
Abstract
Reference spectral library searching, while widely used to identify compounds in other areas of mass spectrometry, is not commonly used in glycomics. Building on a study by Cotter and coworkers on analysis of sialylated oligosaccharides using atmospheric pressure-matrix-assisted laser-induced tandem mass spectrometry (MS/MS), we show that library search methods enable the automated differentiation of such sialylated oligosaccharide isomers using MS/MS derived from electrospray collision-induced dissociation in ion trap and beam-type fragmentation mass spectrometers. We compare MS/MS spectra of five sets of native sialylated oligosaccharide isomers and show a spectral library search method that can distinguish between these isomers using the precursor ion [M+2X-H]+, where X=Li, Na, or K. Sialic acid linkage (α2,3 vs. α2,6) is known to have a dramatic effect on the fragmentation of the sialylated compounds. We found that 2,4A3 cross-ring fragment at the terminal monosaccharide in sialyllactoses, sialyllactosamines, and sialyl pentasaccharides is highly abundant in the MS/MS spectra of [M+2X-H]+ species of α2,6-NeuAc glycans, while (2,4A3-H2O) fragment is highly abundant in α2,3-NeuAc moiety. The 2,4A3-H2O peak is specific to NeuAc-α2,3-Gal-β1,4-Y (Y=GlcNAc or Glc). To our knowledge, this observation was not reported previously. Theoretical calculations reveal major conformational differences between α2,6-NeuAc and α2,3-NeuAc structures that provide reasonable explanations for the observed fragmentation patterns. Other singly-charged ions ([M+X]+) do not show similar cross-ring cleavages. Implemented in a searchable library, these spectral differences provide a facile method to distinguish sialyl isomers without derivatization. We also found good spectral matching across instruments. MS/MS spectra and tools are available at http://chemdata.nist.gov/glycan/spectra.

Graphical Abstract
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sialylated (or sialic acid-containing) oligosaccharides play a role in several biological functions, including host-pathogen interactions, cell protection from membrane proteolysis, cell adhesion, and cell-cell recognition [1, 2]. Sialic acids are nine-carbon acidic monosaccharides typically found in N-glycans, O-glycans, human milk oligosaccharides, and glycosphingolipids. N-Acetylneuraminic acid (abbreviated Neu5Ac, NeuAc or historically, NANA) is the most common sialic acid and is the only sialic acid naturally present in humans [3]. NeuAc is usually attached to terminal galactose (Gal) residues with an α2,3 or an α2,6 linkage or to terminal N-acetylglucosamine (GlcNAc) residues with an α2,6 linkage. In human milk oligosaccharides, NeuAc can also be found attached to the internal Gal or GlcNAc residues [4].
Distinguishing sialyl linkage isomers is critical because most biological activities of sialyl oligosaccharides are structure-dependent [5]. For example, α2,3-sialyllactose, a human milk oligosaccharide, binds to Helicobacter pylori, effectively lessening the adhesion of the bacteria to duodenum-derived human cells [6]. The human influenza virus recognizes α2,6-sialyl linkages; the avian and equine influenza virus recognizes the α2,3-sialyl linkages; and the swine influenza virus seems to recognize both α2,3 and α2,6 sialyl linkages [7]. In biomarker discovery, increased α2,6-sialylation was found to be correlated in the progression of many types of cancer [8].
Using matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) MS in the negative ion mode, Yamagaki and Nakanishi [5] distinguished α2,3 and α2,6 sialyl isomers of sialyllactose (SL) and sialyllactosamine (SLN) using difference in relative intensities of the B1 fragment ion (m/z 290) (for a detailed explanation of the fragmentation nomenclature, see reference [9]). The α2,3-sialic acid cleaves easier in MALDI-post-source decay (PSD) fragmentation, resulting in higher intensities of the B1 ion. Tandem mass spectrometry using soft ionization techniques, such as MALDI and electrospray (ESI), is a powerful tool to analyze glycans. However, sialic acid is labile and may be lost by in-source or metastable decay in MALDI-MS [10]. In positive-ion ESI-MS, the tandem mass spectra of the protonated or sodiated species of underivatized sialylated glycans usually contain very few peaks since the main fragmentation comes from the loss of the sialic acid [11].
The structural characterization of glycans released from proteins follows different approaches, usually implicating labeling, derivatization, and others. Derivatizations, such as permethylation [12, 13], amidation [14], and esterification [15,16,17,18] and are useful in stabilizing sialic acids for analysis in the positive ion mode and when combined with multi-stage MS (MSn), may give valuable cross-ring cleavage peaks and open hydroxyl scars to help elucidate the full glycan structure [19]. However, derivatization may be incomplete and time-consuming. Underivatized sialylated glycans analyzed in the negative ion mode ESI-MS generally produce better fragmentation [20], but the MS analyses are typically harder to optimize. Cotter and coworkers reported that by using infrared-atmospheric pressure (IR-AP) MALDI-ion trap MS with glycerol as matrix, they were able to distinguish cationized sialylated isomers in their underivatized form [11]. They found that doubly sodiated or cobaltinated singly-charged underivatized sialyl glycans produced distinct spectra with cross-ring cleavages.
Building on Cotter’s work, we report spectra with cross-ring cleavages of underivatized sialyl glycans acquired by collision-induced dissociation (CID) in ion trap (IT) and beam-type fragmentation (CID MS/MS, higher-energy collision dissociation (HCD) MS/MS, and CID MSn) at several collision energies using ESI-Orbitrap and ESI-quadrupole TOF (QTOF) MS instruments. We surveyed several precursor ions and confirmed that the [M + 2X-H]+ (where X = Li, Na, or K) precursor ions produce cross-ring-rich tandem mass spectra, as Cotter reported earlier using AP MALDI MS [11]. We show that using ESI ionization, these fragment ions help differentiate sialyl isomers without the need for derivatization or online purification. The signals are intense with minimal adverse effects of the metal ion salts on the ion signals. We find good spectral matching in the IT CID, QTOF, and HCD Orbitrap MS/MS data across several collision energies. Thus, the analysis of [M + 2X-H]+ ions with library searching could be used to differentiate sialyl isomers without derivatization. However, such ions are generally lesser in abundance compared to [M + Na]+ ions, so further work on optimizing the ionization efficiency of these ions is necessary.
