in addition to other immunosuppressive agents related to HSCT, should be considered.
Since UVA-1 and UVB-based phototherapy do not require oral psoralen and are less associated with skin cancer compared with psoralen plus ultraviolet A (PUVA), they should be prioritized for the treatment of acute GVHD. If available, UVA-1 therapy is the modality of choice in these cases as the target structure is the inflammatory infiltrate in the upper dermis. Schlaak et al. retrospectively evaluated 70 acute GVHD patients treated with UVA-1 therapy and evidenced 70 and 24.3% complete and partial response rates, respectively [103]. Furthermore, Feldstein et al. reported 57% complete response rates and 21% partial response rates in steroid-refractory and steroid-dependent cutaneous acute GVHD patients, respectively, treated with narrowband UVB [104]. Using flow cytometry, Ivama et al. demonstrated that NB-UVB therapy induces expansion of regulatory T cells in the peripheral blood of patients [105].
6.2 Chronic GVHD Treatment
6.2.1 Skin-Direct Therapy
For mild chronic GVHD, topical therapies to relieve symptoms are usually prioritized, mainly if a GVL effect is sought [107]. Systemic steroids alone may be considered if the organs involved, such as the liver and fascia, are not reached by topical agents, or in the setting of thrombocytopenia or direct progression from acute GVHD, which is related to worse outcome [60, 107].
For cutaneous chronic GVHD, several skin-direct therapies are available and may be indicated as monotherapy or as adjuvant treatment for more severe cases in order to allow faster tapering and withdrawal of systemic immunosuppression by improving local responses [81, 84, 107,108,109,110,111,112].
For intact skin, lubrication is helpful in relieving itching and preventing skin breakdown. Moisturizers containing urea and glycerol are very effective for skin hydration but may not be well-tolerated [84]. Despite the lack of randomized trials regarding the use of topical steroids, they are the mainstay of therapy for mild chronic GVHD with skin involvement [81, 84, 108]. From the neck down, mid-strength, potent, or very potent topical steroids should be prescribed twice daily for both lichenoid and sclerodermoid eruptions. Lower potency steroids are preferred for the face, axillae, and groin due to the risk of skin atrophy and striae [112]. The evidence for topical CNIs is supported by case reports and case series, and they are of special relevance in areas where high-potency topical steroids should be avoided, such as the face and intertriginous areas, and for steroid-dependent individuals [113]. Choi and Nghiem reported that tacrolimus ointment 0.1% was effective in treating itching and erythema in 13 of 18 patients (72%) with chronic GVHD [114]. Although topical CNIs as monotherapy are usually reserved for mild chronic GVHD, Ziemer et al. reported successful treatment with topical pimecrolimus alone in an infant with extensive chronic GVHD [115].
For nonintact skin, tissue cultures and other methods for ruling out infectious causes or other differential diagnosis should be performed when indicated. Wound care follows the same recommendations for patients without chronic GVHD [84].
When an extensive BSA is involved, ointments may not be feasible and phototherapy may be a good option, both as monotherapy or adjuvant treatment for cutaneous steroid-refractory and steroid-dependent patients [107,108,109,110,111,112]. Grundmann-Kollmann et al. reported on a steroid-refractory chronic sclerodermoid patient successfully treated with UVA-1 in combination with mycophenolate mofetil [110]. In a retrospective study of 16 cutaneous chronic GVHD patients treated with PUVA or UVB narrowband, Ballester-Sánchez et al. evidenced nine and seven patients achieving complete and partial response rates, respectively [111].
Regarding the oral mucosa, high-potency steroid preparations are the mainstay of topical therapy [84, 116]. Solutions are usually prioritized for diffuse involvement, whereas creams, ointments, and gels are preferred for localized lesions [84]. In a randomized, double-blind clinical trial, Noce et al. concluded that topical clobetasol was significantly more effective than topical dexamethasone for symptomatic oral chronic GVHD [116]. Intralesional injections of triamcinolone steroids may be helpful for ulcerative disease, and topical analgesia may be required when painful lesions interfere with patient food intake [84]. Topical tacrolimus is of particular relevance in chronic cases due to its steroid-sparing effects [84, 117]. Successful treatments with phototherapy were also reported in the literature for oral involvement [117, 118].
6.2.2 Systemic Therapy
For moderate and severe chronic GVHD, standard first-line therapy is 1 mg/kg day of prednisone alone or in combination with a CNI, which is particularly important in severe cases due to its steroid-sparing effect [107]. Steroid dose must be maintained for 2 weeks and then reduced to 1 mg/kg on alternate days over a period of 6–8 weeks. The dose may then be tapered by 10–20% monthly, or sustained for 2 or 3 months, depending on the clinical response [119]. Although corticosteroids are established as the mainstay therapy, Solomon et al., in a prospective phase II trial, showed 88% response rates for chronic GVHD patients initially treated with a combination of rituximab, mycophenolate mofetil, and either tacrolimus or sirolimus [120]. For sclerodermatous GVHD, or in cases with fascia involvement, physiotherapy must be considered as adjuvant therapy [108, 112].
To date, limited evidence exists supporting the use of second-line agents for steroid-refractory and steroid-dependent patients, and no consensus has been reached in the recommendation for the best option in these cases.
ECP has been widely used as second-line therapy for mucocutaneous chronic GVHD, particularly for sclerotic lesions, with high response rates in the literature (60–80%). The best evidence is supported by Flowers et al. in a multicenter, phase II, randomized study that demonstrated a significant steroid-sparing effect. No statistically significant improvement in the total skin score at week 12 was observed, likely due to the short duration of treatment; however, unblinded investigators evidenced significant higher response rates (both complete and partial) in the ECP arm compared with the control group [121]. In a crossover, randomized study, Greinix et al. showed progressive improvement in cutaneous and extracutaneous chronic GVHD after a 24-week course of ECP, supporting previous evidence suggesting that prolonged ECP may be necessary for optimal therapeutic effects [122].
