
Abstract
The novel SARS-CoV-2 (severe acute respiratory syndrome coronavirus-2) is spreading, as the causative pathogen of coronavirus disease-19 (COVID-19). It has infected more than 1.65 billion people all over the world since it was discovered and reported 3.43 million deaths by mid of May 2021. SARS-CoV-2 enters the host cell by binding to viral surface glycoprotein (S protein) with human ACE2 (angiotensin-converting enzyme2). Spike protein (contains S1 and S2 sub-domains) molecular interaction with the host cells is considered as a major step in the viral entry and disease initiation and progression and this identifies spike protein as a promising therapeutic target against antiviral drugs. Currently, there are no efficient antiviral drugs for the prevention of COVID-19 infection. In this study, we have analyzed global 8719 spike protein sequences from patients infected with SAR-CoV-2. These SAR-CoV-2 genome sequences were downloaded from the GISAID database. By using an open reading frame (ORF) tool we have identified the spike protein sequence. With these, all spike protein amino acid sequences are subjected to multiple sequence alignment (MSA) with Wuhan strain spike protein sequence as a query sequence, and it shows all SAR-CoV strain spike proteins are 99.8% identical. In the mutational analysis, we found 639 mutations in the spike protein sequence of SARS-CoV-2 and identified/highlighted 20 common mutations L5F, T22I, T29I, H49Y, L54F, V90F, S98F, S221L, S254F, V367F, A520S, T572I, D614G, H655Y, P809S, A879S, D936Y, A1020S, A1078S, and H1101Y. Further, we have analyzed the crystal structure of the 2019-nCoV chimeric receptor-binding complex with ACE2 (PDB ID: 6VW1) as a major target protein. The spike receptor binding protein (RBD) used as target region for our studies with FDA-approved drugs for repurposing, and identified few anti-SARS-CoV2 potential drugs (Silmitasertib, AC-55541, Merimepodib, XL413, AZ3451) based on their docking score and binding mode calculations expected to strongly bind to motifs of ACE2 receptor and may show impart relief in COVID-19 patients.
Similar content being viewed by others


Introduction
COVID-19, an infectious disease caused by coronavirus which is newly discovered in the year 2019. COVID-19 was first begun to emerge in the Wuhan seafood market in the Hubei province of China, it is caused by the SARS-CoV-2 (severe acute respiratory syndrome coronavirus-2). The coronavirus is a single-strand RNA virus (+ ssRNA) that can cause a severe respiratory syndrome in humans [31]. COVID19 is clinically characterized by fever, slight pain in the neck, headache, dry cough, breathing problem, vomit, and it might lead to death through multi-organ failure. COVID-19 has emerged as a severe epidemic declared by WHO and causing more than 3.43 million deaths worldwide between December 2019 to mid-May 2021 in around 200 countries (https://www.worldometers.info/coronavirus/) [33, 40].
Two types of coronavirus were reported, α-coronavirus that causes mild infections whereas, β-coronaviruses including SARS-CoV, and MERS-CoV (Middle East respiratory syndrome coronavirus) that caused pandemic in the recent past [18]. There were about 8000 SARS-CoV infected cases reported in the year 2002 and emerged as an epidemic in China [34]. Later, SARS-CoV, and MERS-CoV were reported as responsible for causing infection in humans [8]. The SARS-CoV-2 re-emergence in December 2019, has kept the world under high alert and has made a severe situation demanding fast treatment to prevent the infected patients [20, 32]. Despite expensive efforts globally by the researchers, there are no antiviral drugs or other treatments available to treat COVID-19 infected people. The current prevention methods are directed on quarantine and containment of infected patients for preventing human-to-human transmission along with vaccinations.
The spike protein (S) is a glycoprotein of SARS-CoV-2 and utilizes ACE2 for the host infection [33]. In the case of SARS-CoV, the spike protein on the virion surface mediates receptor identification as a class-1 fusion protein [7, 39]. During viral infection, the trimeric spike protein will cleavage into two subunits, S1 and S2. However, S1 subunits contain longer N-terminal and a receptor binding domain (RBD), while S2 subunit contains C-terminal membrane fusion domain followed by the two heptad regions (HR1 and HR2), the transmembrane (TM) domain and cytosolic tail [2, 27,28,29]. S2 is highly responsible for membrane fusion and contains the most conserved region. S1 directly binds to the host ACE2 receptor and resulting in a wide open of cleavage site on S2 by host proteases; this process is critical for the viral infection. [22, 26, 39].
The human ACE2 receptor plays a major role that facilitates the viral entry in to the host and infection. Recent studies on the structure of the spike protein of SARS-CoV-2 revealed that spike protein binds to the Peptide Domain of ACE2 with a dissociation constant (Kd) of ~ 15 Nm [10, 15, 17, 35]. ACE2 is widely expressed in the lungs, heart, intestine, and kidneys [6, 19, 37]. Angiotensin, endogenous ligand of ACE2, is a peptide hormone that regulates vasoconstriction and in turn blood pressure. Many cardiovascular diseases are associated with the decreased expression of ACE2 [5, 23, 41].
The structure of the ACE2-PD alone is complex with the RBD of the spike protein of SARS-CoV. The ACE2 structural information is limited to the protein domain. The human ACE2 structure is combined with a membrane protein that is also called chaperones B0AT1 (SLC6A19). This structure of full-length ACE2 with cryo-electron microscopy revealed the presence of ACE2 with neutral amino acid transporter B0AT1 with RBD. Moreover, X-ray crystallography studies at a resolution of 2.9 Å of SARS-CoV-2 reported the structure of spike protein [3, 9, 11, 12, 14, 25, 30, 36]. The RBD is identified by the extracellular peptide domain of ACE2 through polar residues. The ACE2 and SARS-CoV-2 surface spike protein assembles into heterodimers with ACE2 homodimers. These findings will give major information insights into the molecular level for coronavirus recognition and infection. In this complex ACE2 is a dimer and further structure forms a basis for the development of novel therapeutic targets to prevent the viral infection.
The details of molecular interactions and characterization of these interactions between S-RBD and the ACE2 receptor of SARS-CoV-2 are crucial to develop the vaccine and therapeutic drug candidates for the prevention of the viral infection. The structural-based virtual screening of small molecules with RBD, spike protein of SARS-CoV-2 and ACE2 (PDB ID: 6VW1) will provide us with the potential drug candidates. Eventually, structure-based molecular docking was used to identify the inhibitor molecules to target SARS-CoV-2 virus. Recently, the crystal structure of the ACE2 complex was resolved along with the whole genome sequence of 2019-nCoV. In this study, we identified the binding interaction of potential drug candidates and validated the drug candidates using computational methods which lead a way for in vitro and in vivo studies.
Material and Methods
Data availability
The 8,719 SARS-CoV-2 isolates' whole-genome sequences were downloaded from the GISAID (https://www.gisaid.org/) virus database. The sample data was recorded up to date. The whole-genome data was downloaded in FASTA format and each isolate has a unique number for easy identification.
Open reading frame (ORF) identification
ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/) was used to search DNA sequences for potential protein translation. This is the web version; here the query sequence range is 50 kb in length and can identify the spike protein sequence of SARS-CoV-2 for the further alignment process.
Spike protein multiple sequence alignment (MSA) analysis
The amino acid sequences from the different isolates of the SARS-CoV-2 were aligned by the multiple sequence alignment (MSA) method using ClustalW2 program from EMBL-EBI, https://www.ebi.ac.uk/Tools/msa/clustalw2/server and analyzed with default parameter to identify the mutations.
In silico structural characterization of SARS-CoV-2 spike protein
The 3-dimensional structure of the 2019-nCoV chimeric receptor-binding domain (RBD) complexed with its receptor human ACE2 was retrieved from the Protein Data Bank (www.rcsb.org) (PDB ID: 6VW1, 2.68 Å resolution). Structural information was examined and analyzed by the PDBsum online server (https://www.ebi.ac.uk/pdbsum). It includes the secondary structure, tertiary structure, bonds, and angles, dihedral angles of each amino acid, helix-helix, helix-beta, beta-beta, hydrogen bond, disulfide bonds, and covalent bond interactions. Apart from these studies, with the help of Maestro (Schrödinger), further studies like receptor preparation, the binding cavity of the protein analysis, were done.
Molecular modeling and energy minimization of drug library from different database
Total 28 potential anti-viral compounds were retrieved from PubChem (https://pubchem.ncbi.nlm.nih.gov/) and Zinc databases (http://zinc.docking.org/) are summarized in Table 1. All retrieved compounds were converted into a 3-dimensional structure and subjected to energy minimized using the OPLS-2005 force field with the help of Maestro, a visualization software. This process of energy minimization was continued until the molecular conformations got stable or completely energy minimized.
Molecular docking studies of antiviral compounds with SARS-CoV-2 RBD protein
Molecular docking is a popular technique in structural bioinformatics to resolve problems related to protein and ligand interaction. In the context of docking, energy evaluations are usually carried out with the help of binding affinity (Gibbs free energy, ΔG), and structure-based drug design (SBDD).
All energetically minimized molecules were subjected to docking with SARS-CoV-2 RBD protein using FlexX software [https://www.biosolveit.de/FlexX/]. A grid box was defined based on the inhibitory site (Fig. 1) by considering vital residue coordinates by literature. Grid center was defined for SARS-CoV-2 RBD protein as per the functional and structural information obtained from various databases. FlexX supports SDF (Standard Database Format) file format for ligand and PDB format for the receptor. In FlexX the scoring is done using a modified Böhm scoring function including the loss of entropy upon ligand binding; hydrogen bonding; ionic, electrostatic interactions, aromatic interactions, and lipophilic interactions, Molecular docking interactions were analyzed using Maestro. Based on the high negative binding affinity score, the best confirmation of each ligand was used for further analysis.

Binding Pocket of the SARS-CoV-2 Spike Protein RBD region: The protein backbone shown in ribbon form and the cavity of the biding pocket shown in Red color. Amino acid residues that are contributing to the binding site of spike RBD region are shown blue color (Color figure online)
Molecular simulation and dynamic studies of antiviral compounds with SARS-CoV-2 RBD protein
The stability of docked complexes and interaction of spike proteins with the antiviral lead compounds was observed by running 100 ns of molecular dynamic (MD) and simulation using the Desmond Molecular Dynamic System (D.E. Shaw Research, New York, NY, 2017). The complex system was solvated using the explicit solvent (TIP3P) system in the size of 10 Å cubic box with periodic boundary condition (PBC) box and the complex system was neutralized by adding counter ions using the System Setup panel of Desmond. The structural changes of the SARS-CoV-2 RBD (C-α backbone atoms) upon binding of lead compounds were compared with the docked structure of the respective complex in terms of RMSD (Root Mean Square Deviation). Also, the amino acids fluctuations during 100 ns MD simulation were computed and presented as RMSD (Root Mean Square Fluctuation) Plot.
Results
Sequence alignment
In the present study we have analyzed 8,719 spike protein sequences from all over the globe using the GISAID database. The data were analyzed and the number of global mutations of spike protein were shown in Table 2. The infection of COVID-19, male-to-female ratio was 3:2 and with a median age range between 7 and 90 years old. We have identified more novel mutations using Multiple Sequence Alignment and mutational analysis. We have presented in Fig. 2 country wise total mutations and frequent mutations occurred in the SARS-CoV-2 Spike Protein RBD region. The total mutations and frequent mutations were listed in Table 3. Our study revealed that a total of 639 mutations were found globally. The frequent mutations in different isolates are L5F, T22I, T29I, H49Y, L54F, V90F, S98F, S221L, S254F, V367F, A520S, T572I, D614G, H655Y, P809S, A879S, D936Y, A1020S, A1078S, and H1101Y. Moreover, our study found common mutations and the data was presented in Table 4; L5F mutation was identified in the USA, Scotland, Japan, South Africa, France, India, Italy, Norway, and Peru. T22I mutation was observed in the USA, India, Germany, and Iran. The T29I mutation variant is present in Spain and Germany, whereas H49Y is the most common mutation in the USA, India, China, and Mexico. The L54F mutation was present in the USA, China, South Africa, France, and India. V90F mutation was observed in the USA and Scotland. The other mutations, S98F mutation (USA, China, and Spain), S221L mutation (USA and South Africa), S254F and V367F mutations (USA and France), A520S mutations (USA and India), T572I mutation (USA, India and along with France) were observed in various countries. However, D614G mutations are high in worldwide isolates. In the USA, the total sequences that we have analyzed are 2,583 and the number of D614G mutations are 2025 (82%). Similarly in Scotland out of 1134 sequences, the number of D614G mutations are 847 (75%). Out of total 734, 625, 592, 556, 542, 462, 339, sequences from China, Japan, South Africa, France, India, Spain and Germany, D614G mutations are 43 (6%), 331 (53%), 579 (98%), 517(93%), 179 (33%), 276 (60%) and 292 (86%) respectively. Out of total Finland sequences 263, D614G are 253 (96%); Russia sequences 240, D614G are 211 (88%); Brazil sequences 220, D614G are 187 (85%); UK sequences 141, D614G 84 (60%); Italy sequences 128, D614G are 120 (95%); Norway sequences 53, D614G are 43 (81%); Australia sequences 48, D614G are 13 (27%); Peru sequences 34, D614G are 29 (85%); Mexico sequences 20 of D614G are 9 (45%). H655Y mutation was observed in the USA, Scotland, China, South Africa while P809S mutation was present in the USA, Scotland, South Africa, Germany. A879S mutation was present in the USA, Scotland, Japan, India, Finland, UK. And A1020S and A1078S mutations are present in the USA and South Africa and Spain. H1101Y mutations are present in the USA, Scotland, and India.

Global numbers of the Spike protein mutations: The country wise total mutations (blue) and frequent mutations (orange) occurred in the SARS-CoV-2 Spike Protein RBD region is shown in the histogram (Color figure online)
Predicted mutations did not occur in the ACE2 binding site and the role of mentioned mutations is yet to be predicted. All these mutations are based on a comparison with the Wuhan genome sequence (NC_045512.1). These spike protein mutations are present at both upstream and downstream of the receptor-binding domain (RBD) and the mutation Q271R has been found to affect the secondary structure of the S1 domain. The other major mutation at D614G at the position 614 with glycine instead of aspartic acid in the spike protein. Generally, all the amino acids can occur in two isomeric forms except glycine. This could be one of the reasons for spread of the disease faster. Hence, these mutations are unique and might impact the spike receptor interaction with changing of its conformations in the protein. Very importantly, positions of these mutations might be important to find target sites for COVID-19 treatment. Based on the literature review we identified few immune-promoting compounds (Silmitasertib, AC-55541, Merimepodib, XL413, and AZ3451) which could show effectiveness to treat SARS-CoV-2.
Structural analysis
The PROMOTIF (Hutchinson, 1996) program provides information about the secondary structure of a given protein for analysis. As for this analysis, PDB ID: 6VW1 contains chain A and chain B having 596 amino acids with two sheets, two beta hairpins, one beta bulge, six strands, thirty-two helices, fifty six helix-helix interaction, thirty one beta turns, five gamma turns and three disulfides. Other E and F chains contain 193 amino acids with three sheets, one beta bulge, nice strands, seven helices, three helix-helix interaction, thirteen beta turns, three gamma turns and four disulfides.
Docking with SARS-CoV-2 RBD protein
All the designed analogs were docked with SARS-CoV-2 RBD using FlexX software, The docking result shows that the binding score for all the compounds is highly negative suggesting that their binding affinity is greater towards SARS-CoV-2 RBD protein. The 28 analogs were screened for the interaction with the amino acid of the human ACE2 receptor which are FDA approved drugs for the treatment of different cancers and immune promotion, may work against SARS-CoV-2 Silmitasertib, AC-55541, Merimepodib, XL413, and AZ3451 demonstrated the best docking score with hydrogen bonding and hydrophobic interaction with amino acid residues in active site of RBD protein and selected compounds were analyzed using Maestro. Interacting amino acids, bond types, and their bond distances were noted in Table 5.
We have come to a conclusion based on docking studies that the Silmitasertib showed more affinity towards SARS-CoV-2 RBD protein compared with other compounds. Silmitasertib has a docking score of − 26.7713 and also shown a hydrogen bond at a distance 1.85 Å with Phe342, four aromatic hydrogens (AH) bonds with Phe343, Asn343 (2AH), Ala344 with their respective bond distances 2.73 Å, 1.81 Å, 2.33 Å, and 2.62 Å. Also, one halogen bond occurs with Asn343 at 3.34 Å bond distance, two pi–pi interactions with Trp436 and bond distance respectively 4.46 Å and 4.78 Å, and one pi-cation with Arg509 at bond distance 5.73 Å (Fig. 3a).

Intermolecular interactions between docked FDA approved drugs candidates vs SARS-CoV-2 spike RBD region: (a) Binding mode of Silmitasertib with stable SARS-CoV-2 spike RBD, (b) Binding mode of AC55541 with stable SARS-CoV-2 spike RBD (c) The binding mode of Merimepodib within the binding site SARS-CoV-2 spike RBD, (d) Binding mode of XL-413 with SARS-CoV-2 spike RBD protein. (e) Binding mode of AZ3451 within the SARS-CoV-2 spike RBD binding cavity. The hydrogen bonds are shown in dashed Black lines whereas, Pi–Pi stacking, Pi-cation interactions, and aromatic hydrogen bonds were represented in Cyan, Green and Teal color, respectively. The SARS-CoV-2 spike RBD protein is shown in the ribbon form, and the interacting amino acids and the drug candidates are represented in line and ball and stick model (Color figure online)
The second lead compound was shown in Fig. 3b i.e., AC55541 have a docking score of − 24.35 which forms three hydrogen bonds with Asn343 (bond distances—1.60 Å, 1.85 Å and 2.42 Å), one pi–pi interaction with Phe373 (bond distance 4.91 Å) and four aromatic hydrogen bonds with Phe373 (2AH) and Phe342 (2AH) (bond distance 2.43 Å, 2.74 Å, 2.23 Å, and 2.78 Å respectively).
The third lead compound i.e., Merimepodib was shown in Fig. 3c having docking score of − 22.20 which forms two equal hydrogen bonds with Phe342 (bond distance 2.08 Å), one hydrogen bond with Thr372 (bond distance of 2.41 Å), one pi–pi interaction with Trp436 (bond distance 5.09 Å), one pi-cation interaction with Arg509 (bond distance 5.59 Å) and two aromatic hydrogen bonds with Asn343, Ala344 (bond distances of 2.44 Å and 2.27 Å).
Fourth lead compound was XL-413 having docking score of − 20.7836 which forms four hydrogen bonds with Ala344 (two bonds), Asn343, and Trp436 (bond distance 1.83 Å, 2.00 Å, 1.59 Å and 1.77 Å respectively), two aromatic hydrogen bonds with Trp436 and Asn437 (bond distance 3.56 Å and 2.66 Å) and one pi–pi interaction with Phe373 (bond length 5.14 Å) (Fig. 3d).
The fifth lead compound AZ3451 shown in Fig. 3e having a docking score of − 20.64 it forms one hydrogen bond with Asn343 (bond distance 1.61 Å) and two aromatic hydrogen bonds with Thr345 and Phe342 (bond distance 2.35 Å and 1.69 Å).
From the literature study, the screened high-affinity lead compounds were reported for the immunomodulatory effects. The screened compounds have shown high docking scores with strong binding energies, and close interaction with conserved catalytic residues. These results indicate that small molecules identified in our study are promising drugs for COVID-19 treatment.
Molecular dynamics and simulation analysis
To explore dynamic perturbation in the conformation of the spike protein and ligand complex molecular dynamic simulation was carried out. The 100 ns simulations have run for the spike (RBD region) receptor, the potential energy tends to decrease which intimate the stabilization of the system. The conformations obtained during the 100 ns simulation for the RBD-ligand complex were analyzed and RMSD (Root mean square deviation) was calculated during the simulation time. The displacement of selected atoms can be calculated in order of the average change for the particular frame concerning the reference frame.
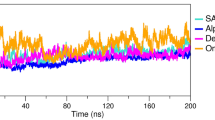
The RMSD plot of RBD and antiviral drugs is shown in Fig. 4a indicating that all the complex systems get equilibrated from 20 to 100 ns MD simulation. In the initial phase (0–20 ns) of MD simulation, deviations in the complex systems were observed and the complex system gets stabilized from 2.5 to 4.0 Å.

Molecular dynamic simulation analysis: a RMSD plot for the SARS-CoV-2 spike RBD (C-α atoms) molecular simulated with antiviral drug candidates within 100 ns time scale. b RMSF plot for the SARS-CoV-2 spike RBD (C-α atoms) docked with antiviral drug candidates within 100 ns time scale in molecular dynamic simulation
Moreover, to probe the structural changes of Spike RBD upon binding of the antiviral compounds, the RMSF (Root mean square fluctuation) for C-alpha atoms of all the residues were compared using trajectories generated during 100 ns molecular dynamic simulations. It is outstanding that the RMSF trajectories of RBD (Fig. 4b) stability of the system is associated with more rigid and stable conformations. The RMSF graph suggests that Spike RBD shows the minimum fluctuation after binding of antiviral lead compounds, only the fluctuation in the region of residues (473–490) fluctuated from 3 to 7 Å. From this MD simulation, we can conclude that the lead compounds form the stable complex with Spike RBD and are thermodynamically favorable.
Discussion
When we embarked the SARS-CoV-2 sequence analysis our interest was turned to identify the mutations that could be a potential target for spike protein to find drug compounds. The GISAID database provides data to look at more deep sequence analysis and evolutionary relationships with many SARS-CoV-2 sequences. In this context, our approach revealed various mutations in the spike protein region, the sequence analysis done by comparing with the initial Wuhan isolate spike protein sequence.
We have analyzed clinical data of spike protein sequences thoroughly and found different mutation in spike region. These mutation help virus to spread fast and increase the virus load [1]. Our studies shown that the spike being manifested among with all sites of the frequent mutations viz. L5F, T22I, T29I, H49Y, L54F, V90F, S98F, S221L, S254F, V367F, A520S, T572I, D614G, H655Y, P809S, A879S, D936Y, A1020S, A1078S, and H1101Y. Among them the most appearance mutation is D614G, identified as high frequent mutation worldwide. D614G is an important mutation that causes structural changes which in turn increase the affinity towards ACE2 and leading to functional changes in SARS-CoV-2 spike protein. While the other mutations may have structural changes nearby the receptor-binding domain in the S1 domain of the spike. This mutation could show an impact on the binding activity of the S1 domain, and it can change secondary structure of the protein [1]. The mutations play an important role in to change evolutionary origins of the new isolates. For viruses like SARS-CoV-2 also the natural law of survival of fittest applies. This leads to the creation of new mutations in spike protein that result in new variants with strong binding to the host receptor to increase the virulence capacity and help in escaping the immune system. The hotspot region of the mutations in the S1 and S2 conserved domain will make a stronger virus to attach the host receptor for viral entry. Hence, exploring this area will help to the best antigen immunogenic region for vaccine development, and other antiviral small molecule drugs. These compounds may be neutralizing the virus potentiality and reduce the infection. For this, we have done a virtual screening of compounds against SARS-CoV-2 binding receptor ACE2.
From the docking analysis, we have selected 5 lead compounds that were having potential inhibitor activity on spike protein RBD binding with ACE2. Among five drugs the recently developed CK2 inhibitor, Silmitasertib, selectively binds and inhibits the enzyme casein kinase II (CK2) in different cancers, also reported anti-fungal activity [21]. Silmitasertib has been tested in phase I/II COVID-19 clinical trials https://clinicaltrials.gov/ct2/show/NCT04668209) and shown highest binding affinity, − 26.44 kcal/mol among all tested compounds. AC-55541 is a potent and selective protease-activated receptor 2 (PAR-2) agonist [24]. Our docking analysis showed binding affinity score − 24.35 kcal/mol. PARs are transmembrane receptors and play a major role in inflammatory responses. They act as a sensor to the proteolytic enzyme formation during viral infection. When tested with human bronchial epithelial cells, the administration of AC-55541 significantly induced the expression of PAR2 and inhibited the Acanthamoeba plasminogen activator.
Merimepodib (MMPD) is a novel selective inhibitor of inosine monophosphate dehydrogenase (IMPDH) and was reported for antiviral effects [38]. Our docking analysis revealed that binding affinity score of MMPD is − 22.20 kcal/mol. The inhibition of IMPDH can reduce the available intracellular guanine nucleotide concentration for viral RNA synthesis. Besides, it is an immune modulator and antiviral agent in combination with interferons (INF) [16]. The significant treatment for viral infections and other clinical benefits of MMPD pegylated interferon may effective for the treatment of COVID-19. XL-413 is a potent and selective ATP competitive CDC7 inhibitor [13]. XL-143 is a novel bezofuropyrimidinone inhibitor with a favorable pharmacokinetics property denoted for the significant tumor growth regression and arrest the cell cycle (S phase to the G2 phase) of tumor cells will prevent viral infections at molecular level [13]. Our docking analysis showed XL-143 has strong binding affinity (− 20.78 kcal/mol) for spike RBD region. AZ3451 is a PAR2 antagonist and binds to the remote allosteric site outer to the helical bunch. Our results showed AZ3451 has stronger binding affinity (− 20.64 kcal/mol) against SARS CoV-2 RBD region. These antagonists prevent structural rearrangements, which required for the receptor activation and signaling cascade mechanisms, and can be effectively used as a therapeutic agent to treat viral infections [4].
In conclusion, we have identified mutations in spike RBD region that lead to structural changes of spike are most important for the interaction with host ACE2 receptor. Our study reveals that Silmitasertib, AC-55541, Merimepodib, XL413 and AZ3451 showed promising results against the novel emerged strain of coronavirus. All these compounds have exhibited excellent binding capacity to SARS-CoV-2 RBD protein. These compounds may be effective to control or stop the viral entry and further infection, as well our study paves a way for further in vivo studies as well clinical trials.
References
Banerjee AK, Begum F, Thagriki D, Tripathi PP, Ray U. Novel mutations in the S1 domain of COVID 19 spike protein of isolate from Gujarat Origin, Western India. Preprints 2020, 2020040450. https://doi.org/10.20944/preprints202004.0450.v1.
Belouzard S, Chu VC, Whittaker GR. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci U S A. 2009;7(106):5871–6. https://doi.org/10.1073/pnas.0809524106.
Bröer A, Klingel K, Kowalczuk S, Rasko JE, Cavanaugh J, Bröer S. Molecular cloning of mouse amino acid transport system B0, a neutral amino acid transporter related to Hartnup disorder. J Biol Chem. 2004;279:24467–76. https://doi.org/10.1074/jbc.M400904200.
Cheng RKY, Fiez-Vandal C, Schlenker O, Edman K, Aggeler B, Brown DG, Brown GA, Cooke RM, Dumelin CE, Doré AS, Geschