
Abstract
Obesity, exposure to stress and inadequate sleep are prevalent phenomena in modern society. In this review we focus on their relationships and critically evaluate causality. In obese individuals, one of the main stress systems, the hypothalamic-pituitary-adrenal axis, is altered, and concentrations of cortisol are elevated in adipose tissue due to elevated local activity of 11β-hydroxysteroid dehydrogenase (HSD) type 1. Short sleep and decreased sleep quality are also associated with obesity. In addition, experimental sleep curtailment induces HPA-axis alterations which, in turn, may negatively affect sleep. These findings implicate that obesity, stress and sleep loss are all related in a vicious circle. Finally, we discuss new strategies to combat obesity through modulating cortisol levels in adipose tissue by 11β-HSD1 inhibitors or by improving sleep duration.
Similar content being viewed by others
Introduction
The National Health and Nutrition Examination Survey reported that in 2009–2010, 33 % of adult Americans were overweight ((body mass index (BMI) of 25.0–29.9 kg/m2)) and 36 % were obese (BMI ≥ 30.0 kg/m2) – a vast increase from 1960 to 1962– when a similar percentage of Americans were overweight (31 %), but only 13 % were obese [1]. Obesity leads to significant health problems impacting nearly every organ system, including the cardiovascular system and the endocrine pancreas, causing type 2 diabetes. In parallel to the obesity epidemic, society has become increasingly fast-paced, presenting us with numerous stressors including lack of restorative sleep. In addition, self-reported sleep duration has declined by 1.5 to 2 h during the last 50 years in America [2]. Epidemiological and experimental research accumulates that both the stress system [3] and sleep quality and duration [4] are affected in obese individuals.
According to a recent meta-analysis including a total of 23,372 individuals, psychological stress is positively correlated with measures of adiposity one to 38 years later [5]. The overall effect size was relatively small (r = 0.014; 0.002–0.025) and effect sizes were larger in men than women (0.024 vs. 0.017), after longer follow up, and in studies of higher quality. The relationship between habitual short sleep (generally less than 5 h per night) and obesity was further demonstrated in a meta-analysis of 45 cross-sectional studies, including 604,509 adults (OR: 1.55; 1.43–1.68) [6]. In addition, sleep curtailment and chronic stress are independently associated. In a sample of 1,300 subjects, perceived chronic stress was negatively associated with self-reported sleep duration [7]. It should be noted however that these observational studies cannot distinguish cause and consequence.
In this review, we will examine respectively the relationship between obesity and the HPA axis, obesity and sleep, and the HPA axis and sleep. For each of the reported associations, we will critically evaluate experimental studies in an attempt to determine causality and its direction. Finally we will assess the potential of strategies that aim to improve disturbances of the HPA-axis and to increase sleep duration for combating obesity.
The HPA-Axis and Obesity
Physiology and Assessment of the HPA-Axis
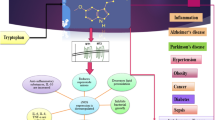
Exposure to stress elicits a highly orchestrated series of physical reactions, mainly operated by the HPA-axis, the sympathetic nervous system, and the sympathoadrenal system (Fig. 1) [8]. The body adapts to repeated and chronic stress via homeostatic mechanisms, adjusting physiological responses and behavior in order to maintain stability [9]. The chronic wear and tear of this accommodation over the lifetime of an organism is referred to as the allostatic load [8]. The parvocellular cells in the paraventricular nucleus of the hypothalamus secrete corticotrophin-releasing hormone (CRH) in response to stressors, including physiological stressors such as fatigue or hunger, as well as stressors of psychological nature. CRH, in turn, stimulates the anterior pituitary gland to secrete adrenocorticotrophic hormone (ACTH), which induces the synthesis and secretion of glucocorticoids, mineralocorticoids and androgenic steroids by the adrenal cortex. Cortisol, the main glucocorticoid in humans, suppresses the immune system and induces glycogenolysis, as well as insulin resistance [10]. CRH and ACTH synthesis and release are inhibited by peripheral negative feedbacks mediated by cortisol (Fig. 1). In addition, CRH activates the sympathetic locus coeruleus-norepinephrine system and cortisol enhances neuronal excitability to norepinephrine. In turn, norepinephrine activates hypothalamic CRH in reverberating circuits [9].