In addition to the identification of specific glycans, a goal of this work to show the effectiveness of a library searching for aiding the identification of glycans, especially by distinguishing isomers having different fragmentation patterns.
Materials and Methods
Materials
Twelve oligosaccharides, such as 3′-sialyllactose (3-SL), 6′-sialyllactose (6-SL), 3′-sialyl-N-acetyllactosamine (3-SLN), 6′-sialyl-N-acetyllactosamine (6-SLN), sialyllacto-N-tetraose a (LSTa), sialyllacto-N-tetraose b (LSTb), sialyllacto-N-tetraose c (LSTc), sialyllacto-N-tetraose d (LSTd), 3′-sialyl-Lewis A tetrasaccharide (SLeA), 3′-sialyl-Lewis X tetrasaccharide (SLeX), sialylated tetraose type 1 (STetra1), and sialylated tetraose type 2 (STetra2), were obtained from Sigma-Aldrich (St. Louis, MO), V-labs (Covington, LA), and Elicityl (Crolles, France). All materials were used without additional purification.
Mass Spectrometry of Oligosaccharides
The underivatized glycans were prepared with, and without, dopants. Cation-doped diluted solution (75:25 water/methanol) were prepared by adding lithium iodide, sodium chloride, or potassium iodide to obtain lithiated, sodiated, or potassiated precursor ions, respectively, prior to MS analysis. Samples were analyzed by direct infusion using a nano-ESI LTQ-Orbitrap Velos or LTQ-Orbitrap Elite MS (Thermo Fisher Scientific, Waltham, MA). Samples were infused at a flow rate of 50 to 100 nL/min using equal volumes of aqueous methanol or aqueous acetonitrile. Data were acquired in the positive ionization mode at a mass range of m/z 100 to m/z 1500. Full-scan Fourier-transform (FT) mass spectra were acquired at a resolution of 30,000. Default values were used for the activation Q and time. CID MS/MS and MSn spectra were obtained at 35% normalized collision energy and with an isolation width of 2 Da. FT CID and HCD MS/MS spectra were obtained at a resolution of 15,000 and 30,000, respectively. HCD MS/MS spectra were acquired at several normalized percent collisional energies ranging from 5 to 180%, but only the useful spectra were included in the library. For example, spectra with a single ion peak are usually not included in the library. Calibration was done using a calibrant mix provided by the manufacturer to allow mass accuracy of 10 ppm (ppm)Footnote 1 or better over the entire m/z range. Oligosaccharides were detected as [M + X]+, [M + 2X-H]+, [M + 2X]2+, [M + X + H]2+, and [M + H]+ precursor ions, where M = molecular ion and X = Li, Na, or K, depending on the dopant.
Similar spectra of a particular glycan acquired using the same instrument, activation method, and collision energy were clustered together using a stringent clustering algorithm in order to generate a consensus spectrum [21, 22]. Several consensus spectra were generated for each glycan. Multi-stage MS (MSn) was performed when further characterization was necessary.
Theoretical Calculations
Three-dimensional (3D) structures were built using GLYCAM-web (www.glycam.org). The structures were preoptimized using molecular mechanics as implemented in Amber16 [23] using the GLYCAM force field [24, 25] with the most recent version of the GLYCAM06 parameters [25]. Although no systematic exploration of the conformational space was performed, the potential surface was spanned starting from various initial configurations. Subsequently, quantum mechanical calculations were performed using the density functional theory (DFT) method with the B3LYP functional and either standard 6-31G(d) or LANLDZ basis sets as implemented in Gaussian 09 [26]. Several relaxed scans were performed for exploring potential fragmentation pathways. Similar calculations have been effectively used in previous studies of ion fragmentation under CID conditions [27, 28]. Frequency analyses at the same level of theory were performed to identify real minima and transition state structures on the potential energy surface, for the model structures, 3-SLN and 6-SLN. Geometry optimization of more complex structures, such as LSTa and LSTc, were performed using the semi-empirical Hamiltonian AM1.
Results and Discussion
3-Sialyllactose (3-SL) Versus 6-SL
We compared the consensus [21, 22] tandem mass spectra of SL isomers shown in Figure 1A and B. The two isomers differ in the sialic acid linkage, with Figure 1A having an α2,3-linked NeuAc (3-SL) and Figure 1B having an α2,6-linked NeuAc (6-SL).