Mycophenolate mofetil has also been successfully used for steroid-refractory chronic GVHD patients. Iida et al. evidenced that 69.1 and 75.9% of chronic GVHD patients treated with mycophenolate mofetil improved symptoms and reduced the dosage of other immunosuppressants, respectively [123], while Baudard et al. reported 9 of 13 cutaneous chronic GVHD patients responded to mycophenolate mofetil, with no difference between lichenoid or sclerodermatous lesions [124].
Imatinib mesylate is an inhibitor of several tyrosine kinases and has recently been reported to be used for the treatment of steroid-refractory sclerotic chronic GVHD due to its inhibitory activity against platelet-derived growth factor. Baird et al. performed an open-label, pilot, phase II trial of imatinib in children and adults with steroid-refractory sclerotic GVHD and showed that 79% of patients experienced improvement in their range of motion [125].
Rituximab is an anti-CD20 monoclonal antibody that has also shown beneficial effects in the treatment of steroid-refractory sclerotic cutaneous chronic GVHD since B cells have been implicated in its pathophysiology. In 2006, Cutler et al. designed a phase I/II study with rituximab for steroid-refractory chronic GVHD patients with cutaneous and musculoskeletal manifestations, and reported a 70% response rate [126]. In a prospective, multicenter, randomized, phase II, crossover trial, Arai et al. recently compared imatinib and rituximab for cutaneous sclerotic GVHD, and concluded that both agents had similar results and low clinical response rates after 6 months (26 and 27%, respectively) [127].
Rapamycin is an inhibitor of the mammalian target of rapamycin kinase that acts as a potent immunosuppressive drug which increases regulatory T cells. Couriel et al. carried out a phase II trial of sirolimus in combination with tacrolimus for steroid-refractory chronic GVHD patients and demonstrated a 65% response rate for cutaneous involvement [128].
Low doses of methotrexate exert an anti-inflammatory effect and it has been shown to be efficacious for chronic GVHD, particularly for skin or sole organ involvement. Wang et al. reported a 90% response rate for cutaneous chronic GVHD patients treated with low-dose methotrexate as first-line therapy in combination with other immunosuppressive agents [129].
6.3 Cutting-Edge Therapy
Recent progress in our understanding of GVHD pathophysiology has led to the identification of new therapeutic targets with the potential to significantly impact disease treatment outcomes. Possible synergistic effects of combining some of these drugs with ECP are presently being explored but are not documented in any extensive manner.
Ibrutinib acts as an irreversible inhibitor of both Bruton’s tyrosine kinase and IL-2-inducible kinase (ITK), enzymes responsible for the phosphorylation and activation of downstream effectors in the B- and T-cell receptor signaling pathways, respectively. The drug is usually used for the treatment of relapsed chronic lymphocytic leukemia (CLL) in the post-HSCT setting [130]. Ryan et al. reported that ibrutinib is related to, and increased, GVL effect by enhancing donor Th1 cell-mediated effects, without causing GVHD due to depletion of pre-germinal B and Th2 cells [130]. These authors evidenced that none of the 27 CLL patients treated with ibrutinib developed GVHD, and one patient with mucocutaneous chronic GVHD improved symptoms during therapy [130]. Schutt et al. showed that ibrutinib was effective in preventing chronic GVHD, with minimal toxicity in mouse models [131], while Dubovsky et al. demonstrated ibrutinib to be remarkably effective in treating both sclerodermatous and nonsclerodermatous features in murine models [132].
Janus kinase (JAK) inhibitors represent a class of immunosuppressive agents, including ruxolitinib (JAK 1/2 inhibitor) and tofacitinib (JAK 3 inhibitor), that act by inhibiting the signal transduction and activation of transcription (STAT) pathway, which is essential for the downstream of growth factors and inflammatory cytokines. Spoerl et al. demonstrated that ruxolitinib could potently reduce proinflammatory cytokine production, T-cell expansion, and differentiation into Th17 subsets, in addition to increasing FoxP3 regulatory cells in mice models [133]. These authors also reported a potent reduction of GVHD symptoms and serum cytokines in six patients with acute and chronic steroid-refractory GVHD [133]. Maffini et al. described a patient with steroid-refractory grade IV acute GVHD treated with ruxolitinib with complete resolution of symptoms [134]. Additionally, Zeiser et al. carried out a retrospective multicenter survey with 95 GVHD steroid-refractory patients treated with ruxolitinib, and showed overall and complete response rates of 81.5 and 46.3%, respectively, for acute GVHD, and 85.4 and 7.3%, respectively, for chronic GVHD [135]. Okiyama et al. suggested that tofacitinib may be a therapeutic option for mucocutaneous GVHD by demonstrating that the drug could reduce expansion and activation of T CD8+ cells in addition to inhibition of the expression of IFNγ-inducible chemoattractants by keratinocytes in murine models [136].
Histone deacetylase inhibitors act by enhancing the expression of indoleamine 2,3 dioxygenase in a STAT3-dependent manner, which consequently suppress the function of APCs, T-regulatory cells, and natural killer cells. Choi et al. conducted a single-arm, phase I/II study and demonstrated a lower incidence of grade II–IV acute GVHD 100 days after HSCT (22%), compared with the literature, by including vorinostat as a prophylactic agent for patients undergoing related donor-reduced intensity conditioning regimen allogeneic HSCT [137].
7 Conclusion
The management of both chronic and acute GVHD is indeed a complex task requiring a high degree of interdisciplinary coordination. Since the cutaneous manifestations of GVHD are at the forefront of its presentation the treating dermatologist must be thoroughly familiar with these manifestations and the most appropriate available therapeutic approaches.