Schematic representation of HPA-axis and sleep alterations in obesity. Plus and minus signs indicate positive and negative effects, respectively. Upward/downward arrows and tildes (~) depict increased/decreased and unaltered levels, respectively, of depicted phenomena in obese individuals. Obese subjects often experience an increased stress exposure, leading to increased activity of the sympathetic nervous system (SNS) and release of corticotrophin-releasing hormone (CRH). CRH triggers release of adrenocorticotropic hormone (ACTH), which levels are elevated in obesity. ACTH stimulates cortisol release into the bloodstream. Cortisol, in turn, inhibits CRH and ACTH release. The HPA-axis and the SNS stimulate each other in a reverberating circuit and are able to affect sleep. Inadequate sleep is more common in obese subjects. Circulating cortisol levels, both in free form or bound to corticosteroid binding globulin (CBG), albumin or sex hormone binding globulin (SHBG), are reported unaltered of decreased in obesity. In tissues, cortisol is converted to inactive cortisone and vice versa by 11β hydroxysteroid dehydrogenase type 2 11β-HSD2 and 11β-HSD1, respectively. 11β-HSD1 is mostly present in the liver and adipose tissue. 11β-HSD2 is present in smaller quantities in these tissues, as depicted by the thinner arrow, whereas it is abundantly expressed in the kidney and colon. Hepatic cortisol levels are decreased in obese subjects, due to decreased 11β-HSD1 levels. In contrast, cortisol levels in adipose tissue are increased due to increased levels of 11β-HSD1. Cortisol and cortisone are metabolized by reductases to the inactive tetrahydrometabolites (THM) of tetrahydrocortisol (THF) and tetrahydrocortisone (THE), respectively. Urinary levels of THM and THE are usually increased in obese subjects
Cortisol depicts circadian rhythmicity, with a peak in the morning, followed by a gradual fall during the day, which is briefly interrupted by meals, and a nadir around 3 am [4]. Only 5–10 % of cortisol circulates in active free form; cortisol binding globulin (CBG) binds most cortisol (80–90 %) with high affinity and 10–15 % is bound to albumin and sex-hormone binding globulin with lower affinity (Fig. 1) [11, 12]. CBG is fully saturated at cortisol levels over 500 nmol/l and also depicts diurnal variability, with a peak in the early afternoon [12, 13].
Cortisol levels can be determined in plasma, saliva and urine [13]. In plasma, usually the total cortisol levels are measured and the unbound, protein-free fraction is indirectly estimated using the Coolens’ equation [12]. Measurements in saliva assess the biologically active, protein-free cortisol; hence the effects of within-subject changes or between-subject differences in CBG are eliminated. Saliva specimens are easier to collect than plasma and, because of the non-invasive collection, they are stress-free. Although urinary free cortisol refers to unconjugated cortisol, its level is proportional to the free (nonprotein-bound) cortisol in blood. Obtaining urine for free, unconjugated cortisol measurement also is stress-free, but the test requires 24-h collection in the presence of acidic preservative. Salivary cortisol levels increase within 5 min after plasma increases in cortisol and although levels are 30–50 % lower, under 500 nmol/l they are generally well correlated with plasma levels [13].
Two NAPD(H)-dependent isoenzymes 11β-hydroxysteroid dehydrogenase (11β-HSD1 and 11β-HSD2) intracellularly convert the inactive cortisone to hormonally active cortisol and vice versa (Fig. 1) [14]. 11β-HSD1 is expressed mainly in the liver and adipose tissue, while 11β-HSD2 exerts its function in the kidney and colon, making cortisol action tissue-specific. Cortisol and cortisone are metabolized to inactive tetrahydrometabolites (THM) by hepatic 5α- and 5β-reductase. Cortisol is converted to cortol and tetrahydrocortisol (THF, 5α-THF and 5β-THF), while cortisone is metabolized to cortolone and tetrahydrocortisone (THE) - metabolites often measured in urine. Alterations in 11β-HSD1 activity are reflected by the urinary (THF + 5α-THF + cortol) / (THE + cortolone) ratio, e.g., decreased 11β-HSD1 activity would lead to elevated THE levels and lower THF levels, thus to a decreased (THF + α-THF) / THE ratio.
Association of the HPA-Axis and Obesity
Morning levels of cortisol in saliva and plasma either show no correlation, or correlate negatively with various measures of adiposity (Fig. 1) [15]. Single-time determinations of cortisol are usually inadequate to infer the functional status of the HPA axis, given its dynamic nature: the cortisol awakening response, that is, the rise in plasma cortisol occuring 20–30 min after the habitual wake up time, is more informative to that end but is highly variable among individuals. Furthermore, cortisol measurements can be confounded by daytime stressors. Twice hourly measured plasma cortisol levels were continuously lower over the course of 24 h in obese individuals (mean BMI 35.4 kg/m2) in comparison with non-obese men (mean BMI 26.8 kg/m2) [16]. In other studies, 24 h urine free cortisol levels did not correlate with BMI or waist circumference, but cortisol’s degradation products (THM) were positively correlated with these anthropometric measurements [17–19]. Urinary 5β-THE were increased in obesity, indicating that increased 5β reduction of cortisone may be the mechanism in underlying increased cortisol clearance rate in obesity [19]. Thus, in obesity there seems to be increased turn-over (production and catabolism of cortisol) without necessarily altering circulating levels or cortisol (Fig. 1).
Variations in tissue-specific cortisol concentrations can exist without any changes in plasma cortisol levels through varying local 11β-HSD activity. In obese subjects, 11β-HSD1 activity has been reported either as normal [18, 19] or impaired [14], as indicated by the urinary cortisol / cortisone ratio – a rather crude and non-specific reflection, since this ratio can be altered by other cortisol metabolizing enzymes. Hepatic 11β-HSD1 activity is impaired in obesity, as indicated by decreased mRNA levels [17] and deuterated cortisol tracer infusion [19] while the enzymatic activity is selectively increased by two- to threefold in the abdominal subcutaneous adipose tissue (Fig. 1) [18]. These human findings are mirrored by observations in animal models (reviewed in [20•]): mice overexpressing 11β-HSD1 in white adipose tissue have unaltered plasma, but elevated corticosterone levels in the adipose tissue, and develop truncal obesity, whereas mice with liver-specific upregulation of 11β-HSD1 do not exhibit increases in adipose mass. Correspondingly, mice with 11β-HSD1 gene deficiency are less prone to develop obesity [20•]. CBG was lower in single measurements in individuals with higher BMI [21] and average daytime concentrations of CBG were decreased in obese vs. lean subjects [12]. However, in a large survey of 477 healthy subjects there was no relationship between plasma CBG levels and BMI or waist circumference [22•]. Individuals with a heterozygous (GTTT)n repeat in intron 1 of the CBG gene were more sensitive to 0.25 mg dexamethasone suppression [23]. Since CBG is expressed locally in the adipose tissue, decreased local CBG levels could enhance free cortisol exposure without changes in circulating cortisol concentrations [21].
Mean 24 h plasma ACTH levels are positively related to BMI, possibly reflecting increased hypothalamic drive and/or reduced negative feedback of cortisol [16, 24]. Individuals with high BMI are less sensitive to cortisol suppression by 4 mg dexamethasone [15], but not by lower doses of dexamethasone (0.0035 mg/kg and 1 mg) [14, 25]. Another study found lower cortisol suppression in women, but not in men, with 0.0035 mg/kg–0.015 mg/kg or a standard 1 mg dose of dexamethasone, indicating the association between obesity and HPA-suppression could be both dose- and gender-dependent [26]. On the other hand, HPA-stimulation with intravenous ovine CRH [16], ACTH [25] or mental stress tests [25] increased cortisol levels more in obese vs. lean individuals. Thus, the HPA-axis appears hyper-responsive to stress in obese individuals, and yet, less responsive to negative feedback, possibly to prevent free cortisol levels to fall below the normal range.
In addition, the sympathetic nervous system seems to be hyperactive in subjects with central obesity, as indicated by higher nervous discharge from the muscle sympathetic activity [27]. Increased adipokine secretion by abdominal fat tissue, including leptin and free fatty acids, may elevate sympathetic activity centrally, as adipokines are often able to cross the blood-brain-barrier. In turn, chronically elevated SNS activity could impair β-adrenergic signaling and deregulate the HPA-axis, contributing to obesity [27].
HPA-Axis Alterations in Obese Subjects: Cause and Effect?
The dramatic effects of altered circulating cortisol levels on body weight are exemplified by Cushing’s syndrome (hypercortisolism due to intake of glucocorticoid drugs or increased endogenous cortisol, ACTH or CRH levels), and Addison’s disease (hypocortisolism due to chronic adrenal insufficiency) presenting with central obesity and weight loss, respectively. Furthermore, visceral fat deposition increased in primates exposed to chronic physical and psychological stress [28]. In the presence of insulin, cortisol promotes triglyceride accumulation, especially in visceral adipocytes thereby leading to an increase in central fat [29]. Additionally, HPA-axis hyperactivation may lead to adiposity through increases in food intake, as artificially-induced stress, exogenous ovine CRH administration and decreased response to dexamethasone suppression led to increases in appetite, especially for more palatable foods [30]. Functional MRI studies indicate that the sensitivity of the central reward system is diminished by stress, possibly upregulating the craving for “comfort foods” [31]. Interestingly, chronic stress may also lead to weight loss in a subset of individuals, possibly mediated by hyperactivation of β-adrenergic lipolytic pathways or differences in dietary restraint or coping mechanisms [3].