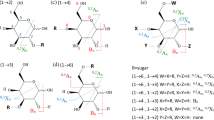
Structures of analyzed sialyloligosaccharides. (a) 3′-Sialyllactose (3-SL), (b) 6′-sialyllactose (6-SL); cartoon representations of (c) 3′-sialyl-N-acetyllactosamine (3-SLN), (d) 6′-sialyl-N-acetyllactosamine (6-SLN); (e) sialyllacto-N-tetraose a (LSTa), (f) sialyllacto-N-tetraose d (LSTd), (g) sialyllacto-N-tetraose c (LSTc), (h) sialyllacto-N-tetraose b (LSTb), (i) 3′-sialyl-Lewis A tetrasaccharide (SLeA), (j) 3′-sialyl-Lewis × tetrasaccharide (SLeX), (k) sialylated tetraose type 1 (STetra1), and (l) sialylated tetraose type 2 (STetra2). Each box is a set of isomers. Symbol nomenclature based on the recommendations by the consortium for functional glycomics (yellow circle: galactose (Gal), blue circle: glucose (Glc), blue square: N-acetylglucosamine (GlcNAc), red triangle: fucose (Fuc), purple diamond: N-acetylneuraminic acid (NeuAc)
In the native form, small oligosaccharides are typically analyzed using the precursor ion [M + Na]+. The CID MS/MS of the [M + X]+ ion (where X = Li, Na, or K) of 3-SL and 6-SL trisaccharides showed primarily the Y2 ion (loss of the labile sialic acid) in all cases and provided minimal differences in the spectra of the isomers (see Supplemental Figure 1). However, the spectra of [M + 2X-H]+ precursor ions of 3-SL and 6-SL isomers gave distinct tandem mass spectra enough to differentiate the isomeric pairs, a difference observed previously by Cotter and coworkers using AP-MALDI ion trap MS [11]. Figure 2A–F shows the MS/MS FT CID spectra of [M + 2X-H]+ ion (where X = Li, Na, K) of 3-SL (left) and 6-SL (right). In all cases, the greatest difference was the intensities of the cross-ring cleavage pair of the reducing glucose, namely 2,4A3 and (2,4A3-H2O). The α2,6-linked SL favors the formation of the 2,4A3 product ion, while the α2,3-linked SL favors the formation of 2,4A3-H2O product ion. Upon closer inspection, we see that the different cation-bound ions show different behaviors. The α2,-6-linked [M + 2Li-H]+ ion favors the formation of the 2,4A3 ion at 100% intensity; the B1 ion is at < 20% intensity (Figure 2B). The α2,3-linked [M + 2Li-H]+ ion favors the formation of the B1 ion (100%); the 2,4A3-H2O ion is at ≈ 50%; the 2,4A3 ion is at > 20%; and the 0,2A3 ion is at ≈ 10% (Figure 2A). The corresponding sodiated ion [M + 2Na-H]+ of the α2,6- and α2,3-linked SL favors the 2,4A3 and 2,4A3-H2O, respectively, at 100% intensity (Figure 2C and D). The [M + 2Li-H]+ ion of the α2,3-linked species favors the 2,4A3-H2O ion formation at 100% (Figure 2E), while the α2,6-linked species only favors the 2,4A3 formation at < 50% (Figure 2F).

FT CID MS/MS spectra of [M + 2X-H]+ ion of 3-SL (left) and 6-SL (right), where X is (a) lithium adducted to 3-SL (m/z 646.24); (b) lithium adducted to 6-SL (m/z 646.24); (c) sodium adducted to 3-SL (m/z 678.18); (d) sodium adducted to 6-SL (m/z 678.18); (e) potassium adducted to 3-SL (m/z 710.13); or (f) potassium adducted to 6-SL (m/z 710.13). Positive FT CID MS3 spectra of (g) 2,4A3-H2O from [M + 2Na-H]+ ion of 3-SL (m/z 540.13) and h2,4A3 from [M + 2Na-H]+ ion of 6-SL (m/z 558.32)
MS3 analysis of the 2,4A3 ion (m/z 540) from [M + 2Na-H]+ of 3-SL reveals primarily the B1 ion (sialic acid), m/z 430, and m/z 522, as shown in Figure 2G. MS3 analysis of 2,4A3-H2O (m/z 558) from [M + 2Na-H]+ of 6-SL (Figure 2H) also gave the B1 ion (sialic acid) as the base peak, but the other small peaks from Figure 2G were not observed, including m/z 430.
The 2,4A3 ion is a cross-ring cleavage in the reducing glucose. The two isomers only differ in the linkage of the sialic acid, a residue that is two monosaccharides away from the cross-ring cleavage site. The fact that the cross-ring cleavages are not observed in the singly sodiated precursor ion means that the two sodium ions and the loss of H+ help in the overall stability of the glycosidic bonds. Without the labile glycosidic bonds, the cross-ring cleavages become more abundant [11]. Most likely, one sodium ion replaces the proton in the carboxylic acid, accounting for one sodium gain and one proton loss. The assignment of the other sodium could be challenging [11]. Most fragments are observed as sodium adducts.
3-Sialyllactosamine (3-SLN) Versus 6-SLN
To verify if the differences in spectra of [M + 2Na-H]+ precursor ions of 3-SL and 6-SL were also applicable to other oligosaccharides, we probed another sialyl trisaccharide pair, 3-SLN (Figure 1C) and 6-SLN (Figure 1D), differing from 3-SL and 6-SL only in the reducing monosaccharide residue, GlcNAc. Figure 3 shows the [M + 2Na-H]+ spectra of 3-SLN (Figure 3A) and 6-SLN (Figure 3B). Table 1 summarizes the fragmentation observations for [M + 2Li-H]+, [M + 2Na-H]+, and [M + 2 K-H]+ ions. Following the same pattern in the SL isomers, the cross-ring cleavage pair in the reducing GlcNAc was observed: the 2,4A3-H2O peak at m/z 540 is ≈ 100% intensity in 3-SLN but is only < 5% intense in 6-SLN and the 2,4A3 peak at m/z 558 is the base peak in 6-SLN but is only < 5% intense in 3-SLN. Also, the B1 ion is the base peak in 3-SL but only ≈ 40% intense in 6-SL.

Consensus FT CID MS/MS spectra of [M + 2Na-H] + ion of SLN isomers (m/z 719.2097): (a) 3-SLN and (b) 6-SLN; and sialyl Lewis isomers (m/z 865.2672): (c) SLeA and (d) SLeX. Consensus spectra from the NIST 17 tandem MS library (http://chemdata.nist.gov)
Figure 4 shows product ion peak intensities as a function of the HCD normalized collision voltage in the MS/MS spectrum of 3-SLN (Figure 4A) and 6-SLN (Figure 4B) in the Orbitrap MS. For both isomers, the two major parallel fragmentation channels are the loss of sialic acid (full blue circles) and the cross-ring cleavages, 2,4A3 (full green triangles), but the latter is accompanied by a simultaneous loss of water (full purple diamonds). The loss of water in the reducing end from the precursor ion [M + 2Na-H]+ at m/z 701 is more intense in 6-SL than in 3-SL, whereas m/z 406 (Y2) is more intense in 3-SL.

