
Abstract
Spatial distribution patterns of As, Ba, Cd, Cr, Cu, Mn, Ni, Pb, U and Zn were determined in topsoil samples collected after 40 years of chemical remediation conducted in the inoperative “Staszic” pyrite–uranium mine in the Holy Cross Mountains, south-central Poland. Soil samples were taken from 58 sites using a systematic random sampling design. Selected samples were subjected to an X-ray diffractometry analysis on bulk soils and separated clay fractions. Hematite, goethite and gypsum are common mineral phases in soil samples. Technogenic soils developed on reclaimed mine spoils show uniform spatial element distribution patterns and additionally a distinct enrichment in As, Pb, Mn, U and Zn. Mineral and chemical composition of soils vs. rocks points to the lithogenic source of the determined elements. The results of chemical analysis have been used for evaluation of geochemical background of trace elements in the study area with the iterative 2σ-technique. This investigation shows that using mean crustal element concentrations (Clarke values) as proxies of threshold values in soils are not useful for determination of strongly positive geochemical anomalies. A modified enrichment factor, i.e. a local enrichment factor, is proposed for identification of sites where soils are contaminated.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Geochemical background is commonly defined as a natural concentration range of a substance in a specific environmental compartment (Matschullat et al. 2000; Gałuszka 2007; Gałuszka and Migaszewski 2011). This term, originally introduced into geochemistry for discrimination between non-anomalous element concentrations and geochemical anomalies (Hawkes and Webb 1962), was adapted to environmental sciences for differentiation between natural and contaminated sites (Förstner and Müller 1981).
An assessment of soil quality requires the knowledge of natural concentrations of elements. The upper limit of geochemical background range is particularly important for evaluation of soil contamination (Baize and Sterckeman 2001). In soils showing naturally elevated levels of elements (representing positive geochemical anomalies) and varied element distribution patterns, geochemical background values are more realistic for assessing soil quality than the regulatory levels of elements, such as soil quality standards (Ungaro et al. 2008; Romano et al. 2015). The use of mean crustal element concentrations or average shale composition as proxies of natural element concentrations in soils affected by multiple small-scale geochemical anomalies is problematic because in this approach a local geologic setting and other environmental factors are not considered. However, many authors use the upper continental crust element concentration for soil quality assessment, especially in calculation of quantitative indices, such as enrichment factor, geoaccumulation index and pollution load index (Dung et al. 2013; Barbieri 2016).
Spatial and temporal distribution of potentially toxic trace elements in soils of the post-mining areas has been a subject of numerous studies (Bačeva et al. 2014; Neiva et al. 2014; Semhi et al. 2014; Kim and Hyun 2015; Wahsha et al. 2016). In contrast, limited data exist on geochemical background of trace metal(loid)s in historic mining areas (Loredo et al. 2006; Martínez et al. 2007; Gałuszka et al. 2015; Gutiérrez et al. 2016). The quality of soils in these areas is very difficult to assess due to overlapping of anthropogenic and natural high concentrations of metal(loid)s and usually a very high variation of metal(loid) distribution patterns in soils (Dołęgowska et al. 2015).
In historic metal ore mining areas, like in other sites with high natural metal(loid) concentrations, it is critical to establish a site-specific geochemical background of potentially toxic trace elements prior to remediating operations (Armiento et al. 2011). Knowledge of geochemical background values is also important for post-remediation assessment of soil quality. Soils of abandoned mining sites pose a serious environmental threat because of potential mobility of pollutants creating a risk of groundwater contamination and health hazards for local communities (Bori et al. 2016; Pérez-Sirvent et al. 2016). The study of soils in the post-mining areas should always include basic geologic, mineralogical and geochemical characteristics of the study area (Gałuszka et al. 2015; Romano et al. 2015).
The main objectives of this study included: (1) evaluation of site-specific geochemical background of As, Ba, Cd, Cr, Cu, Mn, Ni, Pb, U and Zn in topsoils of the inoperative pyrite–uranium mine and a comparison of background to Clarke values; (2) identification of sites highlighted by geochemical anomalies using the upper limits of geochemical background ranges as threshold values; (3) determination of the origin of geochemical trace element anomalies. The general purpose of the study was geochemical characteristics of soil in the historic mining area and delineation of geochemical background from geochemical anomalies of selected trace elements. The novelty of the proposed approach lies in using local geochemical background values for indication of geochemical anomalies and for assessment of the soil quality with the use of a modified enrichment factor.
The soil samples were collected in July 2013 in the reclaimed pyrite–uranium mine in the central part of the Holy Cross Mountains, south-central Poland.