In order to determine the relationship between stress and body weight regulation, the HPA-axis has been examined in obese subjects before and after weight loss. Sixteen weeks of very low caloric diet induced a 29 kg weight loss which was associated with a significant decrease in 11β-HSD1 mRNA expression in adipose tissue [32]. Similarly, a four-fold decrease in 11β-HSD1 mRNA expression was reported in obese women 2 years after gastric-bypass surgery after an average weight loss of 40 kg [33]. In addition, these women had decreased urinary THM and decreased estimated activity of 5α-reducase, indicating that the obesity-associated upregulation of 5α-reductase is an adaptive response to counteract hypercortisolism [33]. Subclinical inflammation in obesity may contribute to alterations of the HPA-axis; for example, obesity is associated with elevated levels of circulating IL-6, which in turn upregulates 11β-HSD1 expression [34]. These findings suggest that the HPA-axis alternations are both cause and effect of obesity and that they are potentially reversible to a point.
Sleep in Obesity
Inadequate sleep has been associated with increased BMI (Fig. 1) [4, 6]. Obese adults (BMI: 41 ± 1 kg/m2) without sleep apnea slept 88 min less than lean subjects, as assessed by polysomnography [35]. In prospective studies, there was a distinct but modest weight gain in short sleepers (less than 5–6 h) who gained 0.4 kg in a 16-year study and 2 kg in a 6-year study [4]. However, individuals with short habitual sleep durations had a 31 % and 35 % greater chance of a clinically significant weight gain (5 kg and 15 kg), suggesting a large inter-individual variability in this association.
Increased appetite may be a mechanism by which short term sleep loss can contribute to obesity. Increase in appetite is usually paralleled by changes in ghrelin and leptin levels, a satiety-inducing hormone secreted from adipocytes and an appetite-stimulating peptide produced in the stomach, respectively. Determining circulating levels of these neuropeptides can therefore serve as an objective measurement to indirectly assess appetite. Short habitual sleep duration, was correlated with lower levels of morning leptin and higher levels of ghrelin, which would presumably lead to increases in appetite via activation of the arcuate nucleus of the hypothalamus and subsequent increases in food intake [36]. In obese individuals however, we found no association between habitual sleep duration and morning leptin levels [37]. Obese subjects are known to display leptin resistance, which may account for these findings. As a word of caution, it should also be noted that the regulation of appetite is complex and redundant involving many neuropeptides influencing appetite, including neuropeptide Y. Short-term sleep deprivation studies can also result in insulin resistance and a pro-inflammatory state with increased plasma levels of cytokines such as IL-6 [4]. Insulin resistance and increased levels of IL-6 are both associated with increased risk of cardiovascular disease and type 2 diabetes [4].
The HPA-Axis and Sleep
HPA-Axis Dynamics and Sleep
A combination of altered HPA-axis function and obesity is often observed in subjects suffering from insomnia, depression or obstructive sleep apnea [10, 16, 38]. A typical 8 h night of sleep typically consists of three to five sleep cycles; every cycle includes non-rapid eye movement (NREM) (stage 1 to 4) and REM sleep [4]. Slow-wave sleep (SWS) encompasses stage 3 and 4 and is most prevalent in the beginning of the night, at the start of the sleep period. Patients suffering from depression often have increased cortisol and ACTH levels, hyperactivation and decreased inhibition of the HPA-axis, while their sleep is often disturbed [10]. Three years after recovery from depression, cortisol levels normalized while sleep alterations persisted [38]. In subjects with insomnia, 24 h plasma cortisol and ACTH and urinary free cortisol levels were all increased [10]. Obstructive sleep apnea is defined as repetitive partial or total cessations of breathing during sleep, inducing frequent micro-awakenings, increases in stage 1 and 2 sleep and reductions in SWS and REM sleep [10]. In obese individuals, morning levels of ACTH and cortisol were positively correlated with the amount of nocturnal apnea events [39•]. Nighttime plasma levels of cortisol are elevated in patients with obstructive sleep apnea vs. weight-matched controls, but the ACTH response to exogenous CRH was increased, suggesting CRH hyposecretion [16]. On the other hand, hypoxia can stimulate ACTH and cortisol levels and treating the sleep apnea with continuous positive airway pressure resulted in normalization of circulating cortisol [16].
Consequences of Inadequate Sleep on the HPA-Axis Function
Sleep onset causes an inhibitory effect on cortisol level, inducing decreased cortisol levels for 1–2 h after sleep onset [10]. This effect was observed when subjects were exposed to a 3-hour sleep-wake cycle or slept during the day. Inhibition of cortisol levels was also observed in “free running” conditions, when individuals have no time cues and sleep is usually initiated at a later phase of the internal circadian cortisol rhythm [10]. In addition, nocturnal awakenings transiently elevate cortisol levels, whereas morning awakening elicits a substantial and more persistent rise in cortisol levels. Slow-wave sleep is correlated with the lowest cortisol levels and lowest cortisol/ACTH response to CRH administration [10].