Product ion peak intensities as a function of the HCD normalized collision energy in the MS/MS spectrum of (a) 3-SLN and (b) 6-SLN. Each curve is identified by the m/z value of the corresponding production
Figure 4A and B demonstrates that around 20 V, the precursor ion intensities for both isomers have been reduced by approximately 50%. At collision energies below this voltage, only the B1 fragmentation is important. Also, the abundance of product ions from the cross-ring cleavages is only significant at normalized collision energies above this voltage. The abundance of cross-ring fragment ions, 2,4A3-H2O, for 3-SLN and 2,4A3 for 6-SLN, can reach up to 15% of the total ion abundance.
An exploratory theoretical study of the gas phase potential energy surface (PES) was conducted to find the most stable locations for the sodium ions using Amber 16. Several local minima were identified and re-optimized using the semi-empirical Hamiltonian AM1. Finally, geometry optimization of the “global” minima for both isomers was performed using DFT calculations. It showed a crucial structural disparity in the conformational structure of both isomers that explains, in part, the differences observed in the fragmentation patterns. Figure 5 shows the optimized structures of the [M + 2Na-H]+ ions of 3-SLN (Figure 5a) and 6-SLN (Figure 5b). It seems that in the gas phase, the positions of the sodium ions are determined by the carboxyl group of the sialic acid. One of the sodium ions is forced in the vicinity of the carboxyl group and coordinating other hydroxyl groups and the other sodium ion is located in the opposite side also coordinating several oxygens. In the 3-SLN isomer, for example, a sodium ion is about the same distance of 2.5 Å from one of the carboxyl oxygen and other three hydroxyl oxygens marked with black asterisks in Figure 5a. The second sodium ion is on the opposite side between the sialic acid and the second-sugar ring making the structure very rigid (the oxygen involved are marked with blue asterisks). A frequency calculation shows weak out-of-plane vibrational modes. On the other hand, in the 6-SLN, both sodium ions are also near the sialic acid end, but the α2,6-sialyl linkage allows the molecule to bend on itself hydrogen-bonding the sugar of the reducing end with the sialic acid. These structures explain the higher stability of the glycosidic bonds and the labile character of the cross-ring fragment. The computational modeling shown in Supplemental Figure 2 shows two major differences in the fragmentation patterns of the 3- and 6-SLN isomers. First, the loss of water from the precursor ions occurs from the C6 branch of the reducing end in both isomers; however, relaxed scans of the C-O bonds corresponding to these water losses show different behaviors for each isomer, consistent with the experimental observations. Whilst the loss of water occurs with a relatively low barrier from 6-SLN (≈ 80 kJ/mol), the stretching of the C▬O bond for the 3-SLN isomer requires more energy (≈ 120 kJ/mol) and at the same time promotes the 2,4A3 cleavage. (It is worth mentioning that our assumption that the loss of water is originated from the C6-branch is based calculations, looking for the minimum energy pathway in all possible water losses; however, it is also consistent with the spectra of both isomers; the B1 peak is less intense for the 6-SLN isomer that experiences the larger water loss.) Second, the 2,4A3 fragmentation of the extended structure of 3-SLN produces an ion that simultaneously experiences loss of water from the generated terminal residue (2,4A3 fragment ion structures are shown in Supplemental Figure 2; red asterisks in Figure 5 mark the oxygen atoms involved in the water losses from the 2,4A3 ions; an approximate planar representation of the structures of the fragment ions is shown in Scheme 1).

DFT optimized structures of 3-SLN (a) and 6-SLN (b), calculated at the B3LYP-LandlDZ level of theory, and showing the 2,4A3 cross-ring fragmentation. Black (blue) asterisks mark the oxygen atoms that are coordinated to the sodium ion in front (behind) the plane of the sugar at the reducing end. Red asterisks mark the oxygen atoms involved in the water losses from the 2,4A3 fragment ions

Planar representation of the 2,4A3 fragment ions from (a) 3-SLN and (b) 6-SLN. Figure 2 of the Supplementary information shows the tridimensional structures of the fragment ions
On the other hand, the 6-SLN’s bent structure favors to retain the water from the generated terminal residue. Of course, this is an oversimplified view of the fragmentation process, but an attempt to include the various contributions of different local minima assuming a Boltzmann distribution complicates matters and sheds no new light on the problem. The optimized coordinates of the molecular geometries are given in Supplemental Table 1. It seems also that the B1 peak is less abundant for the 6-SLN isomer due in part to loss of water from the sialyl moiety.
Sialyl Lewis A (SLeA) Versus SLeX Spectra
Two other isomer pairs, SLeA (Figure 1I) and SLeX (Figure 1J), were examined. Both tetrasaccharide isomers contained an α2,3-linked sialic acid and differed in the lactosamine and fucosyl linkages. In Figure 3C and D, their spectra showed both glycosidic and cross-ring cleavages. The main differences are the intensities of Y2-Na (more abundant in SLeX) and C2α (more abundant in SLeA) peaks and the presence of m/z 623 only in the SLeX spectrum.
The fact that C2α is the base peak in SLeA suggests that the Gal-β1,3-GlcNAc bond is easier to break than the labile fucose or sialic acid. For SLeX, the Gal-β1,4-GlcNAc bond is harder to break. Instead, the labile sialic acid is easier to break, as observed in the base peak intensity of Y2-Na. This also shows that one of the sodium atoms sits on the sialic acid.
The fragmentation spectra of glycans with fucosyl linkages is dominated by glycosidic cleavages and the α2,3 effect is barely observed. The peak at m/z 623 in the spectrum of SLeX corresponds to loss of 242 Da, and it was initially attributed to the neutral ion pair of sodium ion and the product ion generated from cross-ring cleavage of sialic acid (1,5X2). However, the MS3 spectrum of the ion at m/z 623 shows a prominent B1 fragment ion (https://chemdata.nist.gov/glycan/). A previous report [29] showed that protonated native sialyl Lewis tetrasaccharide alkyl glycosides undergo internal-residue rearrangements with the fucose residue migrating toward the non-reducing terminal sialic acid residue. These authors reported two major unexpected peaks in the spectra of SLeA and SLeX at m/z 438 and m/z 600 of the [M + H] + ions, and they also mentioned that the abundance of these ions is significantly reduced for other ion types. In the present study, we observe an unexpected peak at m/z 623 in the spectrum of SLeX, but not in the spectrum of SLeA. Also, the presence of an intact B1 ion in the MS3 spectrum provides strong evidence against the fucosyl rearrangement mechanism. This is an important issue, because rearrangements can isomerize the sequences and lead to errors in interpreting oligosaccharide structures.
Instead of a cross-ring fragmentation of sialic acid or a rearrangement, there is another plausible explanation for the observed peaks in the spectrum of SLeX. The spectrum shows a prominent peak at m/z 701 corresponding to a loss of 164.0681 Da (the exact mass of fucose, 164.0685 Da). It seems that the peak at m/z 623 is originated from this peak by neutral loss of 78 Da {C2H4O2+ H20}. According to Lebrilla et al. [30], this loss is characteristic in the spectra of negative ions of 1–4-linked disaccharides and involves the loss of the anomeric carbon atom and the adjacent carbon atom. This was first suggested by Garozzo et al. [31]. In the present case, the exact mass analysis and theoretical calculations suggests the loss of C2H4O2+ H20 from the ion at m/z 701 (a planar representation of the fragmentation process is included with the supplementary information, Scheme S2). An experimental validation of these findings or an explanation for the observe differences with the SLeA isomer would require further investigation.
MS/MS Spectral Comparison of LSTa, LSTb, LSTc, and LSTd Sialyl Pentasaccharides
Next, we analyzed four isomers of sialylated pentasaccharides (Figure 1E–H). Figure 6 shows the MS2 consensus spectra of the [M + 2Na-H]+ precursor ions at m/z 1043. LSTc (Figure 6C and LSTb (Figure 6B) are both α2,6-linked sialyl oligosaccharide, differing only in the point of attachment of the sialyl residue: the NeuAc residues of LSTc and LSTb are linked to Gal and GlcNAc, respectively. Visually, the spectral peaks of the two isomers differ only in intensities. This is a good example of where in-silico libraries would NOT work because peak intensities could help differentiate the two isomers. Empirical or experimental spectral libraries, such as the NIST MS/MS library take into account all fragmentation pattern differences, not only B1, cross-ring, or theoretical fragmentations as mentioned in previous work [5]. Even the cumulative effect of relatively small peak intensity, differences can render significantly different scores.

Consensus FT CID MS2 spectra of [M + 2Na-H]+ of four LST isomers (m/z 1043.31): (a) LSTa (N = 17), (b) LSTb (N = 33), (c) LSTc (N = 31), and (d) LSTd (N = 49). Consensus spectra from the NIST 17 tandem MS library (http://chemdata.nist.gov)
Moreover, as expected from the cross-ring cleavage patterns of the trisaccharides studied above, the cross-ring fragment of interest is an A cross-ring at the reducing end. For LSTc, this is the 2,4A5 cross-ring ion (Figure 6C) and for LSTb, it is the 2,4A4 ion (Figure 6B), both at m/z 923.
In LSTc shown in Figure 6C, the most intense peak is the 2,4A5 cross-ring ion, followed by another cross-ring fragment 0,2A5 and the loss of sialic acid (Y4-Na). In LSTb (Figure 6B), the most intense peak is the loss of sialic acid followed by the 2,4A4 cross-ring cleavage at the reducing glucose.
Figure 6C and D shows the [M + 2Na-H]+ CID MS2 spectra of LSTa and LSTd, respectively. Both isomers have an α2,3-linked sialic acid and they differ only in one lactosamine linkage: β1,3 for LSTa and β1,4 for LSTd. The peaks mostly differ in intensities except for one peak m/z 540 that only occurs at ≈ 30% intensity in the LSTd spectrum. Further examination of this peak by MS3 at 35 V FT CID (Figure 7A) shows that the spectrum matches the IT CID spectrum of 3-SL m/z 540 in Figure 2G.

MSn of product ions of [M + 2Na-H]+ of LSTd: (a) 35-V CID MS3 spectrum (N = 57) of m/z 540, (b) 18-V CID MS3 spectrum (N = 23) of m/z 540, (c) 25-V CID MS3 spectrum (N = 25) of m/z 540, (d) 20-V CID MS4 spectrum (N = 36) of m/z 336, and (e) 35-V CID MS4 spectrum (N = 59) of m/z 336
The fact that the m/z 540 peak is observed in LSTd and not in LSTa means that this peak is not only specific to an α2,3-sialyl linkage [11], but it is specific to an α2,3-β1,4 linkage. To our knowledge, this observation was not reported previously.
Moreover, the FT CID spectrum of LSTa (Figure 6A) shows the preference of the formation of B3-H2O ion, while the LSTd spectrum (Figure 6D) shows preference of the Y4-Na ion. Following the pattern of SLs and SLNs, the 2,4A5-H2O ion is more abundant than the 2,4A5 ion. However, the difference in intensities is less than for smaller trisaccharides.
The CID MS/MS was activated at lower energies for the same precursor ion in order to understand the product ions. In Figure 7B, MS3 of the precursor ion m/z 540 at 18 V of CID energy shows that the said ion is the base peak, and the B1 ion is the second most abundant peak. We also see that m/z 318 and m/z 430 are starting to form. At 25 V of CID energy in Figure 7C, we see that the B1 ion is now the base peak but m/z 430 and m/z 318 remained at less than 20% intensity.
MS4 of m/z 336 at CID 20 V in Figure 7D shows the formation of B1-H2O and a cross-ring cleavage at m/z 216. Other peaks appear at a higher CID energy of 35 V (Figure 6E).
