
Abstract
Kasumi-1 has played an important role in an experimental model with t(8;21) translocation, which is a representative example of leukemia cell lines. However, previous studies using Kasumi-1 show discrepancies in the genome profile. The wide use of leukemia cell lines is limited to lines that are well-characterized. The use of additional cell lines extends research to various types of leukemia, and to further explore leukemia pathogenesis, which can be achieved by uncovering the fundamental features of each cell line with accurate data. In this study, ten Kasumi cell lines established in Japan, including five that were previously unknown, have been characterized by SNP microarray and targeted sequencing. SNP genotyping suggested that the genetic ancestry in four of the ten Kasumi cell lines was not classified as Japanese but covered several different east-Asian ethnicities, suggesting that patients in Japan are genetically diverse. TP53 mutations were detected in two cell lines with complex array profiles, indicating chromosomal instability (CIN). A quantitative assessment of tumor genomes at the chromosomal level was newly introduced to reveal total DNA sizes and Scales of Genomic Alterations (SGA) for each cell line. Kasumi-1 and 6 derived from relapsed phases demonstrated high levels of SGA, implying that the level of SGA would reflect on the tumor progression and could serve as an index of CIN. Our results extend the leukemia cellular resources with an additional five cell lines and provide reference genome data with ethnic identities for the ten Kasumi cell lines.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Leukemia genomes are primarily characterized by abnormal karyotypes, which often include the formation of a fusion gene [1]. Chromosomal translocations are responsible for leukemia initiation, but are usually not sufficient for further development of the condition [2]. Chromosome instability (CIN) is linked to tumor progression [3] and causes genomic diversity [4]. In addition, recurrent hotspot mutations have been identified in association with leukemogenesis [5]. Both large-scale changes at chromosome level and nucleotide changes at sequence level contribute to a diverse array of leukemia genomes, reflecting a broad range of subtypes [6, 7]. While sequence analysis provides data at the highest resolution, CIN is mostly based on qualitative data. Quantitative analysis of large-scale genomic alterations is required to assess CIN status and would improve the precise disease classification.
Kasumi cell lines have been established at Hiroshima University from patients with leukemia in Japan since 1989. They were named after the location of the laboratory in the Kasumi area of Hiroshima city [8] (Table 1). The aim for establishing these cell lines was to provide a useful research model harboring leukemia-specific chromosomal/gene abnormalities. A fusion gene, RUNX1-RUNX1T1, is well recognized as an important diagnostic and prognostic marker frequently observed in acute myeloid leukemia, according to the French–American–British classification M2 subtype (AML-M2), which was first identified in the Kasumi-1 cell line [9, 10]. Kasumi-1, 2, 3, 4 and 6 had been deposited in three major cell banks, ATCC, DSMZ and JCRB, directly from a laboratory which established the lines. An additional five cell lines have been listed only in the JCRB cell bank (Table 1). Although two comprehensive databases of cancer cell lines, COSMIC and CCLE, are available from the Sanger Institute and the Broad Institute, respectively, information about leukemia cell lines is limited to certain commonly-used cell lines. Kasumi-1 appears in both COSMIC and CCLE, and the latter also includes Kasumi-2 and 6. However, other Kasumi cell lines have not been fully exploited because their cells are poorly characterized.
Cell lines have attractive features, namely their ‘continuous’ or ‘immortalized’ abilities. This enables us to use the same cellular material across different laboratories and allows us to compare results using cell lines. However, three previous studies which analyzed Kasumi-1 by DNA microarray showed discordance in the genome profiles [15,16,17] (Table S1). Analysis of MCF7 strains revealed genetic evolution of cancer cell lines during cell culture [18]. This implies that tumor genomes change during in vitro cell culture, which can be explained by an in vitro clonal evolution model [19]. Analysis of cell lines obtained from a public registry provides results which ensure reproducibility, leading to an accurate reference.
A panel of 100 leukemia and lymphoma cell lines, LL-100, has been reported [20]; however, most of them, 80 of the 100 cell lines, are included in the CCLE and/or COSMIC databases. In addition, all the cell lines have already appeared in publications and no novel cell lines are introduced in the panel. Because the panel does not adequately cover various types of leukemia, additional cell lines are required for further investigation of the underlying molecular mechanisms in leukemogenesis. We performed SNP array and sequence analyses in ten Kasumi cell lines to obtain their genome reference data. Changes at chromosome level in tumor genomes were assessed by measurements of gains, losses and uniparental disomy (UPD), shown as Scales of Genomic Alterations (SGA). Amplicon sequencing detected pathogenic mutations and candidate mutations with their allele frequencies. An RNA sequencing panel identified fusion genes in five cell lines, with accompanying expression levels. Our study demonstrates quantitative assessment of leukemia genomes and adds ethnic information on each cell line.
Materials and methods
Cell lines, cell culture and DNA extraction
Kasumi-1–10 cell lines have been registered with the JCRB cell bank (Table 1) and are available for distribution upon request. When the cells were defrosted, the cells were cultured at a higher concentration, and the culture disk or flask was slanted to achieve a high cell density at the lower end before cells became stable. In addition, increasing the heat-inactivated FBS concentration to 20% in the medium could help to promote growth after defrosting. Because of a lot-to-lot variation in FBS, it has been carefully selected by evaluating a range of different lots (Table S2). It is noted that GM-CSF is required for cultivation of Kasumi-4 and 6. Two normal fibroblast cell lines, SF-TY (JCRB0075) and TIG-7–20 (JCRB0511), were used as controls during sequence variant analysis. Culture conditions and data sets from the standard quality control for each cell line are available in the JCRB Cell Bank website at https://cellbank.nibiohn.go.jp/english. Genomic DNA was extracted from cultured cells using the AllPrep DNA/RNA Mini Kit (Qiagen 80204).
SNP microarray
DNA copy number and genotyping were examined by microarray using a high density chip, CytoScan HD array (Thermo Fisher Scientific). The data analysis was performed based on the GRCh37 (hg19) reference using the Chromosome Analysis Suite software, ChAS 4.0 (Thermo Fisher Scientific). To measure genomic changes at chromosomal level, regions of gains, losses and uniparental disomy (UPD) larger than 1 Mb were counted. Compared with normal diploid male and female DNA sizes of 5977 Mb and 6073 Mb respectively, from the human genome reference hg19, nuclear DNA size of each cell line was estimated from the difference between gains and losses. The sum of sizes from gains, losses and UPD were calculated as the Scale of Genomic Alterations (SGA).
Amplicon sequencing
Mutation analysis by target sequencing was conducted using a multiplex panel, Oncomine™ Myeloid Research Assay (Thermo Fisher Scientific, A36941), consisting of 40 DNA genes analyzed by 526 amplicons and 29 RNA genes representing 700 fusion isoforms found in major myeloid disorders. An On-Demand panel, IAD178152, consisting of 6236 amplicons was designed and applied to AML cell lines and the two normal cell lines. This custom panel covered 286 leukemia-related genes and extended to 1.19 Mb in total (Table S2). Ethnic origin of each cell line was assessed by the Precision ID Ancestry Panel (Thermo Fisher Scientific, A25642). Sequence libraries and templates were prepared using the Ion AmpliSeq Kit for Chef DL8 (Thermo Fisher, A29024) and the Ion PGM Hi-Q View Chef Kit (Thermo Fisher Scientific, A29902), respectively. Sequencing was run on the Ion PGM using the Ion 318 Chip v2 BC (Thermo Fisher Scientific, 4488150). Reads were aligned to the hg19 reference and the analysis was carried out using the Ion Torrent Suite and the Ion Reporter (Thermo Fisher Scientific). To eliminate SNPs from variants, variants specific for each Kasumi cell line were extracted by comparisons with the two normal cell lines. Filter settings used for variant analysis of sequence data from the custom panel are given in Table S3. To obtain biogeographic ancestry information, sequence data from the ID panel were analyzed using the Ion Torrent HID SNP Genotyper Plugin (Thermo Fisher Scientific).
Flow cytometric immunophenotyping
Cells were washed and incubated for 20 min at 4 °C with antibodies and their isotype controls are listed in Table S4. The cells were washed with 5% FBS/PBS twice and resuspended in 4% PFA, and analyzed on a flow cytometer, FACSCanto using the FlowJo software (BD Biosciences).
