
Abstract
The modification of serine and threonine residues in proteins by a single N-acetylglucosamine (O-GlcNAc) residue is an emerging post-translational modification (PTM) with broad biological implications. However, the systematic or large-scale analysis of this PTM is hampered by several factors, including low stoichiometry and the lability of the O-glycosidic bond during tandem mass spectrometry. Using a library of 72 synthetic glycopeptides, we developed a two-stage tandem MS approach consisting of pulsed Q dissociation (PQD) for O-GlcNAc peptide detection and electron transfer dissociation (ETD) for identification and site localization. Based on a set of O-GlcNAc specific fragment ions, we further developed a score (OScore) that discriminates O-GlcNAc peptide spectra from spectra of unmodified peptides with 95% sensitivity and >99% specificity. Integrating the OScore into the two-stage LC-MS/MS approach detected O-GlcNAc peptides in the low fmol range and at 10-fold better sensitivity than a single data-dependent ETD tandem MS experiment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The modification of proteins on serine and threonine residues with β-N-acetylglucosamine (O-GlcNAc) is an emerging and dynamic post-translational modification (PTM) ubiquitously found on metazoan proteins. It was first discovered by Torres and Hart in 1984 [1], and is found on a wide range of cytoplasmic and nuclear proteins [2]. Further, it is known to be associated with several human diseases [3, 4], including neurodegenerative pathologies [3], type II diabetes [3], as well as cancer [4]. Recent technological progress in O-GlcNAc analytics has, by and large, focussed on biochemical enrichment approaches. Notably, this may be achieved by lectin affinity chromatography [5, 6] or using a chemoenzymatic method in which a β-1,4-galactosyltransferase is used to attach a biotinylated galactose to the endogenous O-GlcNAc moiety [7, 8].
Following some form of enrichment, the discovery of O-GlcNAc modified peptides and proteins is greatly aided by tandem mass spectrometry [6, 9], and reports on the discovery of this modification on individual proteins is increasing at a rapid rate. However, despite recent advances in instrumentation, the mass spectrometric analysis of O-GlcNAc peptides is still difficult and mainly hampered by the substoichiometric occupancy of O-GlcNAc sites [10–12] and by the chemical lability of the O-glycosidic bond in the gas phase [13–16]. Under typical collision-induced dissociation (CID) conditions, O-GlcNAc modified peptides readily lose the GlcNAc moiety, and spectra are typically dominated by intense neutral loss species as well as the GlcNAc oxonium ion (m/z 204.0866) and further fragments thereof [14]. The GlcNAc oxonium ion is isobaric to that of other GlcNAc epimers (e.g., GalNAc) and, therefore, commonly referred to as HexNAc oxonium ions. The intense HexNAc oxonium ion has been known for a long time as a diagnostically useful reporter ion [10, 13, 17] and used, e.g., in precursor ion scanning experiments on triple quadrupole [10] and quadrupole-time-of-flight mass spectrometers [18]. Unfortunately, the reporter ion may only be occasionally observed in ion trap CID spectra because of the poor recovery of fragment ions in the low m/z range [19]. This can be overcome by pulsed Q dissociation (PQD) in the ion trap [20] or so-called higher energy collisional dissociation (HCD) in a conventional multipole collision cell [21] on a LTQ Orbitrap XL mass spectrometer. Still, the dominant break of the O-glycosidic bond strongly reduces the occurrence of sequence-informative peptide fragment ions, which in turn impedes peptide identification and O-GlcNAc site localization. Alternative activation methods that enable sequencing the underlying peptide are neutral loss-triggered MS3 (NL-MS3) [5], multistage activation (MSA) [22], electron-capture [23], and electron-transfer dissociation [24] (ECD and ETD, respectively). The latter two preserve labile post-translational modifications, thereby facilitating both the identification of O-GlcNAc modified peptides and localization of the PTM site [5–8, 25]. One published report used a combination of fragmentation methods in which a CID step is followed by ETD fragmentation of the same precursor if the neutral loss of the HexNAc moiety is present in the CID spectrum (NL-ETD) [9]. Similarly, it would be possible to combine CID and HCD (NL-HCD), but this has not yet been published for O-GlcNAc peptides.
In light of the wide range of fragmentation techniques available on a single mass spectrometric platform (i.e., the LTQ Orbitrap XL ETD), it is timely to revisit which fragmentation technique or combination thereof offers particular advantages for the identification and site localization of O-GlcNAc modified peptides. In fact, no systematic study on O-GlcNAc peptides has yet been published using fragmentation techniques available on a hybrid ion trap-Orbitrap instrument. To this end, we evaluated nine different tandem MS acquisition schemes for their ability to identify O-GlcNAc peptides and to localize their PTM sites using a library containing 72 synthetic glycopeptides. As a result of this comparison, we developed a two-stage approach for the analysis of O-GlcNAc peptides, facilitating the detection of such peptides by PQD at low collision energy, and the identification and site localization by ETD. Based on a set of O-GlcNAc-specific fragment ions, we further developed a scoring scheme that is able to discriminate O-GlcNAc peptide spectra from unmodified ones with 95% sensitivity and >99% specificity. The two-stage approach allows detection and identification of O-GlcNAc peptides at the low fmol level in a complex proteomic background and is 10-fold more sensitive than a typical data-dependent ETD experiment.
2 Material and Methods
2.1 O-GlcNAc Standard Peptides and Proteins
Peptides were synthesized on a MultiPep peptide synthesizer (Intavis, Cologne, Germany) using standard Nα-Fmoc solid-phase peptide chemistry. A cytosolic protein extract from exponentially growing E. coli was spiked with different amounts (1:10, 1:100, 1:500, 1:1000 wt/wt) of bovine α-crystalline, followed by trypsin digestion and C18 purification prior to LC-MS/MS analysis. (For details, see supplemental methods.)
2.2 Nano-Liquid Chromatography-Tandem Mass Spectrometry
Mass spectrometry was performed on an LTQ Orbitrap XL ETD mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) connected to a nanoLC Ultra 1D+ liquid chromatography system (Eksigent, Dublin, CA) using in-house packed precolumn (20 mm × 75 μm ReproSil-Pur C18; Dr. Maisch, Germany) and nanocolumn (200 mm × 50 μm ReproSil-Pur C18; Dr. Maisch). The mass spectrometer was equipped with a nano-electrospray ion source (Proxeon Biosystems, DK) and the electrospray voltage was applied via a liquid junction. (For details, see supplemental methods.) All measurements were performed in positive ion mode. Intact peptide mass spectra were acquired at a resolution of 30,000 (at m/z 400) and an automatic gain control (AGC) target value of 106, followed by fragmentation of the most intense ions, a dynamic exclusion of fragmented precursor ions for 20 s, exclusion of singly charged ions and ions without assigned charge state for fragmentation (unless otherwise stated), and internal on-the-fly recalibration using the “lock mass” option. Full scans were acquired in profile mode, whereas all tandem mass spectra were acquired in centroid mode. A complete description of all tandem MS experiments employed in this study can be found in Table S1 in the supplemental methods.
