
Abstract
In recent years, human activity directly and indirectly influenced the demography of moose in Poland. The species was close to extinction, and only a few isolated populations survived after the Second World War; then, unprecedented demographic and spatial expansions had occurred, possibly generating a very complex pattern of population genetic structure at the present-day margins of the species range in Poland. Over 370 moose from seven populations were collected from Poland, and partial sequences of the mitochondrial control region (mtDNA-cr; 607 bp) were obtained. In addition, the entire mtDNA cytochrome b gene (1,140 bp) and Y-chromosome markers (1,982 bp in total) were studied in a chosen set of individuals. Twelve mtDNA haplotypes that all belonged to the European moose phylogroup were recorded. They could be divided into two distinct clades: Central Europe and the Ural Mountains. The first clade consists of three distinct groups/branches: Biebrza, Polesie, and Fennoscandia. The Biebrza group has experienced spatial and demographic expansion in the recent past. Average genetic differentiation among moose populations in Poland at mtDNA-cr was great and significant (Φ ST = 0.407, p < 0.001). Using mtDNA-cr data, four separate groups of population were recognized using spatial analysis of molecular variance and principal coordinate analysis, including a relict population in Biebrza National Park, a reintroduced Kampinos National Park population, as well as populations that were descendants of moose that colonized Poland from the east (Lithuania, Belarus, and Ukraine) and the north (former East Prussia). Among all the sequenced Y-chromosome markers, polymorphisms were found in the DBY14 marker in three populations only; four haplotypes were recorded in total. No significant differentiation was detected for this Y-linked marker among moose populations in Poland. Our mtDNA study revealed that a variety of different factors—bottleneck, the presence of relict, autochthonous populations, translocations, limited female dispersal, and the colonization from the east and north—are responsible for the observed complex pattern of population genetic structure after demographic and spatial expansion of moose in Poland.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Historical processes and geographical factors may strongly affect the genetic structure of mammalian populations (Hundertmark and Bowyer 2004; Pilot et al. 2006). The mechanisms include range shifts, spatial contractions and expansions, the effects of barriers to gene flow, and number and location of refugia (Hewitt 1996, 2000; Excoffier et al. 2009). In recent years, human activity directly and indirectly influenced the demography of free-living animals and, as a result, may have profound consequences for their spatial genetic structure (Nussey et al. 2006; Haanes et al. 2010; Hundertmark and Van Daele 2010). Forest fragmentation and direct persecution by humans have resulted in dramatic reductions of boreal species’ ranges, and in many cases, present-day populations survived as small isolated groups only (Calvignac et al. 2009). Isolated populations, due to small size and strong genetic drift, exhibit reduced levels of genetic variation and may possess unique mtDNA haplotypes (Rowe et al. 2004).
Human impact on the population genetic structure may also result from introductions and translocations of individuals between populations, especially for rare and endangered species as well as for game animals, like roe deer (Capreolus capreolus) and red deer (Cervus elaphus; Randi 2005; Skog et al. 2009). While the majority of species have suffered from recent environmental changes, there are some mammals that greatly benefited from recent human activity and considerably increased in both number and occupied ranges. Best-known examples are among those that are commensal to humans (Schmölcke and Zachos 2005), but many herbivorous species, especially game animals such as roe deer, red deer, and moose (Alces alces), also expanded their ranges during the last century (Vernessi et al. 2002; Charlier et al. 2008; Skog et al. 2009). Factors that allowed such unprecedented range expansions are, for example, climate change, low pressure of predators after the Second World War, and “herbivore-friendly,” present-day management practices in forests (Sommer and Nadachowski 2006; Van Ballenberghe 2006).
Despite the fact that spatial expansions have occurred in the evolutionary history of most species, their impact on population genetic structure has attained surprisingly little attention (Excoffier et al. 2009). The analysis of experimental data as well as simulation study showed that newly colonized areas are expected to be structured into sectors of low genetic diversity separated by sharp allele frequency gradients (Excoffier and Ray 2008). The strong genetic drift usually occurs in populations located on the edge of the expansion; thus, the genetic differentiation among populations should be high. Simulation studies have shown that F ST increases sharply during the colonization phase not only if rare long-distance migration events occurred (Nichols and Hewitt 1994), but complex population genetic structure can also emerge through a continuous range expansion without long-distance dispersal (Excoffier and Ray 2008). For example, Coulon et al. (2006), in an empirical study, documented significant genetic structuring of a roe deer population which recently recolonized a fragmented landscape. Genetic structuring was also found among red deer populations in northeastern Poland and was explained by past human management practices and contemporary natural migrations (Niedziałkowska et al. 2012).
Moose exhibit a notable ability to adapt to changing habitats (boreal forest) and underwent large range shifts associated with climate change (Hundertmark et al. 2002). However, as the species is vulnerable to hunting, at the beginning of the nineteenth century, moose populations declined catastrophically from considerable parts of their distribution range in Europe (Schmölcke and Zachos 2005). In countries where current moose populations are abundant, past intense exploitation by humans almost caused the extirpation of the species. In effect, few small and isolated relict populations were left in western Central Europe (Schmölcke and Zachos 2005), including the Biebrza marshes in northeastern Poland (Brincken 1826; Gębczyńska and Raczyński 2004), East Prussia (Steinbach 2009), and Sweden (Charlier et al. 2008). Such relict moose populations most probably experienced bottlenecks; thus, one should expect low intrapopulation diversity and significant genetic differentiation among them. Indeed, studies of the mitochondrial control region (mtDNA-cr) of moose from Sweden and Poland (Hundertmark et al. 2002, Świsłocka et al. 2008) confirmed a recent bottleneck in these European populations. These data also supported the relict character of both populations as very distinct mtDNA haplotypes were found in these regions at high frequencies.