Materials and methods
Study area
Location and brief characteristics of the study area
The study area is located in the central part of the Holy Cross Mountains, south-central Poland (Fig. 1), in an area of the abandoned “Staszic” mine in Rudki where pyrite and uranium ores were extracted during 1925–1971. This area was reclaimed in the 1970s of the twentieth century. During operation of the mine the post-flotation sludge was stored in 6 ponds taking up an area of 12.5 ha. The total thickness of waste layer was in the range of 4–8 m (Skawina et al. 1974). The sludge consisted of dolomite with an admixture of pyrite and trace amounts of quartz, illite and gypsum. Reclamation of the inoperative mining area was done in three phases. After levelling, the terrain was covered with mine and processing tailings mixed with unslaked lime (5–15 tons//ha), and subsequently with a 30 cm layer composed of clay, unslaked lime and mineral fertilizer. The last phase encompassed planting of this area with vegetation. According to the post-reclamation report, the rehabilitation of the abandoned mining area was successful (Skawina et al. 1974). However, a more recent study has reported that the technogenic soils in Rudki contain very high concentrations of potentially toxic trace elements, including Pb, Ga, As, Tl and U (Uzarowicz 2011). The results of this study indicated that the reclamation was unsuccessful, which was also evidenced by sparsely vegetated habitat with several muddy patches (Fig. 2).

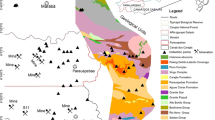
Location of the study area and the sampling sites R1–R58

Muddy unvegetated patches: a close to site R31; b close to site R29
Geology and history of mining in the study area
The study area is located within the Bodzentyn syncline in the north-central part of the Holy Cross Mountains. This structural unit is built of Silurian clayey–muddy shales and sandstones and Lower and Middle Devonian terrigenous–carbonate deposits. The deep-rooted Łysogóry fault extending nearly north–south in the village of Rudki forms an anomalous contact between the Silurian clayey–muddy shales and Lower Devonian dolomites with clayey shale interbeds (Czarnocki 1956). This fault played a critical role in formation of iron (pyrite/marcasite FeS2, siderite FeCO3, hematite (α-Fe2O3) and uranium (pitchblende—partly altered uraninite UO2) ore deposit in Rudki. The uranium ore occurs as an accompanying mineralization of iron ores. At the surface, there is an about 20-m-thick iron weathering cap composed of clays and partly bauxitic clays with limonite/hematite nests (Nieć 1968).
Archaeological artefacts suggest that extraction of hematite in the study area can be dated back to ancient times (Bielenin 1962). During most of the Roman Period (first through fourth century), this area was a major metallurgical centre in Europe, probably supplying German tribes with weaponry. The hematite ore was used as a raw material for smelting of pig iron in numerous primitive furnaces. After the fourth century, this mineral ore deposit was abandoned for sixteen centuries and was rediscovered not until 1922. Extraction of hematite and siderite started in the 1920s of the twentieth century. During 1933–1969 iron sulphides were mined for production of sulphuric acid. Extraction of pitchblende began in 1952 (Szecówka 1987). The mining operations ended in 1971.
Soil sampling
The soils of the study area belong to technogenic soils (Spolic Technosols) developed on mine and processing wastes (Uzarowicz and Skiba 2011; Uzarowicz 2013). Fieldwork was conducted in July 2013 to collect samples for this study. Soils were sampled at 58 locations. Position of sampling sites was determined using a Global Positioning System with a precision of ±4–5 m (Fig. 1). About 2 kg of each soil sample consisting of 5–10 increments (subsamples) was collected at each site from pits that were dug to a depth of about 0.5 m. The collected soil samples were placed in polyethylene bags and transported to the Geochemical Laboratory of the Institute of Chemistry, Jan Kochanowski University, in Kielce on the day of sampling. During sample collection, transport, storage and treatment, procedures were followed to minimize the possibility of contamination (Csuros and Csuros 2002).
Sample treatment and chemical analysis
Soil samples were stored at a room temperature for several days in a separate room with a limited access. After air-drying, they were sieved to obtain <2.0 mm fraction and then disaggregated to pass a <0.063 mm sieve using a Pulverisette 2 Fritsch’s blender and then Analysts 3 Spartan shaker.
The \({\text{pH}}_{{{\text{H}}_{2} {\text{O}}}}\) of soil samples was measured in the laboratory. About 10 g of each sample was placed in a beaker and stirred vigorously with 25 mL of deionized water. The pH measurements were made after 24 h with the use of CP-105 pH meter (Elmetron, Poland).