Salivary and plasma cortisol were elevated in the afternoon and evening following experimental partial and total sleep deprivation, while morning levels were unchanged or even decreased [4]. Mean 24 h plasma cortisol levels measured every 30 min were unchanged [4]. Some studies also report a decreased cortisol peak following sleep deprivation and a slower decline of cortisol levels after its acrophase [4]. In addition, sleep may interfere with the dynamics of the HPA-axis: subjects indicating poor sleep had an exaggerated response to a combined dexamethasone/CRH challenge [40•].
Acute sleep deprivation is a stressor known to activate sympathetic activity, while decreasing the parasympathetic tone, as indicated by increased epinephrine and norepinephrine plasma levels (Fig. 1) [4].
Consequences of HPA Alterations on Sleep
Patients with Cushing’s syndrome often have obstructive sleep apnea, which in turn affects sleep quality [16, 41]. In addition, even patients with ACTH-dependent Cushing’s syndrome without sleep apnea have an 18 % decrease in sleep efficiency (ratio of time asleep and time spent in bed), patients woke up twice as often (6 vs. 3 times) and a 12 % increase in stage 1 sleep compared to normal controls [41]. This demonstrates the direct deleterious effects on sleep of abnormally elevated levels of circulating glucocorticoids. Sleep in patients with Addison’s disease has been less studied but does not present with major sleep disturbances [42, 43]. Upon replacement therapy with hydrocortisone however, REM sleep duration increased in these patients [42].
Intravenous ovine CRH or ACTH administration reduced REM and SWS [43]. Cortisol infusion also reduced REM while increasing SWS, indicating that the HPA-axis effects on REM may be cortisol-mediated, but the increase in SWS in cortisol infusion may be due to negative feedback on CRH [43]. Administration of the synthetic steroid prednisolone reduced REM, but had no effect on SWS [43].
Battling HPA-Axis Alterations and Sleep Loss: Novel Therapies for Obesity?
As both the HPA-axis and sleep alterations appear to influence body weight regulation and composition, targeting them to combat the obesity epidemic appears to be a viable strategy. A recent randomized control weight loss trial reported that overweight or obese African American women with self-reported high levels of stress tended to lose more weight if stress management therapy was added to a 12-week lifestyle program (2.7 vs. 1.4 kg); consistently, morning salivary cortisol tended to decrease to a greater extent in this group [44•]. This small study had limited statistical power with only 22 women per group, but it hints that decreasing overall stress levels may contribute to weight loss.
Novel pharmacological approaches to counteract obesity through manipulations of the HPA-axis have focused on 11β-HSD inhibitors, which in principle would allow tissue-specific alterations of cortisol concentrations, without affecting circulating levels. Carbenoxolone is a non-selective inhibitor of 11β-HSD1 and 11β-HSD2. Some studies using this inhibitor report lower 11β-HSD1 expression in adipose tissue while some do not [19, 20•]. One study reported lower cholesterol levels and reduced glucogenolysis in healthy patients, after 7 days administration of 300 mg carbenoxolone [45]. However, 11β-HSD2 inhibition can lead to cortisol-dependent mineralocorticoid excess, as cortisol can activate the mineralocorticoid receptor in the kidney inducing sodium retention, hypokalaemia and fluid retention.
More selective 11β-HSD1 inhibitors, in theory void of these side effects, have recently been examined in randomized controlled trails. In patients with type 2 diabetes with a BMI between 25 and 45, daily administration of 200 mg INCB13739 for 12 weeks resulted in improved insulin sensitivity and induced a larger decrease in body weight (0.9 vs. 0.2 kg in the placebo group) [46•]. Similarly, daily ingestion of 6 mg MK-0916 and 7 mg MK-0736 for 12 weeks resulted in greater weight loss in patients with a BMI over 27 kg/m2 (1.8 and 1.4 kg more vs. placebo, respectively) [47•]. In addition, MK-0736 improved lipid profile: low- and high-density lipoprotein-cholesterol were reduced by 12 % and 6 %, respectively. As MK-0916 is a CYP3A4 inducer, MK-0736 may be the preferred drug of use. In all three selective 11β-HSD1 inhibitors treatment groups, concentrations of plasma androgen precursors were higher vs. the placebo group, but still within the normal range, suggesting mild HPA-axis hyperactivity. There was no difference in reported adverse effects in treatment vs. placebo groups [46•, 47•]. In 2008–2010, there were over 70 patent submissions for 11-βHSD1 inhibitors [48]. Virtually, most major and several minor pharmaceutical companies are targeting these inhibitors [48]. In addition, there is a magnitude of pre-clinical studies examining other selective 11β-HSD1 inhibitors [20•].