The present-day distribution of moose in western Central Europe is a result of spontaneous demographic expansion and successful reintroductions after the Second World War (Raczyński 2006). This expansion was possible due to the formation of pine stands on huge areas of the former Soviet Union and low pressure of predators. Gębczyńska and Raczyński (2004) suggested that numerous immigrants from the east, the increase of an autochthonous Biebrza population (northeastern Poland, <20 individuals after the Second World War), as well as a very successful reintroduction in the Kampinos National Park near Warsaw that started from five individuals (Dzięciołowski and Pielowski 1993) caused the moose to expand its range in Poland, and some long-distance dispersal events were even documented in Germany (Schönfeld 2009). In addition, numerous moose populations (over 1,500 individuals) survived the Second World War and stayed in 1945 in East Prussia (Steinbach 2009). This population declined catastrophically soon after the Second World War due to poaching; however, some individuals could have survived near the Russian/Polish border and contributed to the present-day gene pool of the species.
If all the aforementioned populations were involved in the spatial expansion of moose, one should expect very complex patterns of population genetic structure with sharp allele frequency gradients at current margins of the species range in Poland.
In this study, we analyzed mitochondrial DNA sequences (control region and cytochrome b gene) as well as nine Y-chromosome markers (Y-chromosome-conserved anchor-tagged sequences (YCATS) and SRY gene) in moose originating from seven populations in Poland, where today the species reaches its natural westernmost edge/range. These sex-linked DNA markers were confirmed to be useful in genetic approaches of mammalian populations (Hellborg and Ellegren 2004).
Our two main objectives were (1) to investigate in more detail phylogenetic relationships among mtDNA haplotypes of moose within the European haplogroup and (2) to reveal the pattern of genetic differentiation among moose populations in Poland after a spontaneous and human-mediated range expansion after the Second World War. We predicted that this expansion from different sources should lead to both high haplotype diversity and high levels of population differentiation. Special emphasis was also given to the autochthonous, possibly relict group of moose from the Biebrza National Park, and we estimated parameters for spatial and demographic expansion of the mtDNA haplotype group that possibly originated in this area.
Material and methods
We analyzed DNA samples of 377 moose collected over the years 2007–2011 from seven localities in Poland (Fig. 1): the Biebrza National Park (53°24′25″ N, 22°47′43″ E), the Knyszyn Forest (53°10′35″ N, 23°48′47″ E), the Augustów Forest (53°58′46″ N, 23°17′23″ E), the Srokowo State Forest (54°12′49″ N, 21°31′15″ E), the Gostynin-Włocławek Forests (52°38′54″ N, 19°04′04″ E), the Kampinos National Park (52°18′00″ N, 20°49′48″ E), and the Polesie National Park (51°23′37″ N, 23°11′41″ E; Table 2). Samples were collected within the moose core area in Poland inhabited by about 70 % of the population. Samples were composed of muscles and skin from individuals killed in car accidents or poached. Stool samples were also collected in winter (individuals were identified using 11 microsatellite loci; data not shown). Samples represented a mixture of males, females, and individuals of unknown sex. They were stored at −20 °C, after which the tissue samples were extracted with the DNeasy Blood and Tissue Kit; for stool samples, the QIAamp DNA Stool Mini Kit (Qiagen, Germany) was used.

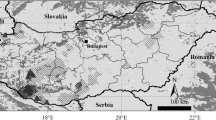
Study area, sampling sites, and frequency of 12 control region mtDNA haplotypes found in the moose populations studied. Gray background on the map shows moose distribution in Poland. GWF Gostynin-Włocławek Forests. *Relict moose population in Biebrza NP. **Translocated moose population in Kampinos NP
Two classes of DNA markers were used in further analyses: mitochondrial DNA and Y-chromosome markers. Amplification of 607 bp of the left hypervariable domain of the mitochondrial control region and a portion of the tRNApro gene was performed using primers LGL283 and ISM015 (Hundertmark et al. 2002; Table 1) and used in phylogenetic and population genetic analyses. The entire mtDNA cytochrome b (1,140 bp long) gene was PCR-amplified in a chosen set of individuals for phylogenetic analyses using primers ML103 and MH104 (Chikuni et al. 1995; Table 1). We used eight universal markers, YCATS, that were previously amplified in males of roe deer and reindeer (Rangifer tarandus; Hellborg and Ellegren 2003). We obtained PCR products for DBY4, DBY7, DBY8, DBY9, DBY14, and UTY11, but not for UBE1Y6 and UTY5 markers (Table 1). In order to verify possible polymorphisms found using universal primers, we designed moose-specific primers with the FastPCR software (Kalendar et al. 2009) to amplify the DBY9 and DBY14 markers (Table 1). Moose-specific primers were also designed to amplify 472 bp of a fragment of the SRY gene for genetic sex identification. These seven Y-chromosome markers were sequenced in 110 males from six populations studied (N ranged from 8 to 37 bulls).
PCR were performed with a GeneAmp PCR System 9700 (Applied Biosystems) in 5-μL reaction volumes containing 2 μL genomic DNA (∼20 ng), 1.7 μL Qiagen Multiplex PCR Master Mix (1×), 0.3 μL mix of primers (0.2 μM of each primer), and 1 μL RNase-free water. The reaction conditions were as follows: 15 min at 95 °C of an initial denaturation, 35 cycles with denaturation at 94 °C for 30 s, annealing at 57 °C (profile A) or 50 °C (profile B; see Table 1) for 90 s, extension at 72 °C for 60 s, and final elongation for 30 min at 60 °C. Sequencing reactions in both directions were performed using the BigDye™ Terminator Cycle Sequencing Kit (Applied Biosystems) following the manufacturer’s protocol. The detection of sequencing reaction products was carried out on an ABI 3130 Genetic Analyzer (Applied Biosystems). Sequences were aligned manually in the BioEdit sequence-editing program (Hall 1999). The sequences of all haplotypes were submitted to GenBank and assigned the following accession numbers: KC337263–KC337269 (for mtDNA-cr), KC337270–KC337273 (for the mtDNA cytochrome b gene), and KC337279–KC337282 (for the DBY14 marker).