For determinations of trace elements by ICP-MS, soil samples (0.5 g) were digested with aqua regia (6 mL HCl + 2 mL HNO3) in a closed microwave system Multiwave 3000 with the use of the following operating conditions: power 1000 W, time 65 min., T 220 °C, p 60 bars, p growth rate 0.3 bar/s. The obtained digests were replenished up to 25 mL and evaporated (160 °C), and the insoluble residue was filtered. In the aqua regia extracts, the following trace elements were determined and quantified: As, Ba, Cd, Cr, Cu, Mn, Ni, Pb, U and Zn using the ICP-MS instrument (model ELAN DRC II, Perkin Elmer). Instrumental and data acquisition parameters of the ICP-MS instrument were as follows: lens voltage—7.50 V, plasma power—1275 W, sweeps/reading—20, readings/replicate—3, replicates—4, nebulizer gas flow—1.03 L/min, plasma gas flow—15 L/min. Measurements were done in the peak hopping mode and the dwell time was set for 50–150 microseconds. Rh and Ir were used as internal standards. The ICP-MS instrument was optimized with a standard daily procedure before measurements (Thomas 2013).
The standard reference materials (SRM) applied for measuring element concentrations were: Certified Reference Material (CRM) NIST 2710a (Montana I Soil) and GSS4 (Chinese Academy of Geological Sciences). Quality control included both accuracy (CRM) and precision (triplicates). The average recovery of elements from the CRM was in the range of 81–96 %. The RSD values were well below 5 % for all the samples analysed.
Petrographic study
Thin polished sections of iron ore collected at the reclaimed Rudki mine tailings pile were examined petrographically in transmitting and reflected light using a polarized microscope Nikon Eclipse LV 100 Pol. The polished thin sections of goethite–hematite–pyrite–marcasite were also studied with an electron microprobe Cameca SX100 equipped with four wavelength dispersive detectors at the Faculty of Geology, University of Warsaw, Poland. The following parameters were set for analyses: signal A = BSE, the accelerating voltage was 15 kV and the probe current was either 10 or 20 nA.
XRD analysis
Bulk soil mineralogy of randomly selected samples (R5, R14, R22, R30, R33, R48, R55 and R58) was examined with the X-ray diffraction method (Thermo ARL SCINTAG X’TRA X-ray diffractometer with Cu Kα irradiation) (Moore and Reynolds 1989). Clay mineral fraction <2 µm was obtained using the sedimentation method (Rowell 2014). The samples were doubly centrifuged for 15 min using a Sigma 4–15 centrifuge. The clay suspensions were pipetted onto a glass slide and left to dry at an ambient temperature. The air-dried, ethylene glycol-saturated and heated (550 °C) samples were used for determination of clay mineral species according to the methods described by Moore and Reynolds (1989).
The measurements were done at 40 kV and 40 mA and scan rates from 3 to 30°2Θ (or from 2 to 30°2Θ) at 0.02°2Θ min−1 step size. Phase identification and peak area determinations were done using the Win XRD software. The XRD analyses were performed in the Institute of Geology, Adam Mickiewicz University in Poznań.
Calculation of geochemical background and enrichment factor
The iterative 2σ-technique (Matschullat et al. 2000) was used for calculation of geochemical background. In this technique, mean and standard deviation are calculated from a dataset representing results of element determinations. This dataset should contain results of analyses of at least 30 samples. All values beyond the mean ±2σ are omitted, and a new mean and standard deviation are calculated from the reduced dataset. If there are still values exceeding mean ±2σ, they are eliminated and this procedure is repeated until all the values lie within this range. At the end of calculation, the reduced dataset represents a normal distribution.
The enrichment factor is calculated with the following formula (Gałuszka and Migaszewski 2011):
where A e is an element concentration in an environmental sample, B e is a reference element concentration in an environmental sample, A c is the Clarke value of the element and B c is the Clarke value of the reference element (McLennan 2001). Ti was used as a reference element (Dragović and Mihailović 2009).
Results and discussion
Ore and gangue mineralogy
Optical microscope and BSE images of mineral ore assemblages are presented in Figs. 3 and 4. The microscopic study indicates that the fine-grained marcasite and subordinate pyrite (FeS2) are the major sulphide minerals. Marcasite with subordinate pyrite form massive aggregates reaching several centimetres in diameter. In contrast, pyrite consists of sub- and euhedral (cubic) grains commonly varying from 0.0X to 0.1 mm across, scattered within a microcrystalline dolomite groundmass (Fig. 3a, b). These two sulphide minerals predominate in goethite (α-Fe3+O(OH)) and hematite (α-Fe2O3) assemblages (Fig. 3c, d). As opposed to uniform hematite, goethite forms porous concentric and locally banded accumulations (Fig. 4b). Both sulphide minerals also occur in the form of individual grains and inclusions in goethite voids (Figs. 3c, d, 4a). Some goethite voids additionally contain tiny galena (PbS) inclusions up to 0.02 mm across (Fig. 4a–d). Galena also forms thin veinlets cutting or abutting marcasite grains (Fig. 4c, d). These veinlets are about 0.05 mm long. Of the other accessory ore minerals, sphalerite (α-ZnS) inclusions about several micrometres in diameter are extremely rare. Similar mineralogical composition of ore samples collected in Rudki was found by Uzarowicz (2013).