2.3 Peaklist Generation and Database Search
Peak processing and peak picking of MS data was performed using Mascot Distiller ver. 2.2.1 (Matrix Science, London, UK). Briefly, (1) un-centroiding of tandem MS spectra and (2) precursor charge state re-calculation were enabled, (3) tandem MS spectra of singly charged precursors were discarded, (4) the minimum number of peaks per tandem MS spectrum was set to three, and (5) isotope fitting was disabled for the mass range below m/z 205. A brief description of the data processing for NL-MS3, NL-HCD, and NL-ETD experiments is available in the supplemental methods. Resulting peaklists were searched using the Mascot search engine ver. 2.2.04 (Matrix Science) against the complete NCBInr database (02/16/2007, 4,626,804 entries) with sequences of synthetic peptides appended. Search parameters included a precursor tolerance of 10 ppm and a fragment tolerance of 0.5 Da for linear ion trap spectra. HCD spectra were searched with a fragment tolerance of 0.05 Da. Enzyme specificity was set to trypsin, and up to two missed cleavage sites were allowed. Further parameters accounted for the misassignment of the monoisotopic peak (up to the second isotopic peak), for variable modifications by O-GlcNAc (203.0794 Da at serine or threonine), methylation (14.0157 Da at the C-terminus), and in case of the α-crystalline spiked E. coli samples by carbamido-methylation (57.0215 Da at cysteine). Except for ETD experiments, the O-GlcNAc modification definition is crucial for the successful Mascot database search of O-GlcNAc peptide spectra. The neutral losses of 203.0794 and 221.0899 Da were defined as both fragment and precursor neutral loss. Moreover, the HexNAc oxonium ion and its fragments (m/z 126.0550, 138.0550, 144.0655, 168.0655, 186.0761, 204.0866) were ignored for Mascot scoring. The database search results were imported into Scaffold ver. 2.6.02 (Proteome Software, Portland, OR).
2.4 Scoring MS Spectra for the Selective Extraction of Candidate O-GlcNAc Precursors
The raw mass spectrometric data of PQD and HCD experiments were processed using the Mascot Distiller software and parameters exactly as described above. For the extraction of potential O-GlcNAc precursors from the resulting mgf file, an in-house written Perl script was utilized. Briefly, the Perl script parses the mgf file and inserts the rank and normalized intensity of every peak in a spectrum. Based on the precursor m/z value and the precursor charge state, the script further calculates the expected m/z values for the neutral loss of the HexNAc moiety (∆m 203.0794 Da) and the loss of the HexNAc oxonium ion (∆m 204.0866 Da, and charge z-1). For the computation of the OScore according to (1), the normalized intensities of the reporter ions (m/z 126.0550, 138.0550, 144.0655, 168.0655, 186.0761, 204.0866) and sugar loss ions are first divided by their intensity rank and then summed up if they are within a user-specified m/z tolerance (e.g., 10 ppm for HCD, 0.3 Da for PQD). The Perl script exports the list of candidate O-GlcNAc precursors along with the OScore, including the accurate precursor mass, charge state, as well as retention time, which was further used to assemble an inclusion list for targeted experiments. Precursors with an OScore better than 2.0 were included in the inclusion list. Probability computations were performed separately using Microsoft Office Excel 2007 (Microsoft Corporation, Redmond, WA).
3 Results
3.1 Systematic Investigation of Tandem MS Methods
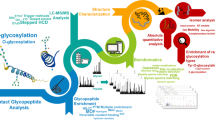
The two-stage LC-MS/MS strategy for the analysis of O-GlcNAc peptides developed in this work is shown in Figure 1. It consists of (1) a discovery LC-MS/MS run for the detection of potential O-GlcNAc peptides using low collision energy PQD, (2) the selective extraction of O-GlcNAc candidates from tandem MS spectra based on a novel spectrum scoring scheme, and (3) a targeted ETD experiment for O-GlcNAc peptide identification and site localization. The strategy was inspired by results from a systematic evaluation of nine different tandem MS methods available on an LTQ-Orbitrap XL ETD instrument. For this investigation, we synthesized a library of glycopeptides with precisely known O-GlcNAc sites using a simple randomization approach (Figure S1). The O-GlcNAc peptide library covers a mass range from 1115 to 1286 Da, and represents a heterogeneous set of doubly-, triply-, and quadruply charged peptides ranging from very hydrophilic to hydrophobic and from highly basic to acidic peptides (Figure S2). According to the extracted ion chromatograms of all 72 O-GlcNAc peptides, the dynamic intensity range of the O-GlcNAc library spans almost three orders of magnitude (Figure S3). Of the 72 possible permutations, 65 glycopeptides could be identified using PQD and ETD tandem MS methods followed by Mascot database search and manual inspection of spectra (see supplemental spectra).

Analytical strategy for detecting and identifying O-GlcNAc-modified peptides
Example tandem mass spectra of the peptide LSGgTYFKAK are depicted in Figure 2 and illustrate the merits of each technique. Owing to the chemical lability of the O-glycosidic bond in the gas phase, CID spectra (Figure 2a) are dominated by three signals; the HexNAc oxonium ion at m/z 203.98, the neutral loss of the HexNAc moiety from the precursor (m/z 493.29), and the charge reduced precursor ion that lost the HexNAc oxonium ion. Although the relative intensities of these three signals may vary substantially between different peptides, they usually represent the most intense fragment ions and typically constitute more than 70% of the entire signal in the tandem mass spectrum. Consequently, little, if any, of the available signal corresponds to sequence-specific peptide fragment ions, which in turn render peptide identification from these spectra very difficult. Furthermore, these peptide fragments do not generally retain the O-GlcNAc moiety, thus eliminating direct evidence for the O-GlcNAc modification and site localization from CID spectra. Further fragmentation of the HexNAc neutral loss by NL-MS3 (Figure 2b) or MSA (Figure 2c) significantly increases the yield of peptide fragment ions. Because the neutral loss of the HexNAc group leaves a plain serine or threonine residue at the previously modified O-GlcNAc site, it is impossible to deduce the modification site from NL-MS3 or MSA fragments should more than one possible site exist within the sequence.