Phylogenetic analyses
To test the phylogenetic relationships among the concatenated control region and cytochrome b haplotypes, we constructed phylogenetic trees with Mega v.5.05 (Tamura et al. 2011) with 1,000 bootstrap replicates used to assess support for tree nodes. The optimal model of substitution (Hasegawa et al. 1985) for our mtDNA sequences was determined using Akaike’s information criterion (Akaike 1973) with jModelTest (Posada 2008) and used to calculate a maximum-likelihood (ML) algorithm. A neighbor-joining (NJ) tree of haplotypes was computed with Kimura’s (1980) model of sequence evolution, which is the closest available MEGA model to HKY. Trees were rooted using mitochondrial sequences from reindeer (AB245426) and water deer (Hydropotes inermis; NC011821), downloaded from GenBank. We also created a descriptive median-joining network with default settings for control region mtDNA haplotypes using the program Network v.4.6.1.0 (Bandelt et al. 1999). To construct a median-joining network, apart from the mtDNA-cr haplotypes found in our study, we used moose haplotypes previously published by Hundertmark et al. (2002), with the following GenBank accession numbers: AF412253 (H23), AF412254 (H24), and AF412261 (H25).
We calculated net pairwise divergence (d A) between mtDNA clades—Central Europe and Ural—and among branches within the clade Central Europe using the program Mega v.5.05. The standard error of estimates was calculated with 1,000 bootstrap pseudo-replicates. We used a Bayesian approach implemented in BEAST v1.7.2 software (Drummond et al. 2012) to calculate the time of divergence between European and Asian moose, and also between two distinct European clades (Central Europe and Ural), and between particular groups/branches from clade Central Europe for concatenated control region and cytochrome b sequences. A strict molecular clock was applied with previously reported mutation rates for control region (4 and 8 % Ma) and cytochrome b (2 % Ma) in ungulates (Randi et al. 1998). Due to the fact that the mutation rate has not been calibrated for moose, we also used a mutation rate of 31.4 % per million years for mtDNA-cr derived for domestic cattle (Bradley et al. 1996).We used different nucleotide substitution models selected under Akaike’s information criterion implemented in jModelTest 0.1.1. They were HKY+I for mtDNA-cr and HKY for cytochrome b, respectively. Logcombiner v1.7.2 (BEAST software) was used to combine the results of independent runs.
To test the hypothesis of recent population growth of the Biebrza mtDNA-cr haplotype branch, we used Fu’s (1997) F S statistics, raggedness index (r; Harpending 1994), Tajima’s D statistic (Tajima 1989), and the pairwise mismatch distribution method. The statistics were calculated in software packages Arlequin v.3.11 (Excoffier et al. 2006). We estimated the demographic and spatial expansion time from the mismatch distribution (τ) as t = τ/2μ, where t is the expansion time in generations and μ is the substitution rate per generation for the whole sequence (Rogers 1995). We assumed a mutation rate of 0.04–0.08 substitutions (Randi et al. 1998) ×10−6 × n nucleotides per generation time, g = 7 years (Gaillard 2007).
Population genetics
We calculated the number of haplotypes (N h), haplotype diversity (h), nucleotide diversity (π, in percent), number of segregating sites (S), and mean number of pairwise differences (PD) for mtDNA-cr and Y-linked markers using the software packages Arlequin and DnaSP v.5 (Librado and Rozas 2009). Genetic differentiation between populations was assessed as pairwise F ST and Ф ST for mtDNA-cr and statistically tested in Arlequin. We also calculated the migration parameter (M = N m for haploid populations), where N m is a number of migrants exchanged between populations. M was calculated using the equation: M = (1 − F ST)/(2F ST) (Excoffier et al. 2006). For this purpose, population-specific F ST indices were calculated with analysis of molecular variance (AMOVA; Excoffier et al. 1992) using Arlequin (with 10,000 permutations). AMOVA was also used to assess structuring within the data, where the sampling sites were grouped as a single population. We also applied the spatial AMOVA procedure using SAMOVA ver. 1.0 (Dupanloup et al. 2002). The significance of Φ statistics was tested using 10,000 permutations for K = 2 to K = 6 partitions of the sampling sites. Principal coordinate analysis (PCA) was performed on mtDNA Φ ST data in GENALEX v.6.0 (Peakall and Smouse 2006).