Microphotographs: a pyrite grains (Py) scattered in a micrograined dolomite matrix (Do), transmitted light, crossed nicols; b same mineral assemblage, reflected light, crossed nicols; c hematite (He) and banded goethite (Goe) with marcasite grains (Ma), transmitted light, crossed nicols; d same mineral assemblage, reflected light, one nicol

Electron microprobe (BSE) images: a pyrite grains (Py) with subordinate galena inclusions (Gal) scattered within a porous concentric goethite assemblage (Goe); b massive hematite (Hem) overgrown with porous concentric goethite (Goe) comprising tiny galena inclusions (Gal); c galena veinlet (Gal) cutting marcasite grain (Ma) within ferric oxyhydroxide groundmass (Fe); d two galena grains (Gal) abutting marcasite grains (Ma) within ferric oxyhydroxide groundmass (Fe)
Mineral composition of soil samples
The results of XRD analysis (Table 1) reveal that the bulk soil samples are composed of quartz and gypsum (samples R5, R22, R33 and R48). Samples from sites R5, R22, R33, R48 and R55 contain goethite and hematite in the bulk fraction. In sample R55 muscovite or illite occurs with goethite and hematite. Dolomite was found in samples R5 and R58. Muscovite and/or kaolinite occur in non-fractionated samples R5, R48 and R55, which may be explained by the predominance of minor fractions over detritic quartz, gypsum, goethite and hematite. Illite and kaolinite prevail in a clay fraction. Bassanite (Ca[SO4]·0.5H2O) is also noted in the EG-treated and heated clay fractions (samples R5, R14, R33, R58). Additionally, in sample R22 traces of mixed-layered minerals, assigned presumably to a kaolinite–smectite group, occur. In fine-grained fractions gypsum and iron oxides and hydroxides are very common. The occurrence of gypsum in carbonate soils has been induced by products of pyrite weathering as a result of carbonate dissolution and hydrolysis of Ca-bearing minerals and desorption of exchangeable Ca (Martín et al. 2008).
The results of these XRD measurements are similar to those reported by Uzarowicz (2013) in a soil sample collected from a depth interval of 0.03–0.3 m close to site R58 in the current study. The presence of gypsum and porous Fe oxides and hydroxide assemblages in Rudki soils gives evidence for weathering of iron sulphides (pyrite and marcasite) that occur in the parent rock (Uzarowicz 2013). Authigenic formation of different secondary Fe minerals (iron oxyhydroxides, jarosite) have been reported in many studies of carbonate soils influenced by sulphide weathering (Martín et al. 2008; Navarro et al. 2008; Lara et al. 2015).
The pH and element concentrations of soils
The pH values and trace element concentrations of soil samples are reported in Table 2. The results show that all the elements, except for Mn and Pb, are rather uniformly distributed in topsoil (Table 3). The extremely low soil pH values (2.0–4.0) occur only in samples R29, R31, R37 and R55. However, the typical pH values in most samples are circumneutral. Acidification of soils results from oxidation of iron sulphides (Jennings et al. 2000; Migaszewski et al. 2015).
Weathering products of marcasite and pyrite have locally been released into soils from the waste material used for remediation of Rudki mining area in the 1970s. The process of sulphide oxidation in soils is rapid as shown by the study of Martín et al. (2008). These authors observed a substantial drop in pH to a value close to 2 after 3 years of experimental deposition of pyrite tailings over a carbonate soil. The presence of dolomite in mine and processing tailings and an application of unslaked lime for soil remediation have increased acid neutralizing capacity in the examined soil and prevented it from acidification at most of the investigation sites. However, the soil pH as a major factor affecting element mobility and cation exchange capacity at extremely acidic sites (pH < 4) may be responsible for soil infertility (Fig. 2) due to partial hydrolysis of primary silicates (Martín et al. 2008) and increased bioavailability of phytotoxic metal(loid)s (Romero-Freire et al. 2016). The highest concentrations of trace elements have been found in samples revealing circumneutral pH (except for sample R55). This can be explained by increased mobility of metal(loid)s in acidic soils and their stabilization at a circumneutral pH (Dorronsoro et al. 2002).