Example tandem MS spectra of the O-GlcNAc peptide LSGgTYFKAK. Fragments of the HexNAc oxonium ion are marked with an asterisk
PQD and HCD provide access to the full fragment mass range on an LTQ Orbitrap instrument, and hence enable detection of the HexNAc oxonium ion (Figure 2d and e, respectively). Overall, PQD and HCD spectra of O-GlcNAc peptides are quite comparable to CID spectra and hence suffer from similar shortcomings with respect to peptide identification and O-GlcNAc site localization. However, HCD and, occasionally, PQD fragmentation give rise to further intense peaks below m/z 204, which are fragments of the HexNAc oxonium ion [26] (Figure S4, Table S2). In contrast to the aforementioned activation types, ETD preserves the O-GlcNAc-modification on every peptide fragment ion, thus allowing direct O-GlcNAc site localization (Figure 2f). However, ETD spectra often exhibit intense non-dissociated electron-transfer products. This can be overcome by supplemental activation of the charge-reduced species [27] resulting in richer spectra than ETD alone. Since the additional radiofrequency pulse does not adversely affect the O-GlcNAc modification, but significantly increases the intensity of peptide fragment ions supporting peptide identification and site localization, supplemental activation was used for all further experiments involving ETD (Figure S5).
Searching triplicate LC-MS/MS data from all nine activation types by Mascot identified 48 O-GlcNAc peptides from the library with Mascot ion scores greater than 25 (Table 1, Tables S4, S5 and S6). However, the success of identification between individual approaches varied significantly. As shown in Table 1, our results indicate superior performance of PQD with 39 O-GlcNAc peptide identifications followed by ETD with 33 identifications, whereas the least successful approaches were those involving Orbitrap detection of fragment ions (HCD and ETD [FT]), which only identified 19 and 10 O-GlcNAc peptides, respectively. In total, 1371 tandem mass spectra were matched to the O-GlcNAc peptide library (Table S6). Of these, 304 spectra point to peptides with multiple serine or threonine residues, which carry the risk of false O-GlcNAc-site assignments. Striking differences of the O-GlcNAc site localization accuracy exist between techniques involving collisional fragmentation and ETD. Both ETD and ETD (FT) achieve the highest accuracy in O-GlcNAc site assignments (90%–100% correct site localization), while the non-ETD approaches lead to a fairly random assignment of O-GlcNAc sites to serine or threonine residues using Mascot (20%–50% correct site localization).
Recently, the Mascot Delta Score (MD-score) has been introduced as a simple method for confident phosphorylation site assignment [28]. Likewise, the MD-score can be applied to O-GlcNAc spectra to increase confidence in O-GlcNAc site assignments (i.e., high MD-score) and to identify O-GlcNAc site assignments for which evidence from tandem mass spectra is lacking (i.e., low MD-score). While the average MD-score for ETD is 21.0, it is only 0.8 for non-ETD approaches. In addition, the MD-score for 44% of all non-ETD spectra is 0, indicating that no decision at all about site localization could be made in these cases. It became clear from this systematic investigation that none of the compared tandem MS approaches was particularly successful in O-GlcNAc peptide identification as well as site localization. We concluded that decoupling O-GlcNAc peptide detection from identification and site localization might improve the analysis of O-GlcNAc peptides because it would allow combining the best features of each acquisition mode.
3.2 Detecting O-GlcNAc Peptides with an Optimized Discovery Experiment
Even though CID-type experiments on a LTQ Orbitrap instrument were inappropriate for site localization, the fragment ions involving the sugar moiety can be highly diagnostic. In particular, PQD and HCD provide access to the full fragment mass range enabling the detection of the HexNAc oxonium ion and its fragments. However, the selectivity of the HexNAc oxonium ion may be compromised by numerous possible interfering peptide fragment ions of very similar mass (Table S3). We synthesized the peptide QCPSYFQAK with or without O-GlcNAc on the serine residue. In addition to the oxonium ion (m/z 204.0866), this peptide can give rise to an a2(QC) fragment ion (m/z 204.0801). This allowed us to investigate how resolution, mass accuracy, and collision energy affect the specificity of detection of the HexNAc oxonium ion in the presence of potential interfering ions. Although sulfhydryl groups are typically blocked by alkylation in proteomics experiments, we have chosen the a2(QC) fragment for investigation because it is (along with the isobaric a3 fragment of the amino acid combination AGC) the only regular peptide fragment within 50 ppm of the mass of the oxonium ion.
Owing to the low resolution of ion trap tandem MS spectra, the a2(QC) and HexNAc oxonium ions cannot be distinguished by PQD (mass difference 32 ppm). In contrast, this is possible by HCD provided that resolution and/or mass accuracy are sufficiently high. It turns out that a 15,000 resolution (FWHM) is insufficient, but a 30,000 resolution separates both ions to near baseline (Figure S6). Because HCD spectra acquired at the lowest possible resolution of an Orbitrap (7500 FWHM) still feature mass accuracy of <10 ppm, unambiguous identification of the oxonium ion is possible by mass accuracy alone provided that one of the two fragment ions has a significantly higher intensity than the other.
Yet another alternative for the selective detection of O-GlcNAc peptides is to control the generation of the HexNAc fragment ions by tuning the collision energy. As depicted in Figure 3a, the HexNAc oxonium ion is already generated at low normalized collision energy (NCE) with maxima at 23% NCE (PQD) and 18% NCE (HCD). Both values are considerably lower than the typical NCE values (35%–40%) used for CID-type experiments. Concomitantly, peptide backbone fragmentation is much reduced at low collision energy, generating spectra that are almost completely devoid of peptide fragments (Figure 3b).

(a) Collision energy characteristic of the HexNAc oxonium ion (solid line) and the interfering a2(QC) fragment ion (dashed line). For PQD, unmodified QCPSYFQAK and modified AEgSFGANAEK were used. HCD was optimized using QCPgSYFQAK. (b) Low collision energy PQD spectrum of the peptide LDGgTYFAAK. Note that almost all signals in this spectrum point to an O-GlcNAc modification of the precursor
HCD detection is inherently less sensitive than PQD, since precursor ions for HCD are isolated in the LTQ, accumulated in the C-trap before they are injected into the collision octopole, and the fragments are then transferred to the Orbitrap for detection. This process is inevitably accompanied by ion losses, which does not apply for PQD. Second, while the electron multipliers of the LTQ are capable of detecting a single ion, the Orbitrap detector requires a minimum of ~20 charges to detect a signal [29, 30]. This necessitates comparatively high AGC target value settings and, consequently, longer accumulation times. HCD is also significantly slower than PQD (sequential versus parallel MS and MS/MS). As expected, a side-by-side comparison of PQD and HCD (in triplicates) using the O-GlcNAc peptide library showed considerable differences in scan speed. While the discovery PQD experiment generated tandem mass spectra at 2.2 Hz, the speed of HCD data acquisition was only 1.1 Hz. Concomitantly, PQD and HCD generated 320 and 183 O-GlcNAc spectra, respectively, within 45 min LC-MS/MS time (Figure S7). All things considered, low energy PQD turned out to be the superior method. It provided sufficient selectivity and was more efficient than HCD for the detection of O-GlcNAc peptides despite its lower mass accuracy and resolution.
3.3 Scoring Tandem MS Spectra for the Presence of O-GlcNAc of Modified Precursors
With an efficient tandem MS method for the generation of diagnostic fragment ions at hand, we went on to develop a simple scoring scheme that differentiates O-GlcNAc from non-O-GlcNAc tandem spectra. We term this scoring scheme OScore because it utilizes and accounts for all spectral features pointing to the O-GlcNAc modification. The OScore S is calculated according to Eq. 1,
where I norm is the normalized intensity (i.e., divided by the sum of all intensities) of up to eight O-GlcNAc-specific spectral features (see experimental procedures and Figure 3b for details) and n is the intensity rank within the tandem mass spectrum. For calculation of the OScore, the fragment intensity is first normalized by the sum of all spectrum features to render the score independent of precursor intensity and, hence, robust against spectra from high abundant precursors. Second, the normalized intensity is further divided by the rank, in order to favour spectra, in which the O-GlcNAc diagnostic fragments are among the most intense peaks. This step concomitantly penalizes spectra that exhibit intense unspecific signals. The logarithmic transformation is used for convenience to rescale the score. The OScore is computed using a Perl script, which parses the peaklist contained in a mascot generic file (mgf) and calculates the rank and normalized intensities of all peaks in a spectrum before calculating the OScore. It requires at least one of the O-GlcNAc features to be present in the peaklist within a user-specified mass tolerance. The OScore script creates a tab-delimited output file containing (among other information) precursor m/z, precursor charge state, as well as retention time, which can be used to build inclusion lists for follow-up targeted experiments.
In order to assess the discriminating power of the scoring scheme, OScores were computed for a test set of low collision energy PQD spectra (approximately 750 O-GlcNAc spectra from the O-GlcNAc peptide library and 11,300 non-O-GlcNAc spectra from a tryptic digest of cytosolic E. coli proteins; Table S7). According to Figure 4a, the bimodal OScore distribution nicely discriminates O-GlcNAc peptides (low OScores) from unmodified peptides (high OScores). We also compared the OScore with other features of O-GlcNAc spectra, which could similarly be used as classifier to group O-GlcNAc and non-O-GlcNAc tandem MS spectra, e.g., the approach employed by Vosseller et al. [5], which utilizes the combination of the HexNAc oxonium ion and the neutral loss, or the HexNAc oxonium ion intensity, its normalized intensity, or the sum of normalized intensities of the oxonium ion and the HexNAc neutral loss. As revealed by a receiver operator characteristic (ROC) analysis, the OScore outperforms alternative classifiers and discriminates O-GlcNAc peptide spectra from spectra of unmodified peptides with 95% sensitivity at 99% specificity (Figure 4b). Furthermore, the area under the ROC curve (AUC) of the OScore is 0.997, indicating very high cumulative accuracy of the classifier.

OScore-enabled detection of O-GlcNAc spectra. (a) OScore distribution of O-GlcNAc spectra (solid line), non O-GlcNAc spectra (dashed line) and positive predictive value (PPV, dotted line). (b) ROC plots of several O-GlcNAc spectrum classifiers
The bimodal distribution of OScores allowed the straightforward calculation of the probability that O-GlcNAc spectrum assignments with a given OScore are correct. Using Bayes’ Law and denoting correct and incorrect assignments as “+” and “–”, respectively, the positive predictive value (PPV) p(+−S) for an OScore S can be calculated according to Eq. 2,
with p(S|+) and p(S|–) being the probabilities of OScores among O-GlcNAc and non-O-GlcNAc peptides, respectively, and p(+) and p(−) being prior probabilities representing the overall proportion of O-GlcNAc and non-O-GlcNAc spectra in the data set. The calculation of a PPV for a given OScore from (2) requires accurate models for the OScore score distributions. The symmetrical distribution of O-GlcNAc spectra was approximated using a Gaussian distribution and the asymmetrically distributed non-O-GlcNAc spectra were modeled on an offset-corrected γ distribution. Both distributions were fitted to the data using the method of least squares. Thus, with calculated mean μ and standard deviation σ, the probability for a correct O-GlcNAc spectrum assignment with an OScore S can be calculated according to Eq. 3,
while the probability for an incorrect assignment can be calculated according to Eq. 4,
with S m being the highest observed OScore and computed parameters α and β. Substitution of p(S|+) and p(S|–) in (2) by the modeled Gaussian and γ distribution along with computed prior probabilities p(+) and p(−) allowed calculation of PPVs (Figure 4a). It should be noted that low OScore values correspond to high probabilities and vice versa. As depicted in Figure S8, the computed PPVs are an accurate estimation of the observed probabilities (i.e., the fraction of correct O-GlcNAc spectrum assignments).
3.4 Identification of O-GlcNAc Peptides in a Complex Proteome
To demonstrate the practical utility of the two-stage LC-MS/MS approach, we compared it side-by-side to a conventional data-dependent ETD experiment using a highly complex tryptic digest of cytosolic E. coli proteins spiked with decreasing amounts of O-GlcNAc modified bovine α-crystalline (1:10, 1:100, 1:500, 1:1,000 wt/wt). Bovine α-crystalline is O-GlcNAc-modified at two sites (serine 162 of chain A, threonine 170 of chain B, see supplemental spectra). Serine 162 of chain A is modified at a stoichiometry of 10% [14] and Thr 170 is barely detectable. Considering that chain A and B are present in 1:1 stoichiometry, the molar content of O-GlcNAc of bovine α-crystalline is in the range of 5%. Hence, the actual spiking ratios in our experiment are in the order of 1:200 to 1:20,000 (wt/wt). The results are summarized in Table 2 and Figure 5. The data-dependent ETD experiment identified the O-GlcNAc peptide AIPVgSREEKPSSAPSS (615.6461 m/z, 3+; 922.9654 m/z, 2+) at a spiking ratio of 1:200. The two-stage approach detects and identifies the O-GlcNAc peptide still in the 1:2000 sample, suggesting an approximately 10-fold increased sensitivity over the conventional data-dependent approach. Both, the limit of detection and the limit of identification are reached at the spiking ratio of 1:2000, corresponding to 70 fmol O-GlcNAc peptide on column. Notably, the increase in sensitivity comes along with a significant increase in Mascot ion score (55 versus 24) at the same spiking ratio, thus providing higher confidence for the O-GlcNAc peptide identification.

Limit of O-GlcNAc peptide identification in a complex proteome. Displayed are (a) the total ion current (TIC) of the 1:2,000 (wt/wt) sample, (b) PQD spectrum of the spiked O-GlcNAc peptide (solid arrows indicate O-GlcNAc ions; dashed arrows point to unexplained signals), and (c) the targeted ETD spectrum leading to the identification of AIPVgSREEKPSSAPSS
4 Discussion
4.1 Systematic Evaluation of Tandem MS Techniques
In light of the poor CID fragmentation of O-GlcNAc peptides, we systematically revisited the numerous fragmentation approaches available on an LTQ Orbitrap XL ETD instrument for their merits in O-GlcNAc peptide identification and site-localization.
Surprisingly, the highest number of O-GlcNAc peptides was identified by PQD (Table 1). ETD, NL-ETD, and NL-HCD also led to a reasonable number of O-GlcNAc peptide identifications. For the latter two, this is reasonable as both spectra are triggered following the detection of a diagnostic neutral loss in the preceding CID spectrum. However, along with other CID-type fragmentation techniques, PQD spectra could not be utilized to localize O-GlcNAc sites reliably. Here, ETD fragmentation offers the distinct advantage that it preserves the O-GlcNAc modification and thus enables the direct inference of the accurate site of modification (Table 1). But ETD has its limitations too: increasing mass and decreasing charge density of the precursor diminish the fragmentation efficiency of ETD [39] and may render the O-GlcNAc site determination with ETD impossible for large peptides. Among the CID-like fragmentation approaches, HCD was the most accurate fragmentation technique with 50% correctly identified O-GlcNAc sites by Mascot. This can be reasoned by the high mass accuracy and high dynamic range of HCD spectra [21], which also allow deducing the O-GlcNAc localization from very low intensity signals.
4.2 Scoring Tandem Mass Spectra for Presence of the O-GlcNAc Modification
The OScore is a conceptually new and straightforward approach to evaluating the presence of an O-GlcNAc modification of potentially modified peptides based on tandem mass spectra. The OScore does not require the detection of sequence-informative peptide fragments but, instead, relies exclusively on the presence and intensity of up to eight different fragments originating from the breakage of the O-glycosidic bond (Figure 3b). Unlike other PTM scores [31–35], the OScore does not contribute any information about the localization of O-GlcNAc modification or the underlying peptide sequence. Instead, it assesses tandem MS spectra of complex peptide mixtures for the presence of the modification (Figure 4a and b). Using Bayesian statistics, the OScore can be further transformed into a positive predictive value for a given OScore (Figure 4a and S7). While these probability computations require a sufficient number of O-GlcNAc spectra to model score distributions of O-GlcNAc and non-O-GlcNAc spectra, the OScore itself is calculated on a single spectrum basis and, as such, indicates the presence of an O-GlcNAc modification by a low OScore irrespective of whether a single or hundreds of O-GlcNAc peptides are present in a sample. By design, the OScore is independent of the precursor signal intensity and, as such, independent of the amount of peptide on column. However, the quality of the OScore will decrease with decreasing signal-to-noise ratio of the precursor ion because the contribution of the chemical noise inevitably increases until the resulting tandem MS spectrum no longer primarily reflects the isolated O-GlcNAc peptide (Figure S9).
Nevertheless, the score is quite robust as exemplified in Figure 5. Apart from diagnostic fragments for O-GlcNAc, this PQD spectrum contains three intense signals (554.53, 729.44, and 831.16 m/z), which cannot be explained by typical fragment ions for the peptide AIPVgSREEKPSSAPSS, but very likely result from co-isolation and co-fragmentation of another peptide. With an OScore of 1.8, the precursor ion generating this mixed tandem mass spectrum is a reasonable candidate for inclusion in a targeted ETD experiment, which confirmed the sequence and modification.
Peptides modified by N- or O-linked glycans will probably also result in fairly low OScores (i.e., high O-GlcNAc probability), as they may lose HexNAc groups from their non-reducing ends. On the other hand, fragmentation of complex glycans will also result in numerous signals that do not indicate the O-GlcNAc modification and, hence, increase the OScore and decrease the probability of a false-positive O-GlcNAc spectrum assignment. The recently reported intracellular single N-linked HexNAc modification presumably resulting from breakdown of glycoproteins [6, 8] will probably not interfere, since the N-glycosidic linkage is more stable than the O-glycosidic bond under CID conditions [36], and the targeted ETD experiment would resolve this particular issue. Future experiments will address if and to which extent this modification may influence the discovery of O-GlcNAc peptides.
In addition to its utility in identifying candidate O-GlcNAc species in complex mixtures, the OScore may also support O-GlcNAc peptide identification by database searching. In particular in large-scale studies, the poor CID fragmentation of O-GlcNAc peptides, along with low search engine scores, is likely to result in both, an unnecessarily high proportion of overlooked (i. e. false-negative) as well as false-positive O-GlcNAc peptide-spectrum matches (PSMs). The OScore provides complementary information to that used by search engines, which may be used to ‘rescue’ genuine O-GlcNAc identifications despite having low search engine scores and to discriminate correct from incorrect O-GlcNAc PSMs. Both ways around, data quality would increase significantly. Another application of the OScore may be the retrospective analysis of existing data sets. This, however, would only work for data sets that were created by tandem MS, including the full mass range to ensure the detection of the HexNAc oxonium ion and its fragments.