Results
The analysis of a 607-bp mitochondrial DNA control region fragment amplified from 377 moose in Poland (Fig. 1) yielded 12 haplotypes (GenBank accession numbers for new haplotypes: KC337263–KC337269) as defined by 27 polymorphic sites (all being transitions). Five haplotypes were found in recent studies, while the other seven were previously unreported. We detected the H1 haplotype (GenBank accession no. EU257814) that was described for the first time from the Biebrza NP (Świsłocka et al. 2008) and the H2–H4 haplotypes reported by Hundertmark et al. (2002) (Finland 1–3; GenBank accession nos. AF412231–AF412233). Interestingly, we also found the haplotype H6 (GenBank accession no. AF412230) that was found by the latter authors in moose from Sweden. The sequence analysis of a whole cytochrome b gene (1,140 bp) in a selected group of moose that had different mtDNA-cr haplotypes revealed in total five synonymous substitutions (all being transitions) that defined four different haplotypes. The phylogenetic analysis (Fig. 2) of the concatenated control region and cytochrome b haplotypes revealed that all mtDNA haplotypes detected in Poland belong to the European haplogroup of moose (Hundertmark et al. 2002). A network (Fig. 3) was also drawn using the median-joining method to identify possible relationships between 12 mtDNA-cr haplotypes found in this study and other control region mtDNA sequences downloaded from GenBank. Two haplotype clades within the European haplogroup of moose were distinguished in Poland: Central Europe and Ural (Figs. 2 and 3). Notably, these clades had high bootstrap support on phylogenetic trees (Fig. 2). In addition, the clade Central Europe consists of three distinct groups/branches: Biebrza (haplotypes H1, H10, and H13); Polesie (haplotypes H12 and H20 in West Polesie Biosphere Reserve); and Fennoscandia (Scandinavian moose—haplotypes H6 and H11 in Kampinos and haplotypes H17 and H22 in former East Prussia). The Fennoscandia group might be further divided into Scandinavian and East Prussia branches (Fig. 2). The net divergence (d A) between clades Central Europe and Ural was 0.7 %, while that among three distinct groups from the clade Central Europe ranged from 0.8 % (Polesie vs. Fennoscandia) to 1.3 % (Biebrza vs. Fennoscandia). Our estimates of divergence based on mutation rates of 4 and 8 % for the control region and 2 % for cytochrome b between European and Asian moose were 0.33 Ma (95% highest posterior density interval (HPD) = 0.20–0.48 Ma), between clades Central Europe and Ural of 0.14 Ma (95% HPD = 0.08–0.21 Ma), and between groups Biebrza and Fennoscandia of 0.13 (95% HPD = 0.07–0.20 Ma). When we applied the mutation rate of 31.4 % per million years for mtDNA-cr derived for domestic cattle, the divergence values between European and Asian moose were 67,000 years ago (95% HPD = 40,800–97,000), between clades Central Europe and Ural of 47,000 years ago (95% HPD = 28,000–69,500), and between groups Biebrza and Fennoscandia of 46,800 years ago (95% HPD = 26,000–69,500).

Neighbor-joining tree, computed with Kimura’s model of sequence evolution, representing phylogenetic relationships among the concatenated control region and cytochrome b sequences. Maximum-likelihood topology computed with HKY85 model of substitution was identical with the NJ tree. Numbers listed at nodes represent percent support for that node from 1,000 bootstrap replicates (above line of a fraction for the NJ tree; below for ML tree). The trees have been rooted with sequences of reindeer (R. tarandus) and water deer (H. inermis)

Median-joining network showing relationships among moose mitochondrial DNA control region haplotypes (607 bp) belonging to the European haplogroup. Twelve mtDNA-cr haplotypes found in our study have numbers from H1 to H22, while haplotypes taken from GenBank (H23–H25) are indicated by italics letters. Missing haplotypes are shown with a gray dot
Demographic expansion was tested using mtDNA-cr sequences and detected in the Biebrza group of moose. Tajima’s neutrality test was negative and significant (D = −1.975, p < 0.001), while Fu’s value was also negative, albeit non-significant (F S = −1.312, p = 0.266). The mismatch distribution fits unimodal curves expected under the sudden expansion model (raggedness index, r = 0.393, p = 0.700). The demographic expansion event occurred at τ = 3.0 and involved a population change from an initial θ 0 = 0 (95% CI = 0–0.007) to a final θ 1 = 0.272 (95% CI = 0–99,999), albeit the 95% CI overlapped. Assuming a mtDNA-cr mutation rate of 0.04–0.08 substitution per site per million years and a generation time of 7 years, the demographic expansion of the Biebrza group took place 4400–8800 BP (95% CI = 490–10,300). The spatial expansion of the Biebrza moose in Poland occurred very recently (τ = 0.109), corresponding to 160–320 years ago (95% CI = 91–4,600).
In the moose populations studied, the number of mtDNA-cr haplotypes ranged from three to six (Fig. 1 and Table 2). The highest genetic diversity was found in Srokowo and Gostynin-Włocławek (h = 0.757 and 0.776; π = 1.41 and 1.30, respectively), whereas the corresponding values were the lowest in the Biebrza and Augustów populations. Despite the fact that the number of haplotypes per population did not exceed six, the number of segregating sites within a given population ranged from 14 to 22, indicating that the majority of haplotypes were not phylogenetically close.
Based on an assessment of the mtDNA-cr haplotype frequencies within the Biebrza population, we distinguished three genetically distinct subpopulations that correspond to three Biebrza valley basins: (1) upper (UBB), (2) middle (MBB), and (3) lower (LBB). All the Biebrza subpopulations differed significantly with respect to haplotype frequencies (at p < 0.05). The corresponding F ST values were: 0.23 (UBB vs. MBB), 0.59 (UBB vs. LBB), and 0.09 (MBB vs. LBB). In the upper Biebrza basin, H1 was the most frequent haplotype. In effect, the UBB subpopulation of moose resembled more the Augustów population than other Biebrza subpopulations (MBB and LBB).
Pairwise genetic differentiation values (mtDNA-cr) between moose populations ranged from 0 to 0.552 (F ST) and from 0 to 0.611 (Φ ST); the majority of comparison values (95 %) were significant (Table 3). The only F ST value that did not differ significantly from zero was between Knyszyn and Polesie populations, indicating their common origin. Non-significant values of Φ ST were also found between Knyszyn and Polesie as well as Srokowo and Gostynin-Włocławek. Especially high F ST and Φ ST values were obtained when the Biebrza and Kampinos populations were compared with the other ones (range = 0.234–0.611; Table 3), indicating great genetic differentiation between moose at these two populations and all others. The populations from Knyszyn and Augustów Forests exhibited great levels of genetic differentiation with respect to the Biebrza population despite their geographic proximity. The average migration parameter per population calculated using population specific F ST values was rather low (M = 1.77, range = 0.53–3.18), and only for the Srokowo population did the 95% CI exceed 10 (Table 2).