Extending sleep duration in chronically sleep-deprived subjects is a challenging task. A small observational study reported that short sleepers (less than 6 h per night; mean BMI 27 kg/m2) who reported having extended their sleep to 7–8 h 6 years later increased one point in BMI, whereas individuals who reported still sleeping less than 6 h per night increased their BMI by 2.1 points [49]. This study was not randomized and assessed sleep duration only at two time-points and with a questionnaire, a subjective measurement. We are currently conducting a large randomized controlled trail (ClinicalTrials.gov, identifier: NCT00261898) to examine the effect of sleep extension in a cohort of short-sleeping obese individuals [50•].
Conclusions
Subjects with obesity display alterations in both the HPA-axis and sleep. Although circulating cortisol levels are often unaltered in obese individuals, the HPA-axis appears to be hyperactive, as there are tissue-specific changes in cortisol concentrations due to differential expression of 11β-HSDs. Short habitual sleep duration and experimental sleep restriction result in bodily changes that may lead to obesity, possibly through increased appetite and food intake, but insufficient sleep also disrupts the HPA-axis. Nevertheless, studies examining the dynamics of the HPA-axis in this condition are scarce, and there is no direct information of the specific correlations of, e.g., 11β-HSD activity, circulating CBG levels and sleep, to the best of our knowledge. In turn, the activation of the HPA-axis may negatively affect sleep. Although an increased prevalence of sleep apnea or increased stress/HPA-activation is reported direct evidence is lacking for the possibility that obesity leads to worsened sleep.
As sleep loss and the HPA-axis appear to be involved in the pathogenesis of obesity, approaches aimed at modulating these systems seem viable. The generation of tissue-specific 11β-HSD1 inhibitors is especially promising, as this may effectively target cortisol in selected tissues without affecting circulating cortisol levels, therefore having limited unwanted side effects. The appeal of a new class of drugs based on the mechanism summarized above is reflected by the magnitude of ongoing research. Clinical studies focusing at reducing overall stress levels or improving sleep hygiene by increasing sleep duration are currently being conducted.
In conclusion, obesity, sleep, and the HPA-axis appear to be related in a deleterious cycle with many complex interactions, including the immune system and appetite-regulation. Additional research in this field is needed to understand the alterations in these systems and to eventually combat obesity.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence and trends in obesity in the distribution of body mass index among US adults, 1999–2010. JAMA. 2012;307:491–7.
National sleep foundation, sleep in America poll. Washington DC; 2005.
Pasquali R: The hypothalamic-pituitary-adrenal axis and sex hormones in chronic stress and obesity: pathophysiological and clinical aspects. Ann N Y Acad Sci 2012;1264:20–35.
Lucassen EA, Rother KI, Cizza G: Interacting epidemics? Sleep curtailment, insulin resistance and obesity. Ann N Y Acad Sci 2012;1264:110–34.
Wardle J, Chida Y, Gibson EL, et al. Stress and adiposity: a meta-analysis of longitudinal studies. Obesity (Silver Spring). 2011;19:771–8.
Cappuccio FP, Taggart FM, Kandala NB, et al. Meta-analysis of short sleep duration and obesity in children and adults. Sleep. 2008;31:619–26.
Vgontzas AN, Lin HM, Papaliaga M, et al. Short sleep duration and obesity: the role of emotional stress and sleep disturbances. Int J Obes (Lond). 2008;32:801–9.
McEwen BS. Central effects of stress hormones in health and disease: understanding the protective and damaging effects of stress and stress mediators. Eur J Pharmacol. 2008;583:174–85.
Chrousos GP, Gold PW. The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA. 1992;267:1244–52.
Balbo M, Leproult R, van Cauter E. Impact of sleep and its disturbances on hypothalamo-pituitary-adrenal axis activity. Int J Endocrinol. 2010;2010:759234.
Cizza G, Rother KI. Cortisol binding globulin: more than just a carrier? J Clin Endocrinol Metab. 2012;97:77–80.
Lewis JG, Möpert B, Shand BI, et al. Plasma variation of corticosteroid-binding globulin and sex hormone-binding globulin. Horm Metab Res. 2006;38:241–5.
Putignano P, Dubini A, Toja P, et al. Salivary cortisol measurement in normal-weight, obese and anorexic women: comparison with plasma cortisol. Eur J Endocrinol. 2001;145:165–71.
Rask E, Olsson T, Söderberg S, et al. Tissue-specific dysregulation of cortisol metabolism in human obesity. J Clin Endocrinol Metab. 2001;86:1418–21.