The soil mineral composition and pH differ from site to site. There is no clear pattern of the element spatial distribution in topsoil and no statistically significant correlations between the determined elements (Table 4). Despite differences in mineral composition of soil samples, there is a lack of spatial diversity in concentrations of As, Ba, Cd, Cr, Cu, Ni, U and Zn.
The problem of soil contamination in Rudki by trace metal(loid)s and radionuclides was first mentioned by Uzarowicz (2011). The concentrations of elements found by this author in the uppermost part of soil profiles in Rudki (site R58 in the present study) were as follows (in mg/kg): As (27.3), Ba (166), Cd (0.5), Cr (52), Cu (20.8), Mn (506), Ni (20.2), Pb (728), U (8.1) and Zn (63) being almost identical to As (27.6), Cd (0.6) and Zn (65) in the present study. The majority of samples show very high Pb and Mn concentrations. This enrichment is a result of complex geochemical processes that occur during sulphide weathering in carbonate soils (Lara et al. 2015). Lead is present as inclusions and veinlets of galena in hematite, goethite and rocks of the study area (Fig. 4). During sulphide weathering in calcareous soils, PbS is first oxidized to anglesite (PbSO4)-like phases that are subsequently converted into cerussite (PbCO3)-like phases (Lara et al. 2011). However, cerussite is the most mobile and bioavailable lead phase in soils, and about 40 years after liming, this mineral must have been removed from soil. The Pb sequestration in soils in the form of plumbojarosite \(\left( {{\text{PbFe}}^{3 + }_{6} \left[ {\left( {\text{OH}} \right)_{12} \left( {{\text{SO}}_{4} } \right)_{4} } \right]} \right)\) has been reported by Root et al. (2015). This mineral is more thermodynamically stable than cerussite (PbCO3) and its occurrence is common in abandoned mining sites (Bosso and Enzweiler 2008). According to Uzarowicz (2013), secondary porous Fe oxyhydroxides formed in the Rudki soils do not show elevated levels of Pb as opposed to a high concentration of Mn. The results of XRD measurements do not allow us to indicate the Pb mineral species that predominates in these soils. Nonetheless, common occurrence of gypsum and the results of the previous studies conducted by Uzarowicz (2013) suggest that plumbojarosite may be the main Pb phase in the Rudki topsoil.
The upper limits of geochemical background ranges are higher than the Clarke values of As, Cu, Mn, Pb, U and Zn (Table 3). Similar enrichment in As, Cu, Pb and Zn has been observed in lime-amended soils of the historic mining sites in Butte, Montana (Davis et al. 1999). The highest number of geochemical anomalies, representing almost 14 % of the results obtained, has been linked to Mn (Table 2). At several sites, multiple anomalies have been found. Three or more elements revealed anomalous concentrations at the following sites R14 (Cd, Mn, Ni), R51 (As, Cr, Mn, U), R52 (As, Mn, Ni), R54 (Mn, Ni, Pb) and R55 (Cu, Ni, Pb, U, Zn).
The enrichment factor (EF) is a popular background enrichment index used in geochemical studies for assessment of soil or sediment enrichment in elements (Dung et al. 2013; Pourret et al. 2016). However, its usefulness for interpretation of the abundance of elements showing strongly positive geochemical anomalies in the study area is debatable because all the calculated EF would indicate extremely high values. Pourret et al. (2016) discussed this issue and concluded that neither concentrations of anomalous element in the Upper Continental Crust nor regulatory permissible levels are adequate for geochemical characteristics of contaminated soils in the Katanga Copperbelt. In our study, Pb shows over hundred times higher background concentration than its Clarke value indicates. According to Reimann and De Caritat (2000) the use of EF by environmental scientists should be abandoned. These authors criticize EF for being based on approximated composition of the upper continental crust (Clarke values). It has been shown that a common practice of using Clarke values for EF calculations introduces a very high uncertainty (of about 100–1000 order) to obtained results (Reimann and De Caritat 2000). Thus, we propose that for pedogeochemical studies of mining and post-mining areas a modified enrichment factor be calculated. The local enrichment factors (LEF), based on site-specific geochemical background of the elements instead of their Clarke values, better characterize local geochemistry. Moreover, these enable us to select sites displaying anomalous element concentrations. This parameter is calculated using the formula below:
where A e is an element concentration in an environmental sample, B e is a reference element concentration in an environmental sample, A BG is the upper limit of geochemical background range of the element and B BG is the upper limit of geochemical background range of the reference element.