4.3 Sensitivity of Detection and Identification
Compared with a conventional data-dependent ETD experiment, the two-stage approach resulted in a 10-fold increased sensitivity and significantly improved Mascot ion scores for the analyzed O-GlcNAc peptide of bovine α-crystalline spiked into a tryptic digest of E. coli proteins (Table 2, Figure 5). These improvements were achieved by decoupling the detection of potential O-GlcNAc precursors (by PQD) from their actual identification and site localization (by ETD), as well as by scoring tandem mass spectra for the presence of the O-GlcNAc moiety. This 10-fold gain in sensitivity comes, however, at the expense of requiring twice the amount of sample and measurement time. The limit of detection and identification determined here for a single peptide spiked into a highly complex background (low fmol range) probably represents a very conservative estimate. For less complex mixtures such as O-GlcNAc enriched proteomes or single O-GlcNAc proteins, one might expect limits of detection and identification in the mid amol range.
The PQD discovery experiment and the classical data-dependent ETD experiment acquired a similar number of tandem MS spectra at each spiking level (Table 2). They, therefore, had the same chance of detecting and identifying an O-GlcNAc modified peptide. However, at the spiking level of 1:2000 (wt/wt), only the PQD discovery experiment, in conjunction with the OScore, allowed the identification of the precursor 615.6461 m/z (3+) as a potentially modified peptide. In contrast, the conventional data dependent ETD experiment did not lead to a successful O-GlcNAc peptide identification at spiking ratios of less than 1:200 (wt/wt). Although the respective precursor ion could still be detected in the full scan spectra at higher spiking ratios, it was no longer among the species selected for fragmentation by PQD in a discovery experiment or a conventional data-dependent ETD experiment (Table 2).
While the PQD discovery experiment as well as the OScore was developed to maximize selectivity and sensitivity, the second-stage experiment focused on the targeted peptide identification and O-GlcNAc site localization. ETD was selected for this purpose because of its outstanding accuracy in O-GlcNAc site identification and its sound peptide identification performance (Table 1). Employing ETD in a targeted fashion enabled the acquisition of multiple ETD spectra across the chromatographic peak, which was accomplished by disabling the monoisotopic precursor selection as well as the rejection of unassigned charge states in the MS acquisition software. Owing to an enhanced signal-to-noise ratio and reliable ion statistics in tandem MS spectra, the acquisition of multiple ETD spectra per chromatographic peak resulted in an increased chance to identify the O-GlcNAc peptide as well as a higher confidence that the O-GlcNAc peptide-spectrum match is correct (Table 2).
4.4 Translation of the Two-Stage Approach to other MS Instruments
The two-stage approach has been developed using an LTQ Orbitrap XL ETD mass spectrometer. However, the approach can be easily translated to any other type of mass spectrometer, which is capable of detecting low-mass ions in tandem mass spectra and is ETD-enabled. When doing so, three aspects have to be considered. First, for the discovery experiment, it is important to adjust the fragmentation amplitude to generate O-GlcNAc spectra showing O-GlcNAc diagnostic fragments while suppressing (interfering) peptide fragment ions. Second, the peak list generated as input for the OScore script has to be converted into the Mascot generic format, e.g., using one of the free available peaklist conversion tools. Third, the resulting OScore distribution of O-GlcNAc and non-O-GlcNAc spectra will likely be different from instrument to instrument. Figure S10 shows the OScore distribution of O-GlcNAc and non-O-GlcNAc spectra acquired on an amaZon ETD ion trap mass spectrometer. As expected, the OScore distributions of PQD (Figure 4a) and the corresponding PAN experiment on the amaZon instrument (Figure S10) are alike, but span a different scale. Consequently, the OScore threshold for O-GlcNAc candidates has to be adjusted. Our approach lends itself to real-time decision-making akin to what has been proposed for the analysis of phosphopeptides [37], and further improvements should arise from increasing scan speed and sensitivity of ion trap-Orbitrap [38] and quadrupole TOF mass spectrometers [39, 40].
5 Conclusion
We believe that the developed analytical strategy has great potential for the broad-scale discovery of O-GlcNAc-containing proteins, particularly if combined with O-GlcNAc-specific enrichment tools. We further expect the OScore to become a valuable tool to improving the quality of O-GlcNAc peptide spectrum assignments. We, finally, anticipate that the increase in the number of documented O-GlcNAc proteins discovered in this or similar ways will shed further light on the functional significance of this emerging intracellular protein modification.
Abbreviations
- AGC:
-
Automatic Gain Control
- ETD (sa):
-
ETD with supplemental activation
- gS:
-
β-O-GlcNAc modified serine residue
- gT:
-
β-O-GlcNAc modified threonine residue
- HCD:
-
Higher Energy C-Trap Dissociation
- NCE:
-
Normalized Collision Energy
- NL-ETD:
-
Neutral Loss-Triggered ETD
- NL-HCD:
-
Neutral Loss-Triggered HCD
- NL-MS3:
-
Neutral Loss-Triggered MS3
- MSA:
-
Multistage Activation
- PQD:
-
Pulsed Q Dissociation
- PSM:
-
Peptide-Spectrum Match
References
Torres, C.R., Hart, G.W.: Topography and polypeptide distribution of terminal N-Acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 259, 3308–3317 (1984)
Love, D.C., Hanover, J.A.: The hexosamine signaling pathway: deciphering the “O-GlcNAc code.” Sci. STKE 2005, re13 (2005)
Dias, W.B., Hart, G.W.: O-GlcNAc modification in diabetes and Alzheimer’s disease. Mol. Biosyst. 3, 766–772 (2007)
Chou, T.Y., Hart, G.W.: O-linked N-acetylglucosamine and cancer: Messages from the glycosylation of c-Myc. Adv. Exp. Med. Biol. 491, 413–418 (2001)