References
Atkinson K, Horowitz MM, Gale RP, van Bekkum DW, Gluckman E, Good RA, et al. Risk factors for chronic graft-versus-host disease after HLA-identical sibling bone marrow transplantation. Blood. 1990;75(12):2459–64.
Jagasia M, Arora M, Flowers ME, Chao NJ, McCarthy PL, Cutler CS, et al. Risk factors for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119(1):296–307.
Molaro GL, De Angelis V. Graft versus host disease after transfusion of blood and its products [in Italian]. Riv Emoter Immunoematol. 1984;31(2):107–23.
Murali AR, Chandra S, Stewart Z, Blazar BR, Farooq U, Ince MN, et al. Graft versus host disease after liver transplantation in adults: a case series, review of literature, and an approach to management. Transplantation. 2016;100(12):2661–70.
Fidler C, Klumpp T, Mangan K, Martin M, Sharma M, Emmons R, et al. Spontaneous graft versus host disease occurring in a patient with multiple myeloma after autologous stem cell transplant. Am J Hematol. 2012;87(2):219–21.
DiRienzo CG, Murphy GF, Jones SC, Korngold R, Friedman TM. T-cell receptor Valpha spectratype analysis of a CD4- mediated T-cell response against minor histocompatibility antigens involved in severe graft-versus-host disease. Biol Blood Marrow Transplant. 2006;12(8):818–27.
Storb R, Prentice RL, Sullivan KM, Shulman HM, Deeg HJ, Doney KC, et al. Predictive factors in chronic graft-versus-host disease in patients with aplastic anemia treated by marrow transplantation from HLA-identical siblings. Ann Intern Med. 1983;98(4):461–6.
Cutler C, Giri S, Jeyapalan S, Paniagua D, Viswanathan A, Antin JH. Acute and chronic graft-versus-host disease after allogeneic peripheral-blood stem-cell and bone marrow transplantation: a meta-analysis. J Clin Oncol. 2001;19(16):3685–91.
James E, Chai JG, Dewchand H, Macchiarulo E, Dazzi F, Simpson E. Multiparity induces priming to male-specific minor histocompatibility antigen, HY, in mice and humans. Blood. 2003;102(1):388–93.
Atkinson K, Farrell C, Chapman G, Downs K, Penny R, Biggs J. Female marrow donors increases the risk of acute graft-versus-host disease: effect of donor age and parity and analysis of cell subpopulations in the donor marrow inoculums. Br J Haematol. 1986;63(2):231–9.
Eibl B, Schwaighofer H, Nachbaur D, Marth C, Gächter A, Knapp R, et al. Evidence for a graft-versus-tumor effect in a patient treated with marrow ablative chemotherapy and allogeneic bone marrow transplantation for breast cancer. Blood. 1996;88(4):1501–8.
Mielcarek M, Martin PJ, Leisenring W, Flowers ME, Maloney DG, Sandmaier BM, et al. Graft-versus-host disease after nonmyeloablative versus conventional hematopoietic stem cell transplantation. Blood. 2003;102(2):756–62.
Filipovich AH, Weisdorf D, Pavletic S, Socie G, Wingard JR, Lee SJ, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant. 2005;11(12):945–56.
Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part I. Pathogenesis and clinical manifestations of graft-versus-host disease. J Am Acad Dermatol. 2012;66(4):515.e1–18 (quiz 533-4).
Jagasia MH, Greinix HT, Arora M, Williams KM, Wolff D, Cowen EW, et al. National Institutes of Health Consensus Development Project on criteria for clinical trials in chronic graft-versus-host disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant. 2015;21(3):389–401.e1.
Zeiser R, Penack O, Holler E, Idzko M. Danger signals activating innate immunity in graft-versus-host disease. J Mol Med (Berl). 2011;89(9):833–45.
MacDonald KP, Shlomchik WD, Reddy P. Biology of graft-versus-host responses: recent insights. Biol Blood Marrow Transplant. 2013;19(1 Suppl):S10–4.
Yu Y, Wang D, Liu C, Kaosaard K, Semple K, Anasetti C, et al. Prevention of GVHD while sparing GVL effect by targeting Th1 and Th17 transcription factor T-bet and RORγt in mice. Blood. 2011;118(18):5011–20.
Sung AD, Chao NJ. Acute graft-versus-host disease: are we close to bringing the bench to the bedside? Best Pract Res Clin Haematol. 2013;26(3):285–92.
Holländer GA, Widmer B, Burakoff SJ. Loss of normal thymic repertoire selection and persistence of autoreactive T cells in graft vs host disease. J Immunol. 1994;152(4):1609–17.
Beres AJ, Haribhai D, Chadwick AC, Gonyo PJ, Williams CB, Drobyski WR. CD8+ Foxp3+ regulatory T cells are induced during graft-versus-host disease and mitigate disease severity. J Immunol. 2012;189(1):464–74.
Trzonkowsk P, Bieniaszewska M, Dobyszuk A, Krzystyniak A, Marek N, Myśliwska J, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127− T regulatory cells. Clin Immunol. 2009;133(1):22–6.
Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117(3):1061–70.
Min CK. The pathophysiology of chronic graft-versus-host disease: the unveiling of an enigma. Korean J Hematol. 2011;46(2):80–7.
Wu T, Young JS, Johnston H, Ni X, Deng R, Racine J, et al. Thymic damage, impaired negative selection and development of chronic graft-versus-host disease caused by donor CD4+ and CD8+ T cells. J Immunol. 2013;191(1):488–99.
Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004;4(8):583–94.
Jakubzick C, Kunkel SL, Puri RK, Hogaboam CM. Therapeutic targeting of IL-4 and IL-13-responsive cells in pulmonary fibrosis. Immunol Res. 2004;30(3):339–49.