The comparison of the EF–LEF values is given in Table 5. The results of one-way ANOVA test show that differences between the EF and LEF values are statistically significant (p < 0.05) for all of the determined elements. The EF values are the highest at sites 54 and 57. Interpretation of these values has been done using a five-category scale (Pourret et al. 2016): EF < 2 indicates no enrichment to minimal enrichment, 2 ≤ EF < 5, moderate enrichment, ≤5 EF < 20, significant enrichment, 20 ≤ EF ≤ 40, very high enrichment, and EF > 40, extremely high enrichment. In case of Pb and to a lesser extent U and Mn, the majority of EF values point out to extremely high enrichment in these elements. On the contrary, the LEF values for Pb, Mn and U enabled us a precise identification of contaminated sites. The proposed approach overcomes the major shortcomings of EF based on Clarke values, such as heterogeneous composition of the upper crust, total concentrations of elements in various crustal rock samples versus pseudo-total concentrations of elements in soil (Reimann and De Caritat 2000). The major advantages of LEF values compared with other indices of soil pollution are: (1) reliable data based on local geochemical conditions, (2) simple calculation and (3) using the same analytical methods for obtaining background and anomalous element concentrations. Methods that are currently in use or are proposed for the study of surface soil pollution with metals are much more complicated, require heavy laboratory workload, expert knowledge or professional software, and they are expensive (Nazarpour et al. 2015; Hu and Cheng 2016; Wu et al. 2016; Xiong and Zuo 2016). This study shows that local geochemical datasets can be a source of reliable knowledge of elements’ distribution in heavily contaminated soils.
Conclusion
Technogenic soils collected from a remediated mining area exhibit elevated levels of As, Pb, Mn, U and Zn. The secondary gypsum that formed in soils after using mine and processing tailings mixed with unslaked lime for site remediation in the 1970s is the evidence for marcasite and pyrite weathering. Hematite and goethite assemblages are genetically related to the parent rock and to subordinate products of pyrite and marcasite oxidation. The upper limits of geochemical background concentration ranges of As, Cu, Mn, Pb, U and Zn are higher than the Clarke values. The site-specific geochemical background values calculated with the iterative 2σ-technique were used for identification of geochemical anomalies of elements in topsoil and for computing local enrichment factors (LEF). This approach enabled us to point out sites with contaminated soil. The present study confirms the importance of site-specific background values for better understanding of spatial distribution of elements and for reliable assessment of soil quality in the highly contaminated areas.
References
Armiento G, Cremisini C, Nardi E, Pacifico R (2011) High geochemical background of potentially harmful elements in soils and sediments: implications for the remediation of contaminated sites. Chem Ecol 27(S1):131–141
Bačeva K, Stafilov T, Šajn R, Tănăselia C, Makreski P (2014) Distribution of chemical elements in soils and stream sediments in the area of abandoned Sb–As–Tl Allchar mine, Republic of Macedonia. Environ Res 133:77–89
Baize D, Sterckeman T (2001) Of the necessity of knowledge of the natural pedo-geochemical background content in the evaluation of the contamination of soils by trace elements. Sci Total Environ 264(1):127–139
Barbieri M (2016) The importance of enrichment factor (EF) and Geoaccumulation Index (Igeo) to evaluate the soil contamination. J Geol Geophys 5:237
Bielenin K (1962) Ancient centre of iron metallurgy in the region of Góry Świętokrzyskie (Świętokrzyskie Mountains). Archaeol Pol 4:221–234
Bori J, Vallès B, Navarro A, Riva MC (2016) Geochemistry and environmental threats of soils surrounding an abandoned mercury mine. Environ Sci Pollut Res 23(13):112941–112953
Bosso ST, Enzweiler J (2008) Bioaccessible lead in soils, slag, and mine wastes from an abandoned mining district in Brazil. Environ Geochem Health 30(3):219–229
Csuros M, Csuros C (2002) Environmental sampling and analysis for metals. CRC Press, Boca Raton
Czarnocki J (1956) Iron ore deposit at Rudki. Mineral raw materials in the holy cross mountains. Pr Geol 5:19–51 (in Polish)