Vosseller, K., Trinidad, J.C., Chalkley, R.J., Specht, C.G., Thalhammer, A., Lynn, A.J., Snedecor, J.O., Guan, S., Medzihradszky, K.F., Maltby, D.A., Schoepfer, R., Burlingame, A.L.: O-Linked N-Acetylglucosamine proteomics of postsynaptic density preparations using lectin weak affinity chromatography and mass spectrometry. Mol. Cell. Proteom. 5, 923–934 (2006)
Chalkley, R.J., Thalhammer, A., Schoepfer, R., Burlingame, A.L.: Identification of protein O-GlcNAcylation sites using eelectron transfer dissociation mass spectrometry on native peptides. Proc. Natl. Acad. Sci. U.S. A 106, 8894–8899 (2009)
Khidekel, N., Ficarro, S.B., Clark, P.M., Bryan, M.C., Swaney, D.L., Rexach, J.E., Sun, Y.E., Coon, J.J., Peters, E.C., Hsieh-Wilson, L.C.: Probing the dynamics of O-GlcNAc glycosylation in the brain using quantitative proteomics. Nat. Chem. Biol. 3, 339–348 (2007)
Wang, Z., Udeshi, N.D., Slawson, C., Compton, P.D., Sakabe, K., Cheung, W.D., Shabanowitz, J., Hunt, D.F., Hart, G.W.: Extensive crosstalk between O-GlcN-Acylation and phosphorylation regulates cytokinesis. Sci. Signal 3, ra2 (2010)
Teo, C.F., Ingale, S., Wolfert, M.A., Elsayed, G.A., Not, L.G., Chatham, J.C., Wells, L., Boons, G.J.: Glycopeptide-specific monoclonal antibodies suggest new roles for O-GlcNAc. Nat. Chem. Biol. 6, 338–343 (2010)
Haynes, P.A., Aebersold, R.: Simultaneous detection and identification of O-GlcNAc-modified glycoproteins using liquid chromatography-tandem mass spectrometry. Anal Chem 72, 5402–5410 (2000)
Hart, G.W., Housley, M.P., Slawson, C.: Cycling of O-linked β-N-Acetylglucosamine on nucleocytoplasmic proteins. Nature 446, 1017–1022 (2007)
Hu, P., Shimoji, S., Hart, G.W.: Site-specific interplay between O-GlcN-Acylation and phosphorylation in cellular regulation. FEBS Lett. 584, 2526–2538 (2010)
Huddleston, M.J., Bean, M.F., Carr, S.A.: Collisional fragmentation of glycopeptides by electrospray ionization LC/MS and LC/MS/MS: Methods for selective detection of glycopeptides in protein digests. Anal. Chem. 65, 877–884 (1993)
Chalkley, R.J., Burlingame, A.L.: Identification of GlcNAcylation sites of peptides and ↦-crystalline using Q-TOF mass spectrometry. J. Am. Soc. Mass Spectrom. 12, 1106–1113 (2001)
Jebanathirajah, J., Steen, H., Roepstorff, P.: Using optimized collision energies and high resolution, high accuracy fragment ion selection to improve glycopeptide detection by precursor ion scanning. J. Am. Soc. Mass Spectrom. 14, 777–784 (2003)
Medzihradszky, K.F., Gillece-Castro, B.L., Townsend, R.R., Burlingame, A.L., Hardy, M.R.: Structural elucidation of O-Linked glycopeptides by high energy collision-induced dissociation. J. Am. Soc. Mass Spectrom. 7, 319–328 (1996)
Carr, S.A., Huddleston, M.J., Bean, M.F.: Selective identification and differentiation of N- and O-Linked oligosaccharides in glycoproteins by liquid chromatography-mass spectrometry. Protein Sci. 2, 183–196 (1993)
Carapito, C., Klemm, C., Aebersold, R., Domon, B.: Systematic LC-MS analysis of labile post-translational modifications in complex mixtures. J. Proteome Res. 8, 2608–2614 (2009)
Cunningham Jr., C., Glish, G.L., Burinsky, D.J.: High amplitude short time excitation: A method to form and detect low mass product ions in a quadrupole ion trap mass spectrometer. J. Am. Soc. Mass Spectrom. 17, 81–84 (2006)
Schwartz, J.C., Syka, J.E., Quarmby, S.T.: Improving the fundamentals of MSn on 2D Ion Traps: New Ion Activation and Isolation Techniques. Proceedings of the 53rd ASMS Conference on Mass Spectrometry, San Antonio, TX, June
Olsen, J.V., Macek, B., Lange, O., Makarov, A., Horning, S., Mann, M.: Higher-energy c-trap dissociation for peptide modification snalysis. Nat. Methods 4, 709–712 (2007)
Schroeder, M.J., Shabanowitz, J., Schwartz, J.C., Hunt, D.F., Coon, J.J.: A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal. Chem. 76, 3590–3598 (2004)
Zubarev, R.A., Kelleher, N.L., McLafferty, F.W.: Electron capture dissociation of multiply charged protein cations. A nonergodic process. J. Am. Chem. Soc. 120, 3265–3266 (1998)
Syka, J.E., Coon, J.J., Schroeder, M.J., Shabanowitz, J., Hunt, D.F.: Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 101, 9528–9533 (2004)
Mirgorodskaya, E., Roepstorff, P., Zubarev, R.A.: Localization of O-Glycosylation sites in peptides by electron capture dissociation in a fourier transform mass spectrometer. Anal. Chem. 71, 4431–4436 (1999)
Chalkley, R.J., Burlingame, A.L.: Identification of novel sites of O-N-Acetylglucosamine modification of serum response factor using quadrupole time-of-flight mass spectrometry. Mol. Cell Proteom. 2, 182–190 (2003)
Swaney, D.L., McAlister, G.C., Wirtala, M., Schwartz, J.C., Syka, J.E., Coon, J.J.: Supplemental activation method for high-efficiency electron-transfer dissociation of doubly protonated peptide precursors. Anal. Chem. 79, 477–485 (2007)
Savitski, M.M., Lemeer, S., Boesche, M., Lang, M., Mathieson, T., Bantscheff, M., Kuster, B.: Confident phosphorylation site localization using the mascot delta score. Mol. Cell. Proteom. in press (2010)
Makarov, A., Denisov, E., Kholomeev, A., Balschun, W., Lange, O., Strupat, K., Horning, S.: Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal. Chem. 78, 2113–2120 (2006)
Bantscheff, M., Boesche, M., Eberhard, D., Matthieson, T., Sweetman, G., Kuster, B.: Robust and sensitive iTRAQ quantification on an LTQ orbitrap mass spectrometer. Mol. Cell. Proteom. 7, 1702–1713 (2008)
Beausoleil, S.A., Villen, J., Gerber, S.A., Rush, J., Gygi, S.P.: A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 (2006)
Savitski, M.M., Nielsen, M.L., Zubarev, R.A.: ModifiComb, a new proteomic tool for mapping substoichiometric post-translational modifications, finding novel types of modifications, and fingerprinting complex protein mixtures. Mol. Cell. Proteom. 5, 935–948 (2006)
Cox, J., Mann, M.: MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies, and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008)