Brüggen MC, Klein I, Greinix H, Bauer W, Kuzmina Z, Rabitsch W, et al. Diverse T-cell responses characterize the different manifestations of cutaneous graft-versus-host disease. Blood. 2014;123(2):290–9.
Zorn E, Kim HT, Lee SJ, Floyd BH, Litsa D, Arumugarajah S, et al. Reduced frequency of FOXP3+ CD4+CD25+ regulatory T cells in patients with chronic graft-versus-host disease. Blood. 2005;106(8):2903–11.
Sarantopoulos S, Stevenson KE, Kim HT, Bhuiya NS, Cutler CS, Soiffer RJ, et al. High levels of B-cell activating factor in patients with active chronic graft-versus-host disease. Clin Cancer Res. 2007;13(20):6107–14.
MacDonald KP, Hil GR, Blazar BR. Chronic graft-versus-host disease: biological insights from preclinical and clinical studies. Blood. 2017;129(1):13–21.
Clancy RM, Buyon JP. Clearance of apoptotic cells: TGF-beta in the balance between inflammation and fibrosis. J Leukoc Biol. 2003;74(6):959–60.
Clancy J Jr, Tonder O, Boettcher CE. The effect of neonatal rat graft-vs-host disease (GVHD) on Fc receptor lymphocytes. J Immunol. 1976;116(1):210–7.
Mathias C, Mick R, Grupp S, Duffy K, Harris F, Laport G, et al. Soluble interleukin-2 receptor concentration as a biochemical indicator for acute graft-versus-host disease after allogeneic bone marrow transplantation. J Hematother Stem Cell Res. 2000;9(3):393–400.
Holler E, Kolb HJ, Moller A, Kempeni J, Liesenfeld S, Pechumer H, et al. Increased serum levels of tumor necrosis factor alpha precede major complications of bone marrow transplantation. Blood. 1990;75(4):1011–6.
Paczesny S, Braun TM, Levine JE, Hogan J, Crawford J, Coffing B, et al. Elafin is a biomarker of graft versus host disease of the skin. Sci Transl Med. 2010;2(13):1–19.
Ahmed SS, Wang XN, Norden J, Pearce K, El-Gezawy E, Atarod S, et al. Identification and validation of biomarkers associated with acute and chronic graft versus host disease. Bone Marrow Transplant. 2015;50:1563–71.
Levine JE, Braun TM, Harris AC, Holler E, Taylor A, Miller H, et al. A prognostic score for acute graft-versus-host disease based on biomarkers: a multicentre study. Lancet Haematol. 2015;2(1):21–9.
Hartwell MJ, Özbek U, Holler E, Renteria AS, Major-Monfried H, Reddy P, et al. An early-biomarker algorithm predicts lethal graft-versus-host disease and survival. JCI Insight. 2017;2(3):e89798.
Pidala J, Sigdel TK, Wang A, Hsieh S, Inamoto Y, Martin PJ, et al. A combined biomarker and clinical panel for chronic graft versus host disease diagnosis. J Pathol Clin Res. 2017;3:3–16.
Byun HJ, Yang JI, Kim BK, Cho KH. Clinical differentiation of acute cutaneous graft-versus-host disease from drug hypersensitivity reactions. J Am Acad Dermatol. 2011;65(4):726–32.
Saliba RM, de Lima M, Giralt S, Andersson B, Khouri IF, Hosing C, et al. Hyperacute GVHD: risk factors, outcomes, and clinical implications. Blood. 2007;109(7):2751–8.
Friedman KJ, LeBoit PE, Farmer ER. Acute follicular graft-vs-host reaction. A distinct clinicopathologic presentation. Arch Dermatol. 1988;124(5):688–91.
Surjana D, Robertson I, Kennedy G, James D, Weedon D. Acute cutaneous graft-versus-host disease resembling type II (atypical adult) pityriasis rubra pilaris. Australas J Dermatol. 2015;56(1):e21–3.
Huang J, Pol-Rodriguez M, Silvers D, Garzon MC. Acquired ichthyosis as a manifestation of acute cutaneous graft-versus-host disease. Pediatr Dermatol. 2007;24(1):49–52.
Matsushita T, Hasegawa M, Shirasaki F, Fujimoto M, Yamazaki H, Sato S, et al. A case of acute cutaneous graft-versus-host disease mimicking psoriasis vulgaris. Dermatology. 2008;216(1):64–7.
Peñas PF, Zaman S. Many faces of graft-versus-host disease. Australas J Dermatol. 2010;51(1):1–10.
Hu SW, Cotliar J. Acute graft-versus-host disease following hematopoietic stem-cell transplantation. Dermatol Ther. 2011;24(4):411–23.
Kaminska-Winciorek G, Czerw T, Kruzel T, Giebel S. Dermoscopic follow-up of the skin toward acute graft-versus-host-disease in patients after allogeneic hematopoietic stem cell transplantation. Biomed Res Int. 2016; Article ID 4535717. doi:10.1155/2016/4535717.
Ruiz-Genao DP, GF-Villalta MJ, Peñas PF, Fraga J, García-Díez A, Fernández-Herrera J. Pustular acral erythema in a patient with acute graft-versus-host disease. J Eur Acad Dermatol Venereol. 2003;17(5):550–3.
Ion D, Stevenson K, Woo SB, Ho VT, Soiffer R, Antin JH, et al. Characterization of oral involvement in acute graft-versus-host disease. Biol Blood Marrow Transplant. 2014;20(11):1717–21.
Saito T, Shinagawa K, Takenaka K, Matsuo K, Yoshino T, Kiura K, et al. Ocular manifestation of acute graft-versus-host disease after allogeneic peripheral blood stem cell transplantation. Int J Hematol. 2002;75(3):332–4.
Vassalo C, Brazzelli V, Zecca M, Locatelli F, Alessandrino PE, Borroni G. Isomorphic cutaneous graft-versus-host disease reaction after ultraviolet exposure: clinical, histological and direct immunofluorescence studies of four allo-transplanted patients. J Eur Acad Dermatol Venereol. 2009;23(8):913–8.
Hillen U, Häusermann P, Massi D, Janin A, Wolff D, Lawitschka A, et al. Consensus on performing skin biopsies, laboratory workup, evaluation of tissue samples and reporting of the results in patients with suspected cutaneous graft-versus-host disease. J Eur Acad Dermatol Venereol. 2015;29(5):948–54.
Lerner KG, Kao GF, Storb R, Buckner CD, Clift RA, Thomas ED. Histopathology of graft-vs-host reaction (GvHR) in human recipients of marrow from HLA-matched sibling donors. Transplant Proc. 1974;6(4):367–71.
Weaver J, Bergfeld WF. Quantitative analysis of eosinophils in acute graft-versus-host disease compared with drug hypersensitivity reactions. Am J Dermatopathol. 2010;32(1):31–4.
Vassallo C, Brazzelli V, Alessandrino PE, Varettoni M, Ardigò M, Lazzarino M, et al. Normal-looking skin in oncohaematological patients after allogenic boné marrow transplantation is not normal. Br J Dermatol. 2004;151(3):579–86.
Przepiorka D, Weisdorf D, Martin P, Klingemann HG, Beatty P, Hows J, et al. 1994 consensus conference on acute GVHD grading. Bone Marrow Transplant. 1995;15(6):825–8.
Patriarca F, Skert C, Sperotto A, Zaja F, Falleti E, Mestroni R, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34(3):389–96.
Martires KJ, Baird K, Steinberg SM, Grkovic L, Joe GO, Williams KM, et al. Sclerotic-type chronic GVHD of the skin: clinical risk factors, laboratory markers, and burden of disease. Blood. 2011;118(15):4250–7.
Spies-Weisshart B, Schilling K, Böhmer F, Hochhaus A, Sayer HG, Scholl S. Lack of association of platelet-derived growth factor (PDGF) receptor autoantibodies and severity of chronic graft-versus-host disease (GvHD). J Cancer Res Clin Oncol. 2013;139(8):1397–404.
Kuzmina Z, Gounden V, Curtis L, Avila D, Rnp TT, Baruffaldi J, et al. Clinical significance of autoantibodies in a large cohort of patients with chronic graft-versus-host disease defined by NIH criteria. Am J Hematol. 2015;90(2):114–9.
Aractingi S, Chosidow O. Cutaneous graft-versus-host disease. Arch Dermatol. 1998;134(5):602–12.
Chosidow O, Bagot M, Vernant JP, Roujeau JC, Cordonnier C, Kuentz M. Sclerodermatous chronic graft-versus-host disease. Analysis of seven cases. J Am Acad Dermatol. 1992;26(1):49–55.
Cornejo CM, Kim EJ, Rosenbach M, Micheletti RG. Atypical manifestations of graft-versus-host disease. J Am Acad Dermatol. 2015;72(4):690–5.
Chasset F, Le Buanec H, Sicre de Fontbrune F, de Masson A, Rivet J, Bergeron A, et al. Evidence of Th1, Th17 and Tc17 cells in psoriasiform chronic graft-versus-host disease. Exp Dermatol. 2016;25(1):64–5.
Jang S, Kim IS, Youn SW. Chronic graft-versus-host disease mimicking psoriasis in a patient with hemophagocytic lymphohistiocytosis. Ann Dermatol. 2016;28(1):90–3.
Čeović R, Desnica L, Pulanić D, Serventi Seiwerth R, Ilić I, Grce M, et al. High frequency of cutaneous manifestations including vitiligo and alopecia areata in a prospective cohort of patients with chronic graft-vs-host disease. Croat Med J. 2016;57(3):229–38.
Brassat S, Fleury J, Camus M, Monégier du Sorbier C, Guillet G. Epidermolysis bullosa acquisita and graft-versus-host disease. Ann Dermatol Venereol. 2014;141(5):369–73.
Hu SW, Myskowski PL, Papadopoulos EB, Busam KJ. Chronic cutaneous graft-versus-host disease simulating hypertrophic lupus erythematosus-a case report of a new morphologic variant of graft-versus-host disease. Am J Dermatopathol. 2012;34(6):e81–3.
Arin MJ, Scheid C, HübeL K, Krieg T, Groth W, Haerrmann G. Chronic graft-versus-host disease with skin signs suggestive of dermatomyositis. Clin Exp Dermatol. 2006;31(1):141–3.
Peñas PF, Jones-Caballero M, Aragüés M, Fernández-Herrera J, Fraga J, García-Díez A. Sclerodermatous graft-vs-host disease: clinical and pathological study of 17 patients. Arch Dermatol. 2002;138(7):924–34.
Skert C, Patriarca F, Sperotto A, Cerno M, Filì C, Zaja F, et al. Sclerodermatous chronic graft-versus-host disease after allogeneic hematopoietic stem cell transplantation: incidence, predictors and outcome. Haematologica. 2006;91(2):258–61.
Kim SJ, Choi JM, Kim JE, Cho BK, Kim DW, Park HJ. Clinicopathologic characteristics of cutaneous graft-versus-host disease: a retrospective study in Korean patients. Int J Dermatol. 2010;49(12):1386–92.
Manalo IF, Miller IA, Davies LS. More immune dysregulation: sarcoidosis and chronic graft-versus-host disease after allogeneic stem cell transplant. JAAD Case Rep. 2016;2(2):138–40.