Davis A, Eary LE, Helgen S (1999) Assessing the efficacy of lime amendment to geochemically stabilize mine tailings. Environ Sci Technol 33(15):2626–2632
Dołęgowska S, Gałuszka A, Migaszewski ZM (2015) Assessing soil sampling uncertainty in heterogeneous historic metal ore mining sites. Accred Qual Assur 20(3):163–170
Dorronsoro C, Martin F, Ortiz I, García I, Simón M, Fernández E, Aguilar J, Fernández J (2002) Migration of trace elements from pyrite tailings in carbonate soils. J Environ Qual 31(3):829–835
Dragović S, Mihailović N (2009) Analysis of mosses and topsoils for detecting sources of heavy metal pollution: multivariate and enrichment factor analysis. Environ Monit Assess 157(1–4):383–390
Dung TTT, Cappuyns V, Swennen R, Phung NK (2013) From geochemical background determination to pollution assessment of heavy metals in sediments and soils. Rev Environ Sci Bio/Technol 12(4):335–353
Förstner U, Müller G (1981) Concentrations of heavy metals and polycyclic aromatic hydrocarbons in river sediments: geochemical background, man’s influence and environmental impact. GeoJournal 5(5):417–432
Gałuszka A (2007) A review of geochemical background concepts and an example using data from Poland. Environ Geol 52(5):861–870
Gałuszka A, Migaszewski Z (2011) Geochemical background—an environmental perspective. Mineralogia 42(1):7–17
Gałuszka A, Migaszewski ZM, Dołęgowska S, Michalik A, Duczmal-Czernikiewicz A (2015) Geochemical background of potentially toxic trace elements in soils of the historic copper mining area: a case study from Miedzianka Mt., Holy Cross Mountains, south-central Poland. Environ Earth Sci 74(6):4589–4605
Gutiérrez M, Wu SS, Rodriguez JR, Jones AD, Lockwood BE (2016) Assessing the state of contamination in a historic mining town using sediment chemistry. Arch Environ Contam Toxicol 70(4):747–756
Hawkes HE, Webb JE (1962) Geochemistry in mineral exploration. Harper, New York
Hu Y, Cheng H (2016) A method for apportionment of natural and anthropogenic contributions to heavy metal loadings in the surface soils across large-scale regions. Environ Pollut 214:400–409
Jennings SR, Dollhopf DJ, Inskeep WP (2000) Acid production from sulfide minerals using hydrogen peroxide weathering. Appl Geochem 15(2):235–243
Kim J, Hyun S (2015) Nonequilibrium leaching behavior of metallic elements (Cu, Zn, As, Cd, and Pb) from soils collected from long-term abandoned mine sites. Chemosphere 134:150–158
Lara RH, Briones R, Monroy MG, Mullet M, Humbert B, Dossot M, Naja GM, Cruz R (2011) Galena weathering under simulated calcareous soil conditions. Sci Total Environ 409(19):3971–3979
Lara RH, Monroy MG, Mallet M, Dossot M, González MA, Cruz R (2015) An experimental study of iron sulfides weathering under simulated calcareous soil conditions. Environ Earth Sci 73(4):1849–1869
Loredo J, Ordóñez A, Alvarez R (2006) Environmental impact of toxic metals and metalloids from the Munon Cimero mercury-mining area (Asturias, Spain). J Hazard Mater 136(3):455–467
Martín F, García I, Díez M, Sierra M, Simón M, Dorronsoro C (2008) Soil alteration by continued oxidation of pyrite tailings. Appl Geochem 23(5):1152–1165
Martínez J, Llamas J, De Miguel E, Rey J, Hidalgo MC (2007) Determination of the geochemical background in a metal mining site: example of the mining district of Linares (South Spain). J Geochem Explor 94(1):19–29
Matschullat J, Ottenstein R, Reimann C (2000) Geochemical background–can we calculate it? Environ Geol 39:990–1000
McLennan SM (2001) Relationships between the trace element composition of sedimentary rocks and upper continental crust. Geochem Geophys Geosyst 2(4). doi:10.1029/2000GC000109
Migaszewski ZM, Gałuszka A, Dołęgowska S, Hałas S, Krzciuk K, Gebus B (2015) Assessing the impact of Serwis mine tailings site on farmer’s wells using element and isotope signatures (Holy Cross Mountains, south-central Poland). Environ Earth Sci 74:629–647
Moore DM, Reynolds RC (1989) X–ray diffraction and the identification and analysis of clay minerals. Oxford University Press, New York
Navarro MC, Pérez-Sirvent C, Martínez-Sánchez MJ, Vidal J, Tovar PJ, Bech J (2008) Abandoned mine sites as a source of contamination by heavy metals: a case study in a semi-arid zone. J Geochem Explor 96(2):183–193
Nazarpour A, Omran NR, Paydar GR (2015) Application of multifractal models to identify geochemical anomalies in Zarshuran Au deposit NW Iran. Arab J Geosci 8(2):877–889