Results
Whole genome profiles
SNP microarray profiles of each cell line are shown in Fig. S1 and described in Table S5. Microarray data, CHP and CEL files, have been submitted to the NCBI repository databases under the BioProject ID PRJNA598005. Copy number alterations were identified in nine of the ten cell lines, with the exception of Kasumi-4. Although the array profile of Kasumi-4 represented an apparently normal female, a translocation, t(9;11;22), which forms the BCR-ABL1 fusion has been reported in Kasumi-4. Kasumi-10 has been established from a female infant with B-cell precursor acute lymphoblastic leukemia (BCP-ALL), which does not show detectable copy number changes in autosomes, but has a balanced translocation, t(11;19)(q23;p13.3). Due to a limitation of this technique [21], array profiles show DNA gross changes, but miss balanced translocations. There was in discordance with the sex chromosomes of Kasumi-10 which showed XXY, which could be explained by disorders of sexual development [22]. Kasumi-1 from a male patient had one copy of X chromosome without a corresponding Y chromosome, consistent with the clinical data, indicating that the loss occurred before cell culture.
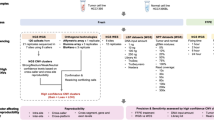
Gains and losses were observed in 6 and 8 cell lines, respectively (Table 2, S6). Trisomy 10 was observed in Kasumi-1 and 6, demonstrating a characteristic feature of AML-M2 [23]. Loss of heterozygosity (LOH) resulting in one copy of alleles did not occur in a whole chromosome but in partial regions. DNA size compared with normal diploid was calculated from 98.4 to 104.7% for the three AML cell lines and from 98.8 to 99.7% for the five BCP-ALL cell lines (Fig. 1a). The largest difference among the 10 cell lines was found in Kasumi-6, which increased its DNA size to 280.2 Mb. SNP profiles revealed that UPD, equivalent to copy neutral LOH consisting of homozygous DNA copies, occurred in 7 cell lines, shown as ‘hmz’ in Table S5. Each case was detected in only parts of chromosomes, which were distinct between samples. The scale of regions involving DNA changes calculated as Scale of Genomic Alterations (SGA) showed that three AML cell lines (Kasumi-1, 3, and 6) had larger sizes of altered regions than ALL cell lines.

Assessment of tumor genomes. a Gross changes of nuclear DNA in each Kasumi cell line are calculated from SNP microarray profiles. Kasumi-4 and 10 have no gross changes. Kasumi-1 and 6 increase the DNA size and others reduce within 100 Mb. b Overall genomic changes are shown by Scale of Genomic Alterations. Although the changes in DNA amounts are apparently small, cell lines with TP53 mutations indicated in red demonstrate high SGA
Because microarray analysis is performed using genomic DNA samples extracted from an admixture of cells, a non-integer copy number appears in the profiles when the cell population is heterogeneous. This was observed at 9q and 18q in Kasumi-9 (Figure S1), which showed copy numbers of 1.3 and indicated mosaic losses, reflecting heterogeneous cell populations.
Cryptic homozygous deletion measured between 300 and 1200 kb at the CDKN2A/p16 locus (9p21.3) was observed in three ALL cell lines, which involved LOH across the boundary of the deletion (Figure S2A-C). Additional losses were detected in other 9p regions, which were different between the three samples. Kasumi-9 had two additional homozygous deletions at 9p23 and 9p21.3 which extended to 5 Mb and 1 Mb, respectively, implying the occurrence of complex rearrangements on its chromosome 9.