The IT CID MS/MS of the [M + 2Na-H] + precursor ion was activated at lower energies in order to understand the formation of the product ions. For example, Supplemental Figure 3 shows the IT CID spectra of m/z 1043 of LSTa at increasing energies. At 22 V (Supplemental Figure 3A), m/z 905 and m/z 683 ions are starting to form. The B3-H2O ion (m/z 683) continues to rise in intensity at increasing energies (Supplemental Figure 3B–F). However, the 2,4A5-H2O (m/z 905) ion increased in intensity gradually near 40% at 25 V energy and then stayed near 20%, even though it started to appear early with m/z 683 at 22 V. The Y4-Na (m/z 730) ion increased in intensity with m/z 905 but surpassed the latter in the 25 V energy spectrum onwards.
We also probed at increasing HCD energies to see if the ions followed the same formation pattern, as shown in Supplemental Figure 4. At 20 V HCD energy (Supplemental Figure 4A), only m/z 905 and m/z 730 are observed at less than 5% intensity. More product ions appear at 25 V HCD energy (Supplemental Figure 4B). At 28 V HCD energy (Supplemental Figure 4C), m/z 730 is more abundant than m/z 683 and m/z 905. This is different from the CID spectrum, where m/z 683 is more abundant than m/z 730. Since the HCD activation fragments all the ions including the product ions, the B1 ion at m/z 336 should be viewed as an ion that comes from other ions that has a sialic acid, including B glycosidic ions and A cross-ring ions (m/z 498, 683, 905, 983). This might explain why m/z 730 (Y4-Na) ion is more abundant than m/z 683 (B3-H2O): some of the m/z 683 ions are further fragmented to produce m/z 336 (B1) ions in HCD.
The CID MS/MS analysis of LSTd and LSTa in the negative mode, shown in Figure 8, also suggests cross-ring cleavages. Of particular interest are m/z 536 and m/z 494 ions that are only present in LSTd (Figure 8A) and not LSTa (Figure 8B). The ions correspond to 0,2A3-2H2O and 2,4A3-H2O, respectively. These cross-ring fragmentations of negative ions are interesting; however, it is expected that the fragmentation mechanisms share not much similarities with the fragmentation mechanism of [M + 2Na-H] + ions, so will not extend the discussion on this topic. However, the presence of similar cross-ring fragmentation processes in the spectra of negative ions indicates that likely the effect of the sodium ions on the mass spectra of [M + 2Na-H] + is more related to the replacement of the acidic hydrogen by a sodium ion than to the promotion of the cross-ring fragmentation itself, whereas a second sodium ion coordinates several hydroxyl groups and adds rigidity to the structure.

Consensus FT CID MS2 spectra of [M-H]- of two LST isomers (m/z 997.34): (a) LSTd (N = 146), (b) LSTa (N = 293)
A similar theoretical modeling strategy was designed for the study of the fragmentation mechanism of the [M + 2Na-H] + ions from LSTa and LSTc isomers. Supplemental Figure 5 shows the optimized structures for the two isomers. The absolute minima for isomer LSTa resulted in an extended structure similar to 3-SLN, and LSTc structure resembled the structure of 6-SLN. Although no further calculations were made for these isomers, a similar explanation probably holds for the observed differences of the 2,4A5 cross-ring fragmentation. It is worth mentioning that there is also some experimental data that supports the simultaneous coordination of cations to the glyceryl side chain and the carboxylate group [32, 33]. In general, the observed cross-ring fragmentation processes in [M + 2Na-H] + ions seem to follow certain general patterns: (i) a stabilizing effect of the gycosidic bond due to the replacement of the acidic hydrogen. Theoretical calculations show that one of the sodium ions is always near the carboxyl group at distances not larger than about 2.5 Å. (ii) The second sodium ion is also nearer to the non-reducing end in between the first and second rings, thus conferring certain rigidity to the structures. (iii) The fact that similar fragmentation processes are observed in the spectra of negative ions, [M-H]−, suggests a charge remote fragmentation mechanism. At this time, there is insufficient experimental and theoretical evidences to fully explain the role of the sodium atoms in the cross-ring fragmentation. (iv) Glycan branching and the presence of fucose in the reducing end inhibit the cross-ring fragmentation and the spectra can exhibit some unexpected peaks.
Lastly, we compared the QTOF CID and Orbitrap HCD MS/MS spectra of these isomers, as shown in Supplemental Figure 6 for LSTd and Supplemental Figure 7 for LSTb. The fragmentation patterns are very similar for most isomers. However, certain comparisons may require constraints, because the spectral differences among isomers are not very pronounced and noisy experimental spectra can match the wrong isomers. Table 2 shows an example of the library search results for the QTOF spectra of the isomer LSTb. It is observed that low collision energy spectra, with low conversion of the parent ion, match the wrong isomers (and with poor scores). A similar problem is observed at high collision energies. On the contrary, QTOF spectra with degree of the parent ion conversion between 5 and 90% match well with the HCD spectra in the library. This is the general finding in the NIST 17 library [34, 35] that spectra of the same precursor on different instruments are generally nearly identical at equivalent extents of decomposition (or effective collision energy). In practice, if library spectra are acquired at over a range of energies the include that of the query spectrum, the closely matching library spectra will appear at the top of the hit list and will have the closest matching fragmentation energy. We also note that when making identification by library searching one typically uses absolute scores (> 800 is usually a good match), differences between the top and the next best hit (100 is usually a good separation) [36, 37], although expertise concerning the fragmentation of the structural class under study is always an essential requirement for a confident identification.
A downloadable NIST Glycan MS/MS library and MS Search software is available at https://chemdata.nist.gov/dokuwiki/doku.php?id=chemdata:glycan-library for enhanced search options. Finally, it is worth mentioning that the NIST library browser provides several tools that enhance the identification capabilities, such as hybrid and exact mass searches. For example, although the number of glycans in the library is relatively small similarity searches can match other related glycans.