Kinsella FA, Amel Kashipaz MR, Scarisbrick J, Malladi R. Donor-derived mycosis fungoides following reduced intensity haematopoietic stem cell transplantation from a matched unrelated donor. BMJ Case Rep. Epub 10 Jan 2017. doi:10.1136/bcr-2016-216331.
Kaffenberger BH, Zuo RC, Gru A, Plotner AN, Sweeney SA, Devine SM, et al. Graft-versus-host disease-associated angiomatosis: a clinicopathologically distinct entity. J Am Acad Dermatol. 2014;71(4):745–53.
Chu GY, Lin HL, Chen GS, Wu CY. Eosinophilic fasciitis following allogeneic bone marrow transplantation in a patient with acute myeloid leukemia. Acta Derm Venereol. 2014;94(2):221–2.
Shulman HM, Cardona DM, Greenson JK, Hingorani S, Horn T, Huber E, et al. NIH consensus development project on criteria for clinical trials in chronic graft-versus-host disease: II. The 2014 Pathology Working Group report. Biol Blood Marrow Transplant. 2015;21(4):589–603.
Greinix HT, Pohlreich D, Maalouf J, Soukup P, Soukup P, Supper V, et al. A single-center pilot validation study of a new chronic GVHD skin scoring system. Biol Blood Marrow Transplant. 2007;13(6):715–23.
Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part II. Management of cutaneous graft-versus-host disease. J Am Acad Dermatol. 2012;66(4):535.e1–16 (quiz 551-2).
Dignan FL, Clark A, Amrolia P, Cornish J, Jackson G, Mahendra P, et al. Diagnosis and management of acute graft-versus-host disease. Br J Haematol. 2012;158(1):30–45.
Couriel D, Carpenter PA, Cutler C, Bolaños-Meade J, Treister NS, Gea-Banacloche J, et al. Ancillary therapy and supportive care of chronic graft-versus-host disease: national institutes of health consensus development project on criteria for clinical trials in chronic Graft-versus-host disease: V. Ancillary Therapy and Supportive Care Working Group report. Biol Blood Marrow Transplant. 2006;12(4):375–96.
Carpenter PA, Kitko CL, Elad S, Flowers ME, Gea-Banacloche JC, Halter JP, et al. National Institutes of Health Consensus Development Project on criteria for clinical trials in chronic graft-versus-host disease: V. The 2014 Ancillary Therapy and Supportive Care Working Group report. Biol Blood Marrow Transplant. 2015;21(7):1167–87.
Kunitomi A, Lida H, Kamiya Y, Hayashi M, Sao H. Successful treatment using tacrolimus ointment for cutaneous graft-versus-host disease. Int J Hematol. 2008;88(4):465–7.
Olson KA, West K, McCarthy PL. Toxic tacrolimus levels after application of topical tacrolimus and use of occlusive dressing in two bone marrow transplant recipients with cutaneous graft-versus-host disease. Pharmacotherapy. 2014;34(6):e60–4.
Rashidi A, DiPersio JF, Sandmaier BM, Colditz GA, Weisdorf DJ. Steroids versus steroids plus additional agent in frontline treatment of acute graft-versus-host disease: a systematic review and meta-analysis of randomized trials. Biol Blood Marrow Transplant. 2016;22(6):1133–7.
Mielcarek M, Storer BE, Boeckh M, Carpenter PA, McDonald GB, Deeg HJ, et al. Initial therapy of acute graft-versus-host disease with low-dose prednisone does not compromise patient outcomes. Blood. 2009;113(13):2888–94.
Weisdorf D, Haake R, Blazar B, Miller W, McGlave P, Ramsay N, et al. Treatment of moderate/severe acute graft-versus-host disease after allogeneic bone marrow transplantation: an analysis of clinical risk features and outcome. Blood. 1990;75(4):1024–30.
MacMillan ML, Weisdorf DJ, Wagner JE, DeFor TE, Burns LJ, Ramsay NK, et al. Response of 443 patients to steroid as primary therapy for acute graft-versus-host disease: comparison of grading systems. Biol Blood Marrow Transplant. 2002;8(7):387–94.
Knobler R, Berlin G, Calzavara-Pinton P, Greinix H, Jaksch P, Laroche L, et al. Guidelines on the use of extracorporeal photopheresis. J Eur Acad Dermatol Venereol. 2014;28(Suppl 1):1–37.
Denney HA, Whittle RJ, Lai J, Jacques RM, Taylor PC. Regulatory T cells in chronic graft-versus-host disease after extracorporeal photopheresis: correlation with skin and global organ responses, and ability to taper steroids. Transplantation. 2017;101(1):204–11.
Greinix HT, Knobler RM, Worel N, Schneider B, Schneeberger A, Hoecker P, et al. The effect of intensified extracorporeal photochemotherapy on long-term survival in patients with severe acute graft-versus-host disease. Haematologica. 2006;91(3):405–8.
Alousi AM, Basset R, Chen J, Overman BJ, Hosing CM, Popat UR, et al. A Bayesian, phase ii randomized trial of extracorporeal photopheresis (ECP) plus steroids versus steroids-alone in patients with newly diagnosed acute graft vs. host disease (GVHD): the addition of ECP improves GVHD response and the ability to taper steroids. Blood. 2015;126:854.
Inagaki J, Kodama Y, Fukano R, Noquchi M, Okamura J. Mycophenolate mofetil for treatment of steroid-refractory acute graft versus host disease after pediatric hematopoietic stem cell transplantation. Pediatr Transplant. 2015;19(6):652–8.
Hattori K, Doki N, Kurosawa S, Hino Y, Yamamoto K, Sakaguchi M, et al. Mycophelonate mofetil is effective only for involved skin in the treatment for steroid-refractory acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Ann Hematol. 2017;96(2):319–21.