Mutational signatures
Sequence variants detected by the Oncomine™ myeloid panel are listed in Table S7. Of them, hotspot mutations were identified in 7 cell lines (Table 3, S8). TP53 mutations were detected in Kasumi-1 and 6, both of them exhibited 17p LOH in the array profiles (Figure S2D), resulting in a 100% allele frequency (Table S8). FMS-like tyrosine kinase 3 internal tandem duplication (FLT3-ITD) was detected in Kasumi-6 and -10, derived from elderly AML and infant ALL patients, respectively. Kasumi-1 has been used as a KIT mutant AML model [24], which was detected at 4q12 with 79.3% variant frequency under 4 copies, indicating duplications of the mutated allele. Additional analysis of six ALL cell lines were done using a custom panel provided variant lists (Table S9) and candidate pathogenic mutations (Table S10). Of them, variants detected in driver genes reported in ALL are shown in Table 3. A low frequency of variants can arise during cell culture. A missense mutation in NSD1 was detected in all of the six cell lines at about a 1:3 frequency ratio, which would be explained by a nucleotide change after the gene duplication.
Detection of fusion genes
Fusion transcripts were detected in five cell lines (Table 3, S11). A very high level of TCF3-PBX1 expression was observed in Kasumi-2, compared with other fusions. NUP98-RAP1GDS1 has been identified in Kasumi-5, but it was not covered as a target in the panel. Although the panel is limited to major fusion genes, these chimeras were undetected in Kasumi-3, 6, 7 and 9, which tested negative for those fusions.
Ethnic background
Analysis of SNP associated with population groups from which the cell lines originated revealed that six of them were classified into the Japanese population and two showed association between Korean and Japanese (Table 3). The other two were related to Han Chinese populations with Korean or Taiwanese. Although the four cell lines have mixed genetic backgrounds from different ancestral populations, pedigree information is not available for these cell lines and the generation of each individual is not clear. Our data show that all donors for the 10 cell lines belong to the east-Asian population. Sequence variations detected by the Precision ID Ancestry Panel are listed in Table S12.
Cell surface markers
Flow cytometric histograms of 20 cell surface markers are shown in Figure S3. Positive cells were calculated from the histograms and listed as a percentage in Table S13. CD33 positive cells appeared in Kasumi-1, 3, 4 and 6, distinguishing them from the other 6 lymphoid cell lines. In contrast to the 4 myeloid cell lines, five BCP-ALL cell lines, Kasumi-2, 7, 8, 9 and 10, were positive for CD19. Although CD22 and CD38 are B-cell markers, these were detected in an AML cell line, Kasumi-3. CD13 expression was reported in the original leukemia cells of Kasumi-1 [8] but was not detected in this study. CD34 is a marker expressed in hematopoietic stem cells, which was present in Kasumi-1 and 3. Because Kasumi-4, diagnosed as chronic myelogenous leukemia blast-crisis (CML-BC), was positive for the CD13 and CD33 characteristics of AML, it could be classified with the AML cells. It is noted that a NK marker, CD56, was detected in Kasumi-3 and 4. CD3 is known as a diagnostic marker for T-cell acute lymphoblastic leukemia (T-ALL), but was absent in Kasumi-5. Kasumi-5 is unique in that it expresses a B-cell marker, CD10, which had been detected during clinical examination. This would be associated with NUP98-RAP1GDS1 [25]. Kasumi-8, 9 and 10 showed a typical expression pattern for BCP-ALL.
Expression of HLA-DR was observed in Kasumi-1, 3 and 6 when established [9, 13, 15], but was weak or negative in our data. HLA-DR is normally expressed in BCP-ALL, and very low levels of expression in Kasumi-2 and -7 indicated the presence of positive cells in subclonal populations. These data imply that loss of HLA-DR expression would occur during cell culture.
Discussion
A series of ten Kasumi cell lines have been evaluated by SNP microarray, each having a unique genome profile. Because gross DNA changes could affect gene expression [26], our quantitative assessment of large-scale genomic changes at chromosome level provides fundamental features for each cell line. DNA size compared to normal genomes can be determined by the differences between gains and losses, corresponding to a DNA index analyzed by a flow cytometer. Although DNA index is an efficient method to determine ploidy changes in tumor cells, chromosome rearrangements involving both gains and losses cannot be accurately shown by the balance of DNA amounts. To assess CIN, we calculated the total of the altered regions and overall size (Table 2). SGA from microarray data would reflect on the amount of chromosome rearrangements and serve as an approach for the quantitation of CIN.