Rutters F, Nieuwenhuizen AG, Lemmens SG, et al. Hypothalamo-pituitary-adrenal (HPA) axis functioning in relation to body fat distribution. Clin Endocrinol (Oxf). 2010;72:738–43.
Vgontzas AN, Pejovic S, Zoumakis E, et al. Hypothalamic-pituitary-adrenal axis activity in obese men with and without sleep apnea: effects of continuous positive airway pressure therapy. J Clin Endocrinol Metab. 2007;92:4199–207.
Baudrant R, Campino C, Carvajal CA, et al. Increased urinary glucocorticoid metabolites are associated with metabolic syndrome, hypoadectinemia, insulin resistance and β cell dysfunction. Steroids. 2011;76:1575–81.
Fraser R, Ingram MC, Anderson NH, et al. Cortisol effects on body mass, blood pressure, and cholesterol in the general population. Hypertension. 1999;33:1364–8.
Sandeep TC, Andrew R, Homer NZ, et al. Increased in vivo regeneration of cortisol in adipose tissue in human obesity and effects of the 11beta-hydroxysteroid dehydrogenase type 1 inhibitor carbenoxolone. Diabetes. 2005;54:872–9.
• Pereira CD, Azevedo I, Monteiro R, Martins MJ: 11β-Hydroxysteroid dehydrogenase type 1: relevance of its modulation in the pathophysiology of obesity, the metabolic syndrome and type 2 diabetes mellitus. Diabetes Obes Metab 2012. doi:10.1111/j.1463-1326.2012.01582.x. Excellent review that combines basic research focusing on physiology of 11β-HSD enzymes and the clinical practice of 11β-HSD1 inhibitors.
Fernandez-Real JM, Pugeat M, Grasa M, et al. Serum corticosteroid-binding globulin concentration and insulin resistance syndrome: a population study. J Clin Endocrinol Metab. 2002;87:4686–90.
• Cizza G, Bernardi L, Smirne N, et al. Clinical manifestations of highly prevalent corticosteroid-binding globulin mutations in a village in southern Italy. J Clin Endocrinol Metab. 2011;96:E1684–93. This study describes novel findings of associations of CBG mutations and responses to stress, and has the potential to help unravel mechanisms in pain/fatigue syndromes that are currently not well understood.
Barat P, Duclos M, Gatta B, et al. Corticosteroid binding globulin gene polymorphism influences cortisol driven fat distribution in obese women. Obes Res. 2005;13:1485–90.
Veldhuis JD, Roelfsema F, Iranmanesh A, et al. Basal, pulsatile, entropic (patterned), and spiky (staccato-like) properties of ACTH secretion: impact of age, gender and body mass index. J Clin Endocrinol Metab. 2009;94:4045–52.
Mårin P, Darin N, Amemiya T, et al. Cortisol secretion in relation to body fat distribution in obese premenopausal women. Metabolism. 1992;41:882–6.
Pasquali R, Ambrosi B, Armanini D, et al. Cortisol and ACTH response to oral dexamethasone in obesity and effects of sex, body fat distribution, and dexamethasone concentrations: a dose-response study. J Clin Endocrinol Metab. 2002;87:166–75.
Smith MM, Minson CT. Obesity and adipokines: effects on sympathetic overactivity. J Physiol. 2012;590:1787–801.
Shively CA, Register TC, Clarkson TB. Social stress, visceral obesity, and coronary artery atherosclerosis: product of a primate adaptation. Am J Primatol. 2009;71:742–51.
Björntorp P. Hormonal control of regional fat distribution. Hum Reprod. 1997;12:21–5.
Cizza G, Rother KI. Was Feuerbach right: are we what we eat? J Clin Invest. 2011;121:2969–71.
Duong M, Cohen JI, Convit A. High cortisol levels are associated with low quality food choice in type 2 diabetes. Endocrine. 2012;41:76–81.
Karlsson EA, Sheridan PA, Beck MA. Differences in associations between HSD11B1 gene expression and metabolic parameters in subjects with and without impaired glucose homeostasis. J Nutr. 2010;140:1691–7.
Rask E, Simonyte K, Lönn L, Axelson M: Cortisol metabolism after weight loss- associations with 11β-HSD type 1 and markers of obesity in women. Clin Endocrinol (Oxf) 2012. doi:10.1111/j.1365-2265.2012.04333.x.
Black PH. The inflammatory consequences of psychologic stress: relationship to insulin resistance, obesity, atherosclerosis and diabetes mellitus, type II. Med Hypotheses. 2006;67:879–91.
Rasmussen MH, Wildschiodtz G, Juul A, Hilsted J. Polysomnographic sleep, growth hormone, insulin-like growth factor-I axis, leptin, and weight loss. Obesity. 2008;16:1516–21.