Yang J, Cheuk DK, Ha SY, Chiang AK, Lee TL, Ho MH, et al. Infliximab for steroid refractory or dependent gastrointestinal acute graft-versus-host disease in children after allogeneic hematopoietic stem cell transplantation. Pediatr Transplant. 2012;16(7):771–8.
Nogueira MC, Azevedo AM, Pereira SC, Ferreira JL, Lerner D, Lobo AM, et al. Anti-tumor necrosis factor-a for the treatment of steroid-refractory acute graft-versus-host disease. Braz J Med Biol Res. 2007;40(12):1623–9.
Motiló C, Ferrà C, Lopez L, Morgades M, Battle M, Ribera JM. Salvage treatment with infliximab of steroid-resistant graft-versus-host disease in allogeneic transplanted patients. Med Clin (Barc). 2011;137(3):115–8.
Tao T, Ma X, Yang J, Zou JY, Ji SM, Tan YS, et al. Humanized anti-CD-25 monoclonal antibody treatment of steroid-refractory acute graft-versus-host disease: a Chinese single-center experience in a group of 64 patients. Blood Cancer J. 2015;5(4):e308.
MacMillan ML, Weisdorf DJ, Davies SM, DeFor TE, Burns LJ, Ramsay NK, et al. Early antithymocyte globulin therapy improves survival in patients with steroid-resistant acute graft-versus-host disease. Biol Blood Marrow Transplant. 2002;8(1):40–6.
Nishimoto M, Nakamae H, Koh H, Nakamae M, Hirose A, Hayashi Y, et al. Response-guided therapy for steroid-refractory acute GVHD starting with very-low-dose antithymocyte globulin. Exp Hematol. 2015;43(3):177–9.
Schlaak M, Schwind S, Wetzig T, Maschke J, Treudler R, Basara N, et al. UVA (UVA-1) therapy for the treatment of acute GVHD of the skin. Bone Marrow Transplant. 2010;45(12):1741–8.
Feldstein JV, Bolaños-Meade J, Anders VL, Abuav R. Narrowband ultraviolet B phototherapy for the treatment of steroid-refractory and steroid-dependent acute graft-versus-host disease of the skin. J Am Acad Dermatol. 2011;65(4):733–8.
Ivama S, Murase K, Sato T, Hashimoto A, Tatekoshi A, Horiguchi H, et al. Narrowband ultraviolet B phototherapy ameliorates acute graft-versus-host disease by a mechanism involving in vivo expansion of CD4+CD25+Foxp3+ regulatory T cells. Int J Hematol. 2014;99(4):471–6.
Garbutcheon-Singh KB, Fernández-Peñas P. Phototherapy for the treatment of cutaneous graft versus host disease. Australas J Dermatol. 2015;56(2):93–9.
Wolff D, Gerbitz A, Ayuk F, Kiani A, Hildebrandt GC, Vogelsang GB, et al. Consensus conference on clinical practice in chronic graft-versus-host disease (GVHD): fist-line and topical treatment of chronic GVHD. Biol Blood Marrow Transplant. 2010;16(12):1611–28.
Marks C, Stadler M, Häusermann P, Wolff D, Buchholz S, Stary G, et al. German–Austria–Swiss Consensus Conference on clinical practice in chronic graft-versus-host disease (GVHD): guidance for supportive therapy of chronic cutaneous and musculoskeletal GVHD. Br J Dermatol. 2011;165(1):18–29.
Lazzeri L, Tripo L, Pescitelli L, Ricceri F, Prignano F. A pediatric case of sclerodermatous graft-versus-host disease responsive to ultraviolet al phototherapy. Pediatr Dermatol. 2016;33(9):e99–102.
Grundmann-Kollmann M, Behrens S, Gruss C, Gottlöber P, Peter RU, Kerscher M. Chronic sclerodermic graft-versus-host disease refractory to immunosuppressive treatment responds to UVA-1 phototherapy. J Am Acad Dermatol. 2000;42(1 Pt 1):134–6.
Ballester-Sánchez R, Navarro-Mira MA, de Unamuno-Bustos B, Pujol-Marco C, Sanz-Caballer J, Botella-Estrada R. The role of phototherapy in cutaneous chronic graft-vs-host disease: a retrospective study and review of the literature. Actas Dermosifiliogr. 2015;106(8):651–7.
Dignan FL, Scarisbrick JJ, Cornish J, Clark A, Amrolia P, Jackson G, et al. Organ-specific management and supportive care in chronic graft-versus-host disease. Br J Haematol. 2012;158(1):62–78.
Elad S, Or R, Resnick I, Shapira MY. Topical tacrolimus—a novel treatment alternative for cutaneous chronic graft-versus-host disease. Transpl Int. 2003;16(9):665–70.
Choi CJ, Nghiem P. Tacrolimus ointment in the treatment of chronic cutaneous graft-vs-host disease: a case series of 18 patients. Arch Dermatol. 2001;137(9):1202–6.
Ziemer M, Gruhn B, Thiele JJ, Elsner P. Treatment of extensive chronic cutaneous graft-versus-host disease in a infant with topical pimecrolimus. J Am Acad Dermatol. 2004;50(6):946–8.
Noce CW, Gomes A, Shcaira V, Corrêa ME, Moreira MC, Silva Júnior A, et al. Randomized double-blind clinical trial comparing clobetasol and dexamethasone for the topical treatment of symptomatic oral chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2014;20(8):1163–8.
Elad S, Levitt M, MYS. Chronic graft-versus-host-disease involving the oral mucosa: clinical presentation and treatment [in Hebrew]. Refuat Hapeh Vehashinayim (1993). 2008;25(4):19–27, 72.
Treister N, Li S, Lerman MA, Lee S, Soiffer R. Narrow-band UVB phototherapy for management of oral chronic graft-versus-host disease. Photodermatol Photoimmunol Photomed. 2015;31(2):75–82.