Geographical structuring among seven moose populations in Poland was supported by the results of AMOVA, where all sampling sites were treated as a single group of populations (Φ ST = 0.407, p < 0.001). Spatial analysis of molecular variance (SAMOVA) was then used to identify the subdivision most likely to explain the observed genetic structure. According to its results, the differentiation at mtDNA-cr among the studied moose populations was best explained assuming four groups: (1) Biebrza, (2) Kampinos, (3) Srokowo and Gostynin-Włocławek, and (4) all other populations from eastern Poland (Augustów, Knyszyn Forests, and Polesie). The percentage of variation was the highest among groups of populations at 35.47 %, while among populations within groups it was 6.71 % and within populations it was 57.82 % (Φ CT = 0.355, p < 0.01; Φ SC = 0.104, p < 0.001; Φ ST = 0.422, p < 0.001). The first and second axes of the PCA (PC1 and PC2) performed on the whole data set (seven populations) explained 48.44 and 31.77 % of the total variability, respectively. The analysis showed that eastern moose populations in Poland (Knyszyn, Augustów Forests, and Polesie) consisted of one group, the Biebrza was the most divergent from all other populations (with respect to both PC1 and PC2), the reintroduced population from Kampinos was distinct with respect to PC2, while Srokowo and Gostynin-Włocławek populations were intermediate (Fig. 4).

Principal coordinate analysis performed on pairwise Φ ST values of the studied moose populations in Poland
Out of nine Y-linked markers studied (1,982 bp in total), two (DBY9 and DBY14) were found to be variable in male moose from Poland using universal primers (Hellborg and Ellegren 2003). However, when we used moose-specific primers for both markers, DBY9 turned out to be monomorphic and DBY14 was the only one variable. For this marker, we found four haplotypes among the 110 studied bulls (GenBank accession nos. KC337279–KC337282) as defined by four polymorphic sites (two transitions and two transversions). The DBY14-H1 haplotype differed by one substitution from the DBY14-H2 haplotype; DBY14-H2 differed by one substitution from DBY14-H4. Two substitutions distinguished DBY14-H1 from DBY14-H3 and DBY14-H1 from DBY14-H4. The DBY14-H1 haplotype was the most common and was fixed in Augustów, Polesie, and Srokowo populations. Two DBY14 haplotypes were present in Biebrza (f(DBY14-H1) = 0.95, f(DBY14-H2) = 0.05) and Knyszyn (f(DBY14-H1) = 0.91 and f(DBY14-H3) = 0.09). Interestingly, as many as four DBY14 haplotypes (H1–H4) were found in the reintroduced Kampinos population: f(DBY14-H1) = 0.88, f(H2, H3, H4) = 0.04 (each). Haplotype (h) and nucleotide (π) diversity in the overall male moose sample in Poland for the DBY14 marker were 0.106 and 0.11 %, respectively. All pairwise genetic differentiation values (both F ST and Φ ST) between moose populations for the DBY14 marker ranged from 0 to 0.05 and were not significantly different from zero.
Discussion
This study using sex-linked markers (mtDNA and YCATS) demonstrated considerable genetic variation of moose in Poland. As many as 12 mtDNA-cr haplotypes were found in a total sample, which corresponds to about 71 % of control region haplotypes recorded in the European phylogroup of the species so far (Hundertmark et al. 2002; M. Niedziałkowska, personal communication). Notably, seven haplotypes were described for the first time, indicating Poland as an important area of moose haplotype diversity. The relatively high mtDNA-cr diversity of moose in Poland may be due to several non-mutually exclusive factors: (1) the large number of founding individuals colonizing Poland from the east (Lithuania, Belarus, Ukraine) after the Second World War; (2) the presence of small local populations at few sites and admixture after spatial expansion; and (3) successful reintroduction(s). Indeed, pine stands planted on huge areas of the former Soviet Union that started soon after the Second World War allowed an unprecedented increase in moose which resulted in spatial expansion to the west (Poland, Czech Republic, and Germany; Gębczyńska and Raczyński 2004; Schönfeld 2009). The colonization from the former Soviet Union is clearly visible by the presence of the H2, H3, and H4 mtDNA-cr haplotypes at relatively high frequencies, especially in populations situated close to the eastern border of Poland (Knyszyn, Augustów, Forests and Polesie; Fig. 1). In addition, large numbers of moose (about 1,500) were present in 1945 in East Prussia (present-day Kaliningrad Region, Russia; Steinbach 2009), and some of them could have colonized Poland by crossing the Russian–Polish border. This was probably evidenced by Lenkowa and Panfil (1973) who reported that, soon after the Second World War, a few individuals were present near Gołdap (northern Poland). Thus, individuals possessing the haplotypes H17 or H22 that were found in the Srokowo State Forest could have partly originated from the East Prussia moose population. Interestingly, haplotypes of these moose formed a branch within Fennoscandian moose that grouped with Scandinavian mtDNA haplotypes. Moose also survived the Second World War in Poland, but their number was very low (<20 individuals at the Biebrza marshes, NE Poland; Tomek 1977; Dzięciołowski and Pielowski 1993). According to Gębczyńska and Raczyński (2004), this population could be a relict group of its previous Holocene range that split early from the main wave of colonization. This was partly confirmed by Świsłocka et al. (2008) who found a unique haplotype (H1), distinct from others, among animals from this area at high frequency. In this study, we not only confirmed the presence of the H1 haplotype at very high frequency in the Biebrza population but also found two other haplotypes (H10 and H13) that clustered together with the H1 haplotype on the neighbor-joining tree with high bootstrap support (Fig. 2). These haplotypes, however, were not present in the Biebrza population, but in the Srokowo State Forest and Gostynin-Włocławek Forests, respectively, indicating they could have belonged to a large autochthonous population that was present in Poland before a severe bottleneck occurred at the end of the nineteenth century (Brincken 1826). Genetic uniqueness of the Biebrza population is somewhat strengthened by the fact that we found the DBY14-H2 allele in 5 % of males; this allele was not recorded in the other studied populations, except for the Kampinos population where four DBY14 haplotypes were found. Generally, low levels of Y-chromosome polymorphism have been recorded in different species of natural mammalian populations (Hellborg and Ellegren 2004). The authors found reduced levels of genetic variability on the Y-chromosome among all studied species, but one of the lowest levels were detected in reindeer and cattle species, as well as lynx (Lynx lynx), where the surveyed Y-chromosome sequences were completely monomorphic. Lack of variation was also found in Y-chromosomal introns of red deer (Barbosa and Carranza 2010). Thus, Y-chromosome polymorphism in moose with four haplotypes that were not randomly distributed in Poland is rather unexpected when compared to other ungulate species.