Taheri S, Lin L, Austin D, et al. Short sleep duration is associated with reduced leptin, elevated ghrelin, and increased body mass index. PLoS Med. 2004;1:e62.
Knutson KL, Galli G, Zhao X, et al. No association between leptin levels and sleep duration or quality in obese adults. Obesity. 2011;19:2433–5.
Kupfer DJ, Ehlers CL, Frank E, et al. Electroencephalographic sleep studies in depressed patients during long-term recovery. Psychiatry Res. 1993;49:121–38.
• de Jonge L, Zhao X, Mattingly M, et al.: Poor sleep quality and sleep apnea are associated with higher resting energy expenditure in obese individuals with short sleep duration. J Clin Endocrinol Metab 2012;97:2881–89. Since most studies are conducted in healthy volunteers, there is a paucity of studies in obese individuals. This study, examining the association between multiple aspects of metabolism and sleep apnea/quality in a large number of obese subjects studied in real life conditions is therefore of great importance.
• Hori H, Teraishi T, Sasayama D, et al. Poor sleep is associated with exaggerated cortisol response to the combined dexamethasone/CRH test in a non-clinical population. J Psychiatr Res. 2011;45:1257–63. This study focuses on the association between alteration of HPA-dynamics and sleep curtailment. Studies focusing on HPA-axis functioning in relation to sleep in detail are lacking; thus, this article may give more insight in the mechanisms of detrimental effects on metabolism of sleep loss.
Shipley JE, Schteingart DE, Tandon R, Starkman MN. Sleep architecture and sleep apnea in patients with Cushing’s disease. Sleep. 1992;15:514–8.
García-Borreguero D, Wehr TA, Larrosa O, et al. Glucocorticoid replacement is permissive for rapid eye movement sleep and sleep consolidation in patients with adrenal insufficiency. J Clin Endocrinol Metab. 2000;85:4201–6.
Steiger A. Neurochemical regulation of sleep. J Psychiatr Res. 2007;41:537–52.
• Cox TL, Krukowski R, Love SJ, et al.: stress management-augmented behavioral weight loss intervention for african american women: a pilot, randomized controlled trial. Health Educ Behav 2012. doi:10.1177/1090198112439411. Although this is a small study and results did therefore not reach statistical significance, it suggests that addition of stress management therapy can enhance weight loss in obese women. If these results can be confirmed in larger trials, this may have practical implications.
Andrews RC, Roovackers O, Walker BR. Effects of the 11 beta-hydroxysteroid dehydrogenase inhibitor carbenoxolone on insulin sensitivity in men with type 2 diabetes. J Clin Endocrinol Metab. 2003;88:285–91.
• Rosenstock J, Banarer S, Fonseca VA. The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care. 2010;33:1516–22. This is a promising 11β-HSD1 inhibitor study, showing beneficial effects on multiple aspects of metabolism with limited side effects.
• Shah S, Hermanowski-Vosatka A, Gibson K, et al. Efficacy and safety of the selective 11b-HSD-1 inhibitors MK-0736 and MK-0916 in overweight and obese patients with hypertension. J Am Soc Hypertens. 2011;5:166–76. Another promising 11β-HSD1 inhibitor study, more focused on improving metabolic profile of obese individuals.
Carpino PA, Goodwin B. Diabetes area participation analysis: a review of companies and targets described in the 2008–2010 patent literature. Expert Opin Ther Patents. 2010;20:1627–51.
Chaput JP, Despres JP, Bouchard C, Tremblay A. Longer sleep duration associates with lower adiposity gain in adult short sleepers. Int J Obes (Lond). 2012;36:752–6.
• Cizza G, Marincola P, Mattingly M, et al. Treatment of obesity with extension of sleep duration: a randomized, prospective, controlled trial. Clinical Trials. 2010;7:274–85. This trial will give us useful clinical insight in the possibility of battling obesity through sleep extension. It could be a step toward implementing individualized sleep extension advice for metabolic abnormalities in the clinical practice.
Acknowledgments
This study was fully supported by the National Institutes of Health (NIH), Intramural Research Program: National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). We would like to thank Dr. Gyorgy Csako for useful suggestions upon reviewing this article.
E.A. Lucassen has received grant support from Fulbright scholarship, VSBfonds scholarship, Leiden University Fund scholarship. G. Cizza has received grant support from ZIA DK047054-04 Chronic sleep deprivation as a risk factor for endocrine and immune changes.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lucassen, E.A., Cizza, G. The Hypothalamic-Pituitary-Adrenal Axis, Obesity, and Chronic Stress Exposure: Sleep and the HPA Axis in Obesity. Curr Obes Rep 1, 208–215 (2012). https://doi.org/10.1007/s13679-012-0028-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13679-012-0028-5