Neiva AMR, Carvalho PCS, Antunes IMHR, Silva MMVG, Santos ACT, Pinto MC, Cunha PP (2014) Contaminated water, stream sediments and soils close to the abandoned Pinhal do Souto uranium mine, central Portugal. J Geochem Explor 136:102–117
Nieć M (1968) Mineralizacja złoża siarczków żelaza i syderytu w Rudkach w Górach Świętokrzyskich. Prace Geologiczne PAN 46. Wydawnictwa Geologiczne, Warszawa
Pérez-Sirvent C, Hernández-Pérez C, Martínez-Sánchez MJ, García-Lorenzo ML, Bech J (2016) Geochemical characterisation of surface waters, topsoils and efflorescences in a historic metal-mining area in Spain. J Soils Sedim 16(4):1238–1252
Pourret O, Lange B, Bonhoure J, Colinet G, Decrée S, Mahy G, Séleck M, Shuchta M, Faucon MP (2016) Assessment of soil metal distribution and environmental impact of mining in Katanga (Democratic Republic of Congo). Appl Geochem 64:43–55
Reimann C, De Caritat P (2000) Intrinsic flaws of element enrichment factors (EFs) in environmental geochemistry. Environ Sci Technol 34(24):5084–5091
Romano E, Bergamin L, Croudace IW, Ausili A, Maggi C, Gabellini M (2015) Establishing geochemical background levels of selected trace elements in areas having geochemical anomalies: the case study of the Orbetello lagoon (Tuscany Italy). Environ Pollut 202:96–103
Romero-Freire A, Fernández IG, Torres MS, Garzón FM, Peinado FM (2016) Long-term toxicity assessment of soils in a recovered area affected by a mining spill. Environ Pollut 208:553–561
Root RA, Hayes SM, Hammond CM, Maier RM, Chorover J (2015) Toxic metal (loid) speciation during weathering of iron sulfide mine tailings under semi-arid climate. Appl Geochem 62:131–149
Rowell DL (2014) Soil science: methods and applications. Routledge, New York
Semhi K, Al Abri R, Al Khanbashi S (2014) Impact of sewage and mining activities on distribution of heavy metals in the water-soil-vegetation system. Int J Environ Sci Technol 11(5):1285–1296
Skawina T, Trafas M, Gołda T (1974) Rekultywacja terenów pogórniczych kopalni pirytu „Siarkopol” w Rudkach k. Kielc. Zeszyty Naukowe AGH. 466. Sozol Sozotech 4:9–21 in Polish, with English abstract
Szecówka M (1987) Uranium mineralization at Rudki near Słupia Nowa (Holy Cross Mountains). Prace Geologiczne PAN, Komitet Nauk Geologicznych Oddział w Krakowie (in Polish)
Thomas R (2013) Practical guide to ICP MS, 3rd edn. CRC Press, Boca Raton
Ungaro F, Ragazzi F, Cappellin R, Giandon P (2008) Arsenic concentration in the soils of the Brenta Plain (Northern Italy): mapping the probability of exceeding contamination thresholds. J Geochem Explor 96(2):117–131
Uzarowicz Ł (2011) Technogenic soils developed on mine spoils containing iron sulfides in select abandoned industrial sites: environmental hazards and reclamation possibilities. Pol J Environ Stud 20(3):771–782
Uzarowicz Ł (2013) Microscopic and microchemical study of iron sulphide weathering in a chronosequence of technogenic and natural soils. Geoderma 197:137–150
Uzarowicz Ł, Skiba S (2011) Technogenic soils developed on mine spoils containing iron sulphides: mineral transformations as an indicator of pedogenesis. Geoderma 163:95–108
Wahsha M, Nadimi-Goki M, Bini C (2016) Land contamination by toxic elements in abandoned mine areas in Italy. J Soils Sedim 16(4):1300–1305
Wu J, Teng Y, Chen H, Li J (2016) Machine-learning models for on-site estimation of background concentrations of arsenic in soils using soil formation factors. J Soils Sedim 16(6):1787–1797
Xiong Y, Zuo R (2016) Recognition of geochemical anomalies using a deep autoencoder network. Comput Geosci 86:75–82
Acknowledgments
The authors acknowledge the assistance of Karina Krzciuk MSc in sampling. Dr. Piotr Dzierżanowski and Ms. Lidia Jeżak are thanked for performing the microprobe analyses. This study was supported by the National Science Center, a research Grant #DEC-2011/03/B/ST10/06328.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial responsibility: J. Aravind.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gałuszka, A., Migaszewski, Z., Duczmal-Czernikiewicz, A. et al. Geochemical background of potentially toxic trace elements in reclaimed soils of the abandoned pyrite–uranium mine (south-central Poland). Int. J. Environ. Sci. Technol. 13, 2649–2662 (2016). https://doi.org/10.1007/s13762-016-1095-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13762-016-1095-z