The Kampinos population arose from a successful introduction of five individuals (two bulls and three females) from Belarus in 1951 (Dzięciołowski and Pielowski 1993). The successful management efforts allowed this population to increase in number and to expand. It is very probable that due to the founder effect, the mtDNA-cr H11 haplotype increased in frequency considerably despite the fact that in Belarus it is very rare (M. Niedziałkowska, personal communication). The presence of six mtDNA-cr and four DBY14 haplotypes in the Kampinos population cannot be explained by the introduction of five individuals only. Indeed, there is also evidence of the translocation of at least a single bull from the Białowieża enclosure in 1955 that was a descendant of Swedish moose, or less likely from Siberia when the Białowieża reintroduction (based on Swedish, Belarusian, and, possibly, Siberian moose; Karpiński 1951) failed. The signs of this theoretically unsuccessful reintroduction are, however, still present as the H6 haplotype, typical for moose in Sweden, was found in the Kampinos population, albeit at a very low frequency (in a single moose). Moreover, long-distance dispersal of both female and male moose was evidenced from the Biebrza to Kampinos population as the mtDNA-cr H1 and DBY14-H2 haplotypes were recorded in this reintroduced population. Thus, our study demonstrated that even reintroduced populations that originated from very limited numbers of founders, due to admixture of individuals from local populations and/or long-distance dispersal, could harbor unprecedentedly high levels of genetic polymorphism. The origin of Polish moose from various sources is supported by the relatively high number of haplotypes and possible presence of two separate mtDNA clades (termed Central Europe and Ural) that all belonged to the European phylogroup (Fig. 2). We also found three distinct branches within the clade Central Europe in our country (Biebrza, Polesie, and Fennoscandia). It is worth stressing that the same tree topology was obtained using the Bayesian method. The topology of haplotype network by possessing long terminal branches and comb-like structure clearly shows signs of their recent expansion. Excoffier and Ray (2008) suggested that recently colonized areas due to spatial bottlenecks might exhibit private phylogenetic lineages. This does not rule out the possibility that some moose have survived in several distinct refuge areas in Europe as paleontological data evidenced that moose was present in the Balkans, Carpathian, and Italian Peninsula refugia during the Last Glacial Maximum (Sommer and Nadachowski 2006). Our estimate of the divergence time for European and Asian moose ranged from 201,800 to 480,400, suggesting that these two haplogroup divergences predated the last glaciation. This is a general phenomenon for species in Europe revealed by mtDNA (Taberlet et al. 1998). The defined clades of moose (Central Europe and Ural) from the European lineage could have evolved during the Riss Glacial Period, as suggested by estimates of divergence times (140,000 years ago). Three distinct branches within the clade Central Europe could have evolved further during the glaciations. Our results are consistent with the divergence times for the different haplogroups detected in red deer (Skog et al. 2009); however, they considerably predate the estimate reported by Hundertmark et al. (2002). The authors reported d A between the European and Asian phylogroups (2.5 %) of moose and postulated that they diverged approximately 34,000 years ago. However, they assumed a mutation rate (μ) of 31.4 % per million years derived for domestic cattle (Bradley et al. 1996). When we applied a mutation rate of 31.4 % per million years, the estimated divergence times for the different lineages/clades detected ranged from 46,800 to 67,000 for the mtDNA-cr data, suggesting that their divergences occurred during the last glaciation. This obvious discrepancy can be solved by further calibration of the mutation rate of mtDNA in moose.
Demographic expansion of the Biebrza group of moose from the clade Central Europe deduced from the mismatch distribution suggests that it occurred soon after moose colonized Poland after the Last Glacial Maximum. It is noteworthy that spatial expansion was much recent as our estimate from mtDNA-cr and its 95% CI suggests that it could have occurred in historical times.
Another noteworthy finding was the occurrence of very high levels of genetic differentiation among moose populations in Poland where the species reaches the natural western edge of its distribution. There are a few potential explanations for the high and significant differentiation of the population of moose. First, the studied moose populations could have experienced bottlenecks in the past that resulted in profound differences in haplotype frequencies among populations. Indeed, severe bottlenecks were documented during historical times in Poland (Brincken 1826), former East Prussia (Steinbach 2009), and even in Sweden (Charlier et al. 2008). Second, the gene flow among moose populations may be restricted (Charlier et al. 2008). This may be due to the moose dispersal pattern. Radio-tracking data indicate that both females and males are philopatric (Cederlund and Sand 1994). High and significant pairwise F ST values between the moose populations studied as well as the estimates of migration parameter, M, deduced from population-specific F ST values seem to confirm limited female dispersal in moose, at least for the majority of populations. Third, Hallatschek et al. (2007) evidenced that range expansion in two dimensions results in an increase in the genetic differentiation among populations, as measured by F ST. The colonization pattern based on moose dispersal from the east, successful reintroduction in Kampinos, and the presence of an autochthonous population in Biebrza, as well as remnants of a large population in East Prussia, could have resulted in spatial structure and the presence of sharp mtDNA haplotype frequency gradients after range expansion that occurred after the Second World War, e.g., about 70 years ago. This finding is, to our knowledge, the first evidence for the great and significant population structuring of an ungulate species that occurred in a recently recolonized area within less than ten decades. Indeed, SAMOVA and PCA showed that the genetic differentiation among the studied moose populations exhibited a clear spatial pattern. The relict, autochthonous Biebrza population was the most genetically divergent from all other ones. The second group was the Kampinos population that experienced a founder effect during the first years of reintroduction; three eastern moose populations in Poland that originated from moose immigrants from the east formed group 3, while the Srokowo and Gostynin-Włocławek Forests populations seemed to be intermediate to the other populations on PCA. They could also possess some signs of introgression from East Prussia. These results are consistent with the proposed colonization pattern of Poland by moose after the Second World War (Gębczyńska and Raczyński 2004).