Flowers ME, Martin PJ. How we treat chronic graft-versus-host disease. Blood. 2015;125(4):606–15.
Solomon SR, Sizemore CA, Ridgeway M, Zhang X, Smith J, Brown S, et al. Corticosteroid-free primary treatment of chronic extensive graft-versus-host disease incorporating rituximab. Biol Blood Marrow Transplant. 2015;21(9):1576–82.
Flowers ME, Apperley JF, van Besien K, Elmaagacli A, Grigg A, Reddy V, et al. A multicenter prospective phase 2 randomized study of extracorporeal photopheresis for treatment of chronic graft-versus-host disease. Blood. 2008;112(7):2667–74.
Greinix HT, van Besien K, Elmaagacl AH, Hillen U, Grigg A, Knobler R, et al. Progressive improvement in cutaneous and extracutaneous chronic graft-versus-host disease after a 24-week course of extracorporeal photopheresis—results of a crossover randomized study. Biol Blood Marrow Transplant. 2011;17(12):1775–82.
Iida M, Fukuda T, Uchida N, Murata M, Aotsuka N, Minagawa K, et al. Mycophenolate mofetil use after unrelated hematopoietic stem cell transplantation for prophylaxis and treatment of graft-vs.-host disease in adult patients in Japan. Clin Transplant. 2014;28(9):980–9.
Baudard M, Vincent A, Moreau P, Kergueris MF, Harousseau JL, Milpied N. Mycophenolate mofetil for the treatment of acute and chronic GVHD is effective and well tolerates but induces a high risk of infectious complications: a series of 21 BM or PBSC transplant patients. Bone Marrow Transplant. 2002;30(5):287–95.
Baird K, Comis LE, Joe GO, Steinberg SM, Hakim FT, Rose JJ, et al. Imatinib mesylate for the treatment of steroid-refractory sclerotic-type cutaneous chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2015;21(6):1083–90.
Cutler C, Miklos D, Kim HT, Treister N, Woo SB, Bienfang D, et al. Rituximab for steroid-refractory chronic graft-versus-host disease. Blood. 2006;108(2):756–62.
Arai S, Pidala J, Pusic I, Chai X, Jaglowski S, Khera N, et al. A randomized phase II crossover study of imatinib or rituximab for cutaneous sclerosis after hematopoietic cell transplantation. Clin Cancer Res. 2016;22(2):319–27.
Couriel DR, Saliba R, Escalón MP, Hsu Y, Ghosh S, Ippoliti C, et al. Sirolimus in combination with tacrolimus and corticosteroids for the treatment of resistant chronic graft-versus-host disease. Br J Haematol. 2005;130(3):409–17.
Wang Y, Xu LP, Liu DH, Chen H, Chen YH, Han W, et al. First-line therapy for chronic graft-versus-host disease that includes low-dose methotrexate is associated with a high response rate. Biol Blood Marrow Transplant. 2009;15(4):505–11.
Ryan CE, Sahaf B, Logan AC, O’Brien S, Byrd JC, Hillmen P, et al. Ibrutinib efficacy and tolerability in patients with relapsed chronic lymphocytic leukemia following allogeneic HCT. Blood. 2016;128(25):2899–908.
Schutt SD, Fu J, Nguyen H, Bastian D, Heinrichs J, Wu Y, et al. Inhibition of BTK and ITK with ibrutinib is effective in the prevention of chronic graft-versus-host disease in mice. PLoS One. 2015;10(9):e0137641.
Dubovsky JA, Flynn R, Du J, Harrington BK, Zhong Y, Kaffenberger B, et al. Ibrutinib treatment ameliorates murine chronic graft-versus-host disease. J Clin Invest. 2014;124(11):4867–76.
Spoerl S, Mathew NR, Bscheider M, Schmitt-Graeff A, Chen S, Mueller T, et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood. 2014;123(24):3832–42.
Maffini E, Giaccone L, Festuccia M, Brunello L, Buondonno I, Ferrero D, et al. Ruxolitinib in steroid-refractory in graft-vs-host disease: a case report. J Hematol Oncol. 2016;9(1):67.
Zeiser R, Burchert A, Lengerke C, Verbeek M, Maas-Bauer K, Metzelder SK, et al. Ruxolitinib in a corticosteroid-refractory graft versus host disease after allogeneic stem cell transplantation: a multicenter survey. Leukemia. 2015;29(10):2062–8.
Okiyama N, Furumoto Y, Villarroel VA, Linton JT, Tsai WL, Gutermuth J, et al. Reversal of CD8-T cell mediated mucocutaneous graft-versus-host-like disease by the JAK inhibitor tofacitinib. J Invest Dermatol. 2014;134(4):992–1000.
Choi SW, Gatza E, Hou G, Sun Y, Whitfield J, Song Y, et al. Histone deacetylase inhibition regulates inflammation and enhances T regs after allogeneic hematopoietic cell transplantation in humans. Blood. 2015;125(5):815–9.
Acknowledgements
Open access funding provided by Medical University of Vienna.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Fusnding
No financial support was received for this study or the preparation of this article.
Conflict of interest
Robert Knobler is a consultant to Mallinckrodt-Therakos, a company specialising in extracorporeal photopheresis systems, used in the treatment of graft-versus-host disease. Karla Strong Rodrigues, Carla Oliveira-Ribeiro, and Silvia de Abreu Fiuza Gomes declare that they have no conflicts of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Strong Rodrigues, K., Oliveira-Ribeiro, C., de Abreu Fiuza Gomes, S. et al. Cutaneous Graft-Versus-Host Disease: Diagnosis and Treatment. Am J Clin Dermatol 19, 33–50 (2018). https://doi.org/10.1007/s40257-017-0306-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-017-0306-9