Complex cytogenetic profiles are strongly associated with TP53 mutations [27], which cause CIN [28] and lead to poor prognosis in leukemia [29]. TP53 mutations occur during the later development of leukemic cells and promote disease progression [30]. Although TP53 alterations can be specified by DNA sequencing, CIN is described qualitatively. Among the 10 cell lines, a high level of SGA, more than 10% of total DNA, is marked in Kasumi-1 and 6 (Fig. 1b), which were established from samples taken when the patients relapsed [8, 14]. These two cell lines exhibit TP53 pathogenic mutations, implying that the extent of tumor progression accompanied with TP53 deficiencies could be estimated from SGA. This will be examined in clinical cases at different phases and SGA could be applied to other cancer types.
Analysis of clinical samples by whole-genome or whole-exome sequencing reveals that AML genomes have the fewest mutations compared with other adult cancers [28]. A mutation in CEBPA, which is not included in the Oncomine™ Myeloid Research Assay, has been reported in Kasumi-6 [14], but it is one of the five genes often mutated in AML [28]. GATA-2 mutation was detected in Kasumi-4, derived from childhood CML in a patient without Down syndrome. It is suggested that mutations in GATA-2 are involved in acute myeloid transformation in CML [31], which could be applicable to Kasumi-4 characterized by CD13 and CD33 expression. Kasumi-4 could be a representative model to explore a possible role of GATA-2 mutation and MECOM activation in leukemogenesis. FLT3 mutations are often found in AML using clinical samples [32] and cell lines [33], but FLT3-ITD was identified in a childhood BCP-ALL cell line, Kasumi-10, which would be a distinctive feature. Because there are few cell lines accompanied with normal samples as tumor-normal pairs, this study employed normal cell lines as controls in the sequence data analysis using a custom panel. However, several variants have been detected, corresponding to differences in the hg19 reference, which cannot be clearly distinguished between SNPs and pathogenic variants. In addition, mutations which occurred during in vitro cell culture have not been identified and excluded. Our variant data can provide candidate causative genes, which would be assessed by future studies using clinical samples.
Most fusion genes in leukemia are repeatedly found [34, 35], allowing targeted sequencing to detect fusions at high rates. Alternative fusion isoforms can be resolved by targeted RNA sequencing with the expression levels [36]. Kasumi-10 has two isoforms of KMT2A-MLLT1 with similar expression levels. Targeted sequencing could work as a first screening of major fusion genes, and minor or novel chimeras could be discovered by whole RNA sequencing. NUP98-RAP1GDS1 is a recurrent fusion gene in T-ALL [25], which has not been identified in cell lines [20, 37], suggesting that Kasumi-5 is a unique model of T-ALL in this fusion.
Homozygous deletion of both p16 and p15 genes at 9p21 are frequently observed in T-ALL, but few mutations were identified in these genes [38]. Analysis of T-ALL reported that p16 inactivation was caused not only by deletions but also by methylation [39]. SNP microarray in Kasumi cell lines revealed that deletions involving p16 varied between samples with additional changes in other 9p regions (Figure S2). Because all four cell lines with 9p changes have p16 alteration, instability of 9p regions would be associated with rearrangements at the p16 locus.
Diversity is found not only in leukemia genomes but also in human populations. It is reported that sensitivity to chemotherapy is sometimes different between ethnic groups [40]. SNP analysis allows us to identify ethnic origin, which has been introduced in forensic and anthropological sciences. This has been applied to cancer cell lines, which classified them into six ancestral origins [41, 42]. Kasumi cell lines have been established from patients admitted to a hospital in Japan. Such cell lines are usually described as Japanese-derived. However, analysis of genetic ancestry may reveal different ethnic origin. Ancestry information which reflects on genetic background could be taken into account to provide precise treatment for each patient.
In contrast to solid tumors, hematopoietic samples are readily available from bone marrow or peripheral blood, allowing diagnosis to be made by direct examination of leukemic cells. Karyotyping can be achieved by a short-term cell culture of these samples, which led to the discovery of disease-specific translocations such as BCR-ABL1 in CML [43] and KMT2A-MLLT3 in ALL [44]. Experimental research on leukemia has been also undertaken using clinical samples obtained from patients. However, a large amount of samples from the same individual are not easily obtained for further analysis. Because Kasumi cells were derived from leukemia patients with distinct features, each Kasumi cell line has the potential to be an in vitro leukemia model.
Cancer cells frequently undergo genomic changes through proliferation, known as clonal evolution, resulting in intra-tumor heterogeneity [45]. As this is observed not only in vivo but also in vitro, tumor cell lines under the same name are not always identical because of changes during cell culture. Clonal evolution of tumor cells in vitro is different from that in in vivo in terms of limited space, and causes the replacement of cell populations during serial passage [19]. This process is specific to the in vitro procedure of subculture by dilutions of cells, which is unavoidable in their growth in culture dishes. Cell lines well-characterized by reproducible data can be used as references in comparison with other samples, leading to an accumulation of additional data and a robust experimental resource. Characterization of cell lines obtained from a public cell bank would assure high reproducibility. Our results provide fundamental genomic profiles, and can serve as reference data for the Kasumi cell lines.
Leukemia cell lines play a key role in drug development [46]; however, there has been a decrease in the establishment of new cell lines since the 1990s [47]. Differing from the LL-100 panel which provides a catalogue comprising of commonly-used cell lines [20], we present the genome profiles of 10 Kasumi cell lines, including five newly characterized ALL cell lines. Kasumi cell lines can be useful models characterized by mutations or fusion genes; c-Kit mutation (Kasumi-1), MECOM mutation/activation (Kasumi-3, 4 and 8), FLT3-ITD (Kasumi-6 and 10), GATA-2 mutation (Kasumi-4), C/EBPα mutation (Kasumi-6), NUP98-RAP1GDS1 fusion (Kasumi-5), and other well-known fusion genes (Kasumi-1, 2 and 10). Our panel of Kasumi cell lines promises to serve as a unique resource, which helps to develop a novel molecular target therapy.
References
Rowley JD. Chromosome translocations: dangerous liaisons revisited. Nat Rev Cancer. 2001;1:245–50. https://doi.org/10.1038/35106108.
Greaves MF, Wiemels J. Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer. 2003;3:639–49.
Geigl JB, Obenauf AC, Schwarzbraun T, Speicher MR. Defining 'chromosomal instability'. Trends Genet. 2008;24:64–9. https://doi.org/10.1016/j.tig.2007.11.006.
Krem MM, Press OW, Horwitz MS, Tidwell T. Mechanisms and clinical applications of chromosomal instability in lymphoid malignancy. Br J Haematol. 2015;171:13–28. https://doi.org/10.1111/bjh.13507.
Naoe T, Kiyoi H. Gene mutations of acute myeloid leukemia in the genome era. Int J Hematol. 2013;97:165–74. https://doi.org/10.1007/s12185-013-1257-4.
Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21. https://doi.org/10.1056/NEJMoa1516192.
Gu Z, Churchman ML, Roberts KG, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51:296–307. https://doi.org/10.1038/s41588-018-0315-5.
Asou H, Tashiro S, Hamamoto K, Otsuji A, Kita K, Kamada N. Establishment of a human acute myeloid leukemia cell line (Kasumi-1) with 8;21 chromosome translocation. Blood. 1991;77:2031–6.
Kozu T, Miyoshi H, Shimizu K, et al. Junctions of the AML1/MTG8(ETO) fusion are constant in t(8;21) acute myeloid leukemia detected by reverse transcription polymerase chain reaction. Blood. 1993;82:1270–6.
Miyoshi H, Kozu T, Shimizu K, et al. The t(8;21) translocation in acute myeloid leukemia results in production of an AML1-MTG8 fusion transcript. EMBO J. 1993;12:2715–21.
Tan KT, Ding LW, Sun QY, et al. Profiling the B/T cell receptor repertoire of lymphocyte derived cell lines. BMC Cancer. 2018;18:940. https://doi.org/10.1186/s12885-018-4840-5.
Asou H, Suzukawa K, Kita K, et al. Establishment of an undifferentiated leukemia cell line (Kasumi-3) with t(3;7)(q27;q22) and activation of the EVI1 gene. Jpn J Cancer Res. 1996;87:269–74.
Asou H, Eguchi M, Suzukawa K, et al. Establishment of a myeloid leukaemia cell line (Kasumi-4) with t(9;22;11)(q34;q11;q13), inv(3)(q21q26) and the EVI1 gene activation from a patient with chronic myelogenous leukaemia in blast crisis. Br J Haematol. 1996;93:68–74.
Asou H, Gombart AF, Takeuchi S, et al. Establishment of the acute myeloid leukemia cell line Kasumi-6 from a patient with a dominant-negative mutation in the DNA-binding region of the C/EBPalpha gene. Genes Chromosom Cancer. 2003;36:167–74.
Pedranzini L, Mottadelli F, Ronzoni S, et al. Differential cytogenomics and miRNA signature of the Acute Myeloid Leukaemia Kasumi-1 cell line CD34+38- compartment. Leuk Res. 2010;34:1287–95. https://doi.org/10.1016/j.leukres.2010.02.012.
Veigaard C, Kjeldsen E. Exploring the genome-wide relation between copy number status and microRNA expression. Genomics. 2014;104:271–8. https://doi.org/10.1016/j.ygeno.2014.08.002.
Peterson JF, Aggarwal N, Smith CA, et al. Integration of microarray analysis into the clinical diagnosis of hematological malignancies: How much can we improve cytogenetic testing? Oncotarget. 2015;6:18845–62.
Ben-David U, Siranosian B, Ha G, et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature. 2018;560:325–30. https://doi.org/10.1038/s41586-018-0409-3.
Kasai F, Hirayama N, Ozawa M, Iemura M, Kohara A. Changes of heterogeneous cell populations in the Ishikawa cell line during long-term culture: proposal for an in vitro clonal evolution model of tumor cells. Genomics. 2016;107:259–66. https://doi.org/10.1016/j.ygeno.2016.04.003.
Quentmeier H, Pommerenke C, Dirks WG, et al. The LL-100 panel: 100 cell lines for blood cancer studies. Sci Rep. 2019;9:8218. https://doi.org/10.1038/s41598-019-44491-x.
Savage MS, Mourad MJ, Wapner RJ. Evolving applications of microarray analysis in prenatal diagnosis. Curr Opin Obstet Gynecol. 2011;23:103–8. https://doi.org/10.1097/GCO.0b013e32834457c7.
Thangaraj K, Gupta NJ, Chakravarty B, Singh L. A 47, XXY female. Lancet. 1998;352:1121.
Ohyashiki K, Kodama A, Nakamura H, et al. Trisomy 10 in acute myeloid leukemia. Cancer Genet Cytogenet. 1996;89:114–7.
Beghini A, Magnani I, Ripamonti CB, Larizza L. Amplification of a novel c-Kit activating mutation Asn(822)-Lys in the Kasumi-1 cell line: a t(8;21)-Kit mutant model for acute myeloid leukemia. Hematol J. 2002;3:157–63.
Hussey DJ, Nicola M, Moore S, Peters GB, Dobrovic A. The (4;11)(q21;p15) translocation fuses the NUP98 and RAP1GDS1 genes and is recurrent in T-cell acute lymphocytic leukemia. Blood. 1999;94:2072–9.
Stranger BE, Forrest MS, Dunning M, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–53.
Cancer Genome Atlas Research Network, Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74. https://doi.org/10.1056/NEJMoa1301689.
Cazzola A, Schlegel C, Jansen I, Bochtler T, Jauch A, Krämer A. TP53 deficiency permits chromosome abnormalities and karyotype heterogeneity in acute myeloid leukemia. Leukemia. 2019;33:2619–27. https://doi.org/10.1038/s41375-019-0550-5.
Stengel A, Kern W, Haferlach T, Meggendorfer M, Fasan A, Haferlach C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: an analysis of 3307 cases. Leukemia. 2017;31:705–11. https://doi.org/10.1038/leu.2016.263.
Guinn BA, Mills KI. p53 mutations, methylation and genomic instability in the progression of chronic myeloid leukaemia. Leuk Lymphoma. 1997;26:211–26.
Zhang SJ, Ma LY, Huang QH, et al. Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proc Natl Acad Sci USA. 2008;105:2076–81. https://doi.org/10.1073/pnas.0711824105.
Kawamura M, Kaku H, Taketani T, Taki T, Shimada A, Hayashi Y. Mutations of GATA1, FLT3, MLL-partial tandem duplication, NRAS, and RUNX1 genes are not found in a 7-year-old Down syndrome patient with acute myeloid leukemia (FAB-M2) having a good prognosis. Cancer Genet Cytogenet. 2008;180:74–8.
Quentmeier H, Reinhardt J, Zaborski M, Drexler HG. FLT3 mutations in acute myeloid leukemia cell lines. Leukemia. 2003;17:120–4.
Mitelman F, Johansson B, Mertens F. Fusion genes and rearranged genes as a linear function of chromosome aberrations in cancer. Nat Genet. 2004;36:331–4.
Wang Y, Wu N, Liu D, Jin Y. Recurrent fusion genes in leukemia: an attractive target for diagnosis and treatment. Curr Genomics. 2017;18:378–84. https://doi.org/10.2174/1389202918666170329110349.
Heyer EE, Deveson IW, Wooi D, et al. Diagnosis of fusion genes using targeted RNA sequencing. Nat Commun. 2019;10:1388. https://doi.org/10.1038/s41467-019-09374-9.
Klijn C, Durinck S, Stawiski EW, et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol. 2015;33:306–12. https://doi.org/10.1038/nbt.3080.
Ohnishi H, Kawamura M, Ida K, et al. Homozygous deletions of p16/MTS1 gene are frequent but mutations are infrequent in childhood T-cell acute lymphoblastic leukemia. Blood. 1995;86:1269–75.
Kawamura M, Ohnishi H, Guo SX, et al. Alterations of the p53, p21, p16, p15 and RAS genes in childhood T-cell acute lymphoblastic leukemia. Leuk Res. 1999;23:115–26.
O'Donnell PH, Dolan ME. Cancer pharmacoethnicity: ethnic differences in susceptibility to the effects of chemotherapy. Clin Cancer Res. 2009;15:4806–14. https://doi.org/10.1158/1078-0432.CCR-09-0344.
Dutil J, Chen Z, Monteiro AN, Teer JK, Eschrich SA. An interactive resource to probe genetic diversity and estimated ancestry in cancer cell lines. Cancer Res. 2019;79:1263–73. https://doi.org/10.1158/0008-5472.CAN-18-2747.
Kessler MD, Bateman NW, Conrads TP, Maxwell GL, Dunning Hotopp JC, O'Connor TD. Ancestral characterization of 1018 cancer cell lines highlights disparities and reveals gene expression and mutational differences. Cancer. 2019;125:2076–88. https://doi.org/10.1002/cncr.32020.
Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–30.
Super HJ, Martinez-Climent J, Rowley JD. Molecular analysis of the Mono Mac 6 cell line: detection of an MLL-AF9 fusion transcript. Blood. 1995;85:855–6.
Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–13. https://doi.org/10.1038/nature10762.
Barata JT, Boussiotis VA, Yunes JA, et al. IL-7-dependent human leukemia T-cell line as a valuable tool for drug discovery in T-ALL. Blood. 2004;103:1891–900.
Drexler HG, Macleod RA. History of leukemia-lymphoma cell lines. Hum Cell. 2010;23:75–82. https://doi.org/10.1111/j.1749-0774.2010.00087.x.
Acknowledgements
Authors would like to thank Dr. Ryoka Miki (Thermo Fisher Scientific, Japan) for her technical support on sequence data analysis, and Michael Martin for his proofreading.
Author information
Authors and Affiliations
Contributions
FK designed the study with input from YK and MK. HA established cell lines. MO and AK contributed to sample preparation. FK and MO performed experiments and analysis. FK, HA, KK, HK, MS, YK and MK contributed to interpretation of data. FK wrote the manuscript in consultation with HA, YK and MK.
Corresponding author
Ethics declarations
Conflict of interest
We have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kasai, F., Asou, H., Ozawa, M. et al. Kasumi leukemia cell lines: characterization of tumor genomes with ethnic origin and scales of genomic alterations. Human Cell 33, 868–876 (2020). https://doi.org/10.1007/s13577-020-00347-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13577-020-00347-5