Mortensen, P., Gouw, J.W., Olsen, J.V., Ong, S.E., Rigbolt, K.T., Bunkenborg, J., Cox, J., Foster, L.J., Heck, A.J., Blagoev, B., Andersen, J.S., Mann, M.: MSQuant, an open source platform for mass spectrometry-based quantitative proteomics. J. Proteome Res 9, 393–403 (2010)
Ruttenberg, B.E., Pisitkun, T., Knepper, M.A., Hoffert, J.D.: PhosphoScore: An open-source phosphorylation site assignment tool for MSn data. J. Proteome Res. 7, 3054–3059 (2008)
Medzihradszky, K.F.: Characterization of protein N-glycosylation. Methods Enzymol 405, 116–138 (2005)
Swaney, D.L., McAlister, G.C., Coon, J.J.: Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat. Methods 5, 959–964 (2008)
Olsen, J.V., Schwartz, J.C., Griep-Raming, J., Nielsen, M.L., Damoc, E., Denisov, E., Lange, O., Remes, P., Taylor, D., Splendore, M., Wouters, E.R., Senko, M., Makarov, A., Mann, M., Horning, S.: A dual pressure linear ion trap orbitrap instrument with very high sequencing speed. Mol. Cell. Proteom. 8, 2759–2769 (2009)
Ow, S.Y., Noirel, J., Salim, M., Evans, C., Watson, R., Wright, P.C.: Balancing robust quantification and identification for iTRAQ: Application of UHR-TOF MS. Proteomics 10, 2205–2213 (2010)
Ibrahim, Y.M., Prior, D.C., Baker, E.S., Smith, R.D., Belov, M.E.: Characterization of an ion mobility-multiplexed collision induced dissociation-tandem time-of-flight mass spectrometry approach. Int. J. Mass Spectrom. 293, 34–44 (2010)
Acknowledgments
The authors are indebted to Simone Lemeer, Kurt Fellenberg, and Andrea Hubauer for valuable support in aspects of mass spectrometry, Perl programming, and solid-phase peptide synthesis. The authors gratefully acknowledge the Studienstiftung des deutschen Volkes e. V. for a Ph.D. fellowship to H.H., and the support of the Faculty Graduate Center Weihenstephan of TUM Graduate School at the Technische Universität München, Germany.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1
O-GlcNAc peptide library synthesized using a sequence randomization approach. The nona-peptide LX1GX2YFX3AK was randomized during solid-phase peptide synthesis at positions 2, 4, and 7 by in-situ mixing of six amino acid derivatives at position 2 and 7 and two O-GlcNAc amino acid derivatives at position 4 giving rise to 72 possible sequence permutations. (PDF 141 kb)
Figure S2
Predicted properties of O-GlcNAc library peptides. The synthesized O-GlcNAc peptide library covers a limited mass range from 1115 to 1286 Da, which is due to the fact that all O-GlcNAc peptides in the library have the same length. However, the library shows substantial heterogeneity in hydrophilicity (GRAVY -0.76) 1.10) and isoelectric points (4.4 and 10.3). (PDF 38 kb)
Figure S3
XIC intensity of O-GlcNAc library peptides displayed as a function of isoelectric point, hydrophobicity and neutral mass. The XIC intensity of the O-GlcNAc library peptides spans almost 3 orders of magnitude. (PDF 91 kb)
Figure S5
ETD spectra of AEgSFGANAEK without (A, ETD) and with supplemental activation (B, ETD [sa]). (PDF 25 kb)
Figure S6
HCD spectra of QCPgSYFQAK acquired with 15,000 and 30,000 resolution (FWHM at m/z 400). (PDF 19 kb)
Figure S7
Comparison of PQD and HCD acquisition speed using the O-GlcNAc peptide library. (PDF 16 kb)
Figure S8
Accuracy of computed probabilities. OScores of the test data set were sorted into bins of width 0.1, and the actual probabilities (i. e. the fraction of O-GlcNAc spectrum assignments that are correct) as well as the computed probabilities (PPVs) were plotted. The expected probability is indicated as a dashed line. (PDF 32 kb)
Figure S9
OScore of the peptide AEgTFGANAEK undiluted and diluted 1:10 (w/w), 1:100 (w/w), and 1:500 (w/w) into a tryptic E. coli digest. O-GlcNAc-specific ions are indicated (red peaks). (PDF 27 kb)
Figure S10
OScore distribution of O-GlcNAc (O-GlcNAc peptide library) and non-O-GlcNAc spectra (E. coli digest) acquired on an amaZon ETD ion trap mass spectrometer (Bruker Daltonics, Bremen) using the PAN scan mode. Like PQD, the PAN fragmentation technique enables the detection of low m/z fragments in ion trap tandem mass spectra. (PDF 20 kb)
Table S2
Fragments of the GlcNAc oxonium ion (PDF 13 kb)
Table S3
List of exact masses and amino acid combinations giving rise to unmodified a-, band y-type peptide fragments interfering with the HexNAc oxonium ion and its fragments (< 100 ppm). We did not include any covalent modifications, the 13C isotopes or the loss of water or ammonia during the CID process, but all these are likely to expand the list of interfering peptide fragment ions. (PDF 19 kb)
Table S4
Summary statistics (part I) for the comparison of nine different activation types displaying each replicate separately. (PDF 27 kb)
Table S5
Summary statistics (part II) for the comparison of nine different activation types displaying the sum of three replicates. (PDF 25 kb)
Table S6
O-GlcNAc peptide identification from nine different fragmentation techniques with three replicates each. (XLS 3774 kb)
Table S7
This worksheet contains the test data used for OScore evaluation including 754 O-GlcNAc scans and >11,000 scans from an E. coli digest after running the OScore script on the test data mgf files. (XLS 3774 kb)
ESM 1
(PDF 273 kb)
ESM 2
(PDF 2992 kb)
ESM 3
OScore script (ZIP 3 kb)
Appendix A
Appendix A
The OScore script is available as a supplemental zip file along with an executable batch file and a short description. Mass spectrometry raw data as well as peaklist files, Mascot search results, and result files of the OScore algorithm may be downloaded from ProteomeCommons.org Tranche using the following hash: Whv45wINDEKo3Y7unLODHeVlsHKXkqiMZX+lcQ5clZ0e1BtwGIzl0YovNPA9M7EElnIJwlQMjy0lJ4I2saM6ouz15c0AAAAAAABNpw==.
Rights and permissions
About this article
Cite this article
Hahne, H., Kuster, B. A Novel Two-Stage Tandem Mass Spectrometry Approach and Scoring Scheme for the Identification of O-GlcNAc Modified Peptides. J. Am. Soc. Mass Spectrom. 22, 931–942 (2011). https://doi.org/10.1007/s13361-011-0107-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-011-0107-y