The relict Biebrza population is not only the most genetically different from the other populations but also, as our results revealed, is composed of three distinct subpopulations occurring in three basins. The presence of clearly differentiated subpopulations is most probably related to strong female philopatry. On the other hand, the high frequency of the haplotype H2 (mtDNA-cr) in the subpopulation from the upper basin clearly shows that its origin is from the neighboring Augustów population and that female dispersal between Biebrza and the neighboring Knyszyn populations is limited.
In conclusion, this study emphasizes the existence of high levels of genetic differentiation among moose populations in Poland for mtDNA-cr, but not for Y-linked markers. It also suggests a variety of factors that may underlie this phenomenon, including bottlenecks, the presence of relict populations, reintroductions, limited female dispersal, as well as the colonization pattern. A complete elucidation of how these different factors define the patterns of genetic variability in moose will require more intensive sampling of animals from larger geographical areas and a wider range of nuclear markers.
References
Akaike H (1973) Information theory and an extension of the maximum likelihood principle. Second International Symposium on Information Theory, Akademiai Kiado, Budapest, pp 267–281
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Barbosa MA, Carranza J (2010) Lack of geographic variation in Y-chromosomal introns of red deer (Cervus elaphus). J Neg Res Ecol Evol Biol 7:1–4
Bradley DG, MacHugh DE, Cunningham P, Loftus RT (1996) Mitochondrial diversity and the origins of African and European cattle. Proc Natl Acad Sci U S A 93:5131–5135
Brincken J (1826) Memoire descriptif sur la foret imperiale de Białowieża en Lithuanie. N. Glückserg, Warsaw (in French)
Calvignac S, Hughes S, Hänni C (2009) Genetic diversity of endangered brown bear (Ursus arctos) populations at the crossroads of Europe, Asia and Africa. Divers Distrib 15:742–750. doi:10.1111/j.1472-4642.2009.00586.x
Cederlund G, Sand H (1994) Home-range size in relation to age and sex in moose. J Mammal 75:1005–1012
Charlier J, Laikre L, Ryman N (2008) Genetic structure and evidence of a local bottleneck in moose in Sweden. J Wild Manag 72:411–415. doi:10.2193/2007-122
Chikuni K, Mori Y, Tabata T, Saito M, Monma M, Kosugiyama M (1995) Molecular phylogeny based on the kappa-casein and cytochrome b sequences in the mammalian suborder Ruminantia. J Mol Evol 41:859–866
Coulon A, Guillot G, Cosson JF, Angibault JMA, Aulagnier S, Cargnelutti B, Galan M, Hewison AJM (2006) Genetic structure is influenced by landscape features: empirical evidence from a roe deer population. Mol Ecol 15:1669–1679. doi:10.1111/j.1365-294X.2006.02861.x
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973. doi:10.1093/molbev/mss075
Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define the genetic structure of populations. Mol Ecol 11:2571–2581
Dzięciołowski R, Pielowski P (1993) Alces. Anton-5 Sp. z o.o., Warsaw, pp 1–208 (in Polish)
Excoffier L, Laval G, Schneider S (2006) ARLEQUIN ver 3.1: an integrated software package for population genetics date analysis. Genetics and Molecular Genetics Population Lab, Institute of Zoology, University of Geneva, Switzerland
Excoffier L, Ray N (2008) Surfing during population expansions promotes genetic revolutions and structuration. Trends Ecol Evol 23:347–351. doi:10.1016/j.tree.2008.04.004
Excoffier L, Foll M, Petit RJ (2009) Genetic consequences of range expansions. Annu Rev Ecol Evol Syst 40:481–501. doi:10.1146/annurev.ecolsys.39.110707.173414
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Gaillard JM (2007) Are moose only a large deer? Some life history considerations. Alces 43:1–11
Gębczyńska Z, Raczyński J (2004) Moose in the Biebrza River valley. In: The situation of moose populations in Poland. Biebrza National Park Press, Osowiec, pp 5–19 (in Polish)
Haanes H, Røed KH, Mysterud A, Langvatn R, Rosef O (2010) Consequences for genetic diversity and population performance of introducing continental red deer into the northern distribution range. Conserv Genet 11:1653–1665. doi:10.1007/s10592-010-0048-1
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41:95–98
Hallatschek O, Hersen P, Ramanathan S, Nelson DR (2007) Genetic drift at expanding frontiers promotes gene segregation. Proc Natl Acad Sci U S A 104:19926–19930. doi:10.1073/pnas.0710150104
Harpending HC (1994) Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum Biol 66:591–600
Hasegawa M, Kishino H, Yano T (1985) Dating of the human–ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174
Hellborg L, Ellegren H (2003) Y chromosome conserved anchored tagged sequences (YCATS) for the analysis of mammalian male-specific DNA. Mol Ecol 12:283–291. doi:10.1046/j.1365-294X.2003.01702.x
Hellborg L, Ellegren H (2004) Low levels of nucleotide diversity in mammalian Y chromosomes. Mol Biol Evol 21(1):158–163. doi:10.1093/molbev/msh008
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–276. doi:10.1111/j.1095-8312.1996.tb01434.x
Hewitt GM (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913. doi:10.1038/35016000
Hundertmark KJ, Bowyer RT (2004) Genetics, evolution, and phylogeography of moose. Alces 40:103–122
Hundertmark K, Van Daele LJ (2010) Founder effect and bottleneck signatures in an introduced, insular population of elk. Conserv Genet 11:139–147. doi:10.1007/s10592-009-0013-z