Conclusion
The fragmentation pattern of the [M + 2X-H]+ ion of sialyl isomers allowed us to differentiate spectra of underivatized sialyl isomers. The MS/MS spectra suggest that the main contribution to the spectral differentiation derive from an A cross-ring cleavage in the GlcNAc residue. We found that the differentiation is specific not only to the α2,3 and α2,6 NeuAc acid linkages, but to NeuAc-α2,3-Gal-β1,4-GlcNAc (or NeuAc-α2,3-Gal-β1,4-Glc) linkage, as we showed in the sialyllactose, sialylactosamine, and penta-sialyl isomers. To our knowledge, this is the first time that this observation is reported. Theoretical calculations show that the [M + 2Na-H]+ ions derived from glycans with α2,3 and α2,6 NeuAc acid linkages produce different conformational structures. The former linkage leads to rigid and extended structures, while the latter allows the molecule to bend on itself forming compact structures. These structural differences are consistent with the observed disparities between fragmentation patterns.
The NIST library of glycans contains MS/MS spectra from a broad range of collision energies and allows users to perform an exhaustive comparison and analysis of the differences between spectra, including relative small peak intensity differences. Consensus spectra searched against the library showed that the differentiation of isomers, at least in the case of [M + 2Na-H]+ ions of pentasaccharide sialyl isomers, was possible. It was also shown that for an effective comparison, the degree of parent ion conversion must be between 5 to 90%. Moreover, the CID QTOF and HCD Orbitrap MS/MS spectra of oligosaccharides showed good spectral match regardless of the type of mass spectrometer.
Notes
ppm is conventionally used in mass spectrometry and corresponds to (μg/g)/charge.
References
Nie, H., Li, Y., Sun, X.: Recent advances in sialic acid-focused glycomics. J. Proteome. 75(11), 3098–3112 (2012)
Varki, A., Schauer, R.: Sialic acids. In: Varki, A., Cummings, R. D., Esko, J. D., Freeze, H. H., Stanley, P., Bertozzi, C. R., Hart, G. W., Etzler, M. E. (eds.) Essentials of glycobiology, 2nd edn. Cold Spring Harbor Laboratory Press (2009) Chapter 14
JJantscher-Krenn E., Bode L.: Human Milk Oligosaccharides: Role in Infant Health. In: Watson R., Grimble G., Preedy V., Zibadi S. (eds.) Nutrition in Infancy, pp. 209-219. Nutrition and Health. Humana Press, Totowa, NJ (2013)
Kunz, C., Rudloff, S., Baier, W., Klein, N., Strobel, S.: Oligosaccharides in human milk: structural, functional, and metabolic aspects. Annu. Rev. Nutr. 20, 699–722 (2000)
Yamagaki, T., Nakanishi, H.: A new technique distinguishing alpha2-3 sialyl linkage from alpha2-6 linkage in sialyllactoses and sialyl-N-acetyllactosamines by post-source decay fragmentation method of MALDI-TOF mass spectrometry. Glycoconj. J. 16(8), 385–389 (1999)
Simon, P.M., Goode, P.L., Mobasseri, A., Zopf, D.: Inhibition of Helicobacter pylori binding to gastrointestinal epithelial cells by sialic acid-containing oligosaccharides. Infect. Immun. 65(2), 750–757 (1997)
Skehel, J.J., Wiley, D.C.: Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69, 531–569 (2000)
Alley Jr., W.R., Novotny, M.V.: Glycomic analysis of sialic acid linkages in glycans derived from blood serum glycoproteins. J. Proteome Res. 9(6), 3062–3072 (2010)
Domon, B., Costello, C.E.: A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconjugate. 5, 397–409 (1988)
Selman, M.H., Hoffmann, M., Zauner, G., McDonnell, L.A., Balog, C.I., Rapp, E., Deelder, A.M., Wuhrer, M.: MALDI-TOF-MS analysis of sialylated glycans and glycopeptides using 4-chloro-α-cyanocinnamic acid matrix. Proteomics. 12, 1337–1348 (2012)
Von Seggern, C.E., Zarek, P.E., Cotter, R.J.: Fragmentation of sialylated carbohydrates using infrared atmospheric pressure MALDI ion trap mass spectrometry from cation-doped liquid matrixes. Anal. Chem. 75(23), 6523–6530 (2003)
Kang, P., Mechref, Y., Klouckova, I., Novotny, M.V.: Solid-phase permethylation of glycans for mass spectrometric analysis. Rapid Commun. Mass Spectrom. 19(23), 3421–3428 (2005)
Mechref, Y., Kang, P., Novotny, M.V.: Differentiating structural isomers of sialylated glycans by matrix assisted laser desorption/ionization time-of-flight/time-of-flight tandem mass spectrometry. Rapid Commun. Mass Spectrom. 20(8), 1381–1389 (2006)
Sekiya, S., Wada, Y., Tanaka, K.: Derivatization for stabilizing sialic acids in MALDI-MS. Anal. Chem. 77(15), 4962–4968 (2005)
Egge, H., Dell, A., Von Nicolai, H.: Fucose containing oligosaccharides from human milk. I. Separation and identification of new constituents. Arch Biochem Biophys. 224(1), 235–253
Reiding, K.R., Blank, D., Kuijper, D.M., Deelder, A.M., Wuhrer, M.: High-throughput profiling of protein N-glycosylation by MALDI-TOF-MS employing linkage-specific sialic acid esterification. Anal. Chem. 86(12), 5784–5793 (2014)
Reiding, K.R., Lonardi, E., Hipgrave Ederveen, A.L., Wuhrer, M.: Ethyl esterification for MALDI-MS analysis of protein glycosylation. In: Reinders, J. (ed.) Proteomics in systems biology. Methods in molecular biology, p. 1394. Humana Press, New York (2016)
de Haan, N., Reiding, K.R., Haberger, M., Reusch, D., Falck, D., Wuhrer, M.: Linkage-specific sialic acid derivatization for MALDI-TOF-MS: profiling of IgG glycopeptides. Anal. Chem. 87(16), 8284–8291 (2015)