Hundertmark KJ, Shields GF, Udina IG, Bowyer RT, Danilkin AA, Schwartz CC (2002) Mitochondrial phylogeography of moose (Alces alces): Late Pleistocene divergence and population expansion. Mol Phyl Evol 22:375–387. doi:10.1006/mpev.2001.1058
Kalendar R, Lee D, Schulman AH (2009) FastPCR software for PCR primer and probe design and repeat search. Gene Genomes Genom 3:1–14
Karpiński JJ (1951) Moose (Alces alces L.) in Bialowieza primeval forest. Chrońmy Przyr Ojcz 11–12:40–48 (in Polish)
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Lenkova A, Panfil J (1973) Moose, Alces alces on the Polish land. Studia Naturae Seria B, Warsaw
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi:10.1093/bioinformatics/btp187
Nichols RA, Hewitt G (1994) The genetic consequences of long distance dispersal during colonization. Heredity 72:312–317. doi:10.1038/hdy.1994.41
Niedziałkowska M, Jędrzejewska B, Wójcik JM, Goodman SJ (2012) Genetic structure of red deer population in northeastern Poland in relation to the history of human interventions. J Wild Manag 76:1264–1276. doi:10.1002/jwmg.367
Nussey DH, Pemberton J, Donald A, Kruuk LEB (2006) Genetic consequences of human management in an introduced island population of red deer (Cervus elaphus). Heredity 97:56–65. doi:10.1038/sj.hdy.6800838
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295. doi:10.1111/j.1471-8286.2005.01155.x
Pilot M, Jędrzejewski W, Branicki W, Sidorovich VE, Jędrzejewska B, Stachura K, Funk SM (2006) Ecological factors influence population genetic structure of European grey wolves. Mol Ecol 15:4533–4553. doi:10.1111/j.1365-294X.2006.03110.x
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256. doi:10.1093/molbev/msn083
Raczyński J (2006) Moose in Poland—a current state and perspectives. In: Is there a place for moose? Uroczysko Supraśl, pp 24–38 (in Polish with English summary)
Randi E (2005) Management of wild ungulate populations in Italy: captive-breeding, hybridisation and genetic consequences of translocations. Vet Res Commun 29:71–75. doi:10.1007/s11259-005-0025-1
Randi E, Pierpaoli M, Danilkin A (1998) Mitochondrial DNA polymorphism in populations of Siberian and European roe deer (Capreolus pygargus and C. capreolus). Heredity 80:429–437. doi:10.1046/j.1365-2540.1998.00318.x
Rogers A (1995) Genetic evidence for a Pleistocene population explosion. Evolution 49:608–615
Rowe KC, Heske EJ, Brown PW, Paige KN (2004) Surviving the ice: Northern refugia and postglacial colonization. PNAS 101:10355–10359. doi:10.1073/pnas.0401338101
Schmölcke U, Zachos FE (2005) Holocene distribution and extinction of the moose (Alces alces, Cervidae) in Central Europe. Mamm Biol 70:329–344. doi:10.1016/j.mambio.2005.08.001
Schönfeld F (2009) Presence of moose (Alces alces) in Southeastern Germany. Eur J Wildl Res 55:449–453. doi:10.1007/s10344-009-0272-5
Skog A, Zachos FE, Rueness EK, Feulner PGD, Mysterud A, Langvatn R, Lorenzini R, Hmwe SS, Lehoczky I, Hartl GB, Stenseth NC, Jakobsen KS (2009) Phylogeography of red deer (Cervus elaphus) in Europe. J Biogeogr 36:66–77. doi:10.1111/j.1365-2699.2008.01986.x
Sommer RS, Nadachowski A (2006) Glacial refugia of mammals in Europe: evidence from fossil records. Mammal Rev 36:251–265. doi:10.1111/j.1365-2907.2006.00093.x
Steinbach HJ (2009) Moose survived in East Pussia. Wild und Hund 17:17–21 (in German)
Świsłocka M, Ratkiewicz M, Borkowska A, Komenda E, Raczyński J (2008) Mitochondrial DNA diversity in moose, Alces alces from Northeastern Poland: evidence for admixture in bottlenecked relic population in the Biebrza valley. Ann Zool Fenn 45:360–365
Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF (1998) Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7:453–464
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi:10.1093/molbev/msr121
Tomek A (1977) The occurrence and some ecological parameters of the moose in Poland. Acta Theriol 22:485–508
Van Ballenberghe V (2006) Predator control, politics, and wildlife conservation in Alaska. Alces 42:1–11
Vernesi C, Pecchioli E, Caramelli D, Tiedemann R, Randi E, Bertorelle G (2002) The genetic structure of natural and reintroduced roe deer (Capreolus capreolus) populations in the Alps and Central Italy, with reference to the mitochondrial DNA phylogeography of Europe. Mol Ecol 11:1285–1297. doi:10.1046/j.1365-294X.2002.01534.x
Acknowledgments
We are grateful to the Head of the Polish State National Forest Holding and the Biebrza National Park for kindly allowing us to collect moose stool samples from Poland and P. Rode for drawing the figures. The study was financed by the Ministry of Science and Higher Education, the University of Bialystok (NN304 024134 and BST-117, respectively) for the Biebrza NP population, and by the National Fund for Environmental Protection and Water Management for the other populations (project no. 326/09/Wn50/NE-PR-IX/D).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Kris Hundertmark
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Świsłocka, M., Czajkowska, M., Duda, N. et al. Complex patterns of population genetic structure of moose, Alces alces, after recent spatial expansion in Poland revealed by sex-linked markers. Acta Theriol 58, 367–378 (2013). https://doi.org/10.1007/s13364-013-0148-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13364-013-0148-7