Wheeler, S.F., Domann, P., Harvey, D.J.: Derivatization of sialic acids for stabilization in matrix-assisted laser desorption/ionization mass spectrometry and concomitant differentiation of α(2 → 3)- and α(2 → 6)-isomers. Rapid Commun. Mass Spectrom. 23, 303–312 (2009)
Wheeler, S.F., Harvey, D.J.: Negative ion mass spectrometry of sialylated carbohydrates: discrimination of N-acetylneuraminic acid linkages by MALDI-TOF and Esi-TOF mass spectrometry. Anal. Chem. 72(20), 5027–5039 (2000)
Yang, X., Neta, P., Stein, S.E.: Quality control for building libraries from electrospray ionization tandem mass spectra. Anal. Chem. 86(13), 6393–6400 (2014)
Lam, H., Deutsch, E.W., Eddes, J.S., Eng, J.K., Stein, S.E., Aebersold, R.: Building consensus spectral libraries for peptide identification in proteomics. Nat. Methods. 5(10), 873–875 (2008)
Case, D.A., Ben-Shalom, I.Y., Brozell, S.R., Cerutti, D.S., Cheatham III, T.E., Cruzeiro, V.W.D., Darden, T.A., Duke, R.E., Ghoreishi, D., Gilson, M.K., Gohlke, H., Goetz, A.W., Greene, D., Harris, R., Homeyer, N., Izadi, S., Kovalenko, A., Kurtzman, T., Lee, T.S., LeGrand, S., Li, P., Lin, C., Liu, J., Luchko, T., Luo, R., Mermelstein, D.J., Merz, K.M., Miao, Y., Monard, G., Nguyen, C., Nguyen, H., Omelyan, I., Onufriev, A., Pan, F., Qi, R., Roe, D.R., Roitberg, A., Sagui, C., Schott-Verdugo, S., Shen, J., Simmerling, C.L., Smith, J., Salomon-Ferrer, R., Swails, J., Walker, R.C., Wang, J., Wei, H., Wolf, R.M., Wu, X., Xiao, L., York, D.M., Kollman, P.A.: AMBER. University of California, San Francisco (2016)
Singh, A., Tessier, M.B., Pederson, K., Wang, X., Venot, A.P., Boons, G., Prestegard, J.H., Woods, R.J.: Extension and validation of the glycam force field parameters for modeling glycosaminoglycans. Can. J. Chem. 94(11), 927–935 (2016)
Kirschner, K.N., Yongye, A.B., Tschampel, S.M., González-Outeiriño, J., Daniels, C.R., Foley, B.L., Woods, R.J.: Glycam06: a generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 29(4), 622–655 (2008)
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.J.A., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Keith, T., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, O., Foresman, J.B., Ortiz, J.V., Cioslowski, J., Fox, D.J.: Gaussian 09, Revision A.02. Gaussian, Inc., Wallingford (2016)
Neta, P., Godugu, B., Liang, Y., Simón-Manso, Y., Yang, X., Stein, S.E.: Electrospray tandem quadrupole fragmentation of quinolone drugs and related ions. On the reversibility of water loss from protonated molecules. Rapid Commun. Mass Spectrom. 24(22), 3271–3278 (2010)
Simón-Manso, Y., Neta, P., Yang, X., Stein, S.E.: Loss of 45 Da from A2 ions and preferential loss of 48 Da from A2 ions containing methionine in peptide ion tandem mass spectra. J. Am. Soc. Mass Spectrom. 22(2), 280–289 (2011)
Ernst, B., Müller, D.R., Richter, W.J.: False sugar sequence ions in electrospray tandem mass spectrometry of underivatized sialyl-Lewis-type oligosaccharides. Int J Mass Spectrom Ion Proc. 160, 283–290 (1997)
Carroll, J.A., Ngoka, L., Beggs, C.G., Lebrilla, C.B.: Liquid secondary ion mass spectrometry/Fourier transform mass spectrometry of otigosaccharide anions. Anal. Chem. 65, 1582–1587 (1993)
Garozzo, D., Giuffrida, M., Impallomeni, G., Ballistreri, A., Montaudo, G.: Determination of linkage position and identification of the reducing end in linear oligosaccharides by negative ion fast atom bombardment mass spectrometry. Anal. Chem. 62, 279 (1990)
Jaques, L.W., Brown, E.B., Barrett, J.M., Brey Jr., W.S., Weltner Jr., W.: Sialic acid. A calcium-binding carbohydrate. J Biol Chem. 252(13), 4533–4538 (1977)
Behr, J., Lehn, J.: Stability constants for the complexation of alkali and alkaline-earth cations by N-acetyl-neuraminic acid. FEBS Lett. 22(2), 178–180 (1972)
Yang, X., Neta, P., Stein, S.E.: Extending a tandem mass spectral library to include MS2 spectra of fragment ions produced in-source and MSn spectra. J. Am. Soc. Mass Spectrom. 28(11), 2280–2287 (2017)
NIST Tandem mass spectral library. (2017) http://chemdata.nist.gov/mass-spc/msms-search/ Accessed 4 July 2018
Stein, S.E.: Estimating probabilities of correct identification from results of mass spectral library searches. J. Am. Soc. Mass Spectrom. 5(4), 316–323 (1994)
Stein, S.: Mass spectral reference libraries: an ever-expanding resource for chemical identification. Anal. Chem. 84(17), 7274–7282 (2012)
Acknowledgements
The authors are grateful to Drs. P Neta, X Yang, and Y Liang for valuable assistance and to Dr. B L Foley for her suggestions on how to implement the molecular mechanic calculations using the GLYCAM force field in AMBER16. RJW thanks the NIH for financial support (U01 CA207824, P41 GM103390).
Disclaimer
Reference to commercial equipment or supplies does not imply endorsement by the National Institute of Standards and Technology, nor does it imply that the products identified are necessarily the best available for the purpose.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that there is no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOCX 1518 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
De Leoz, M.L.A., Simón-Manso, Y., Woods, R.J. et al. Cross-Ring Fragmentation Patterns in the Tandem Mass Spectra of Underivatized Sialylated Oligosaccharides and Their Special Suitability for Spectrum Library Searching. J. Am. Soc. Mass Spectrom. 30, 426–438 (2019). https://doi.org/10.1007/s13361-018-2106-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-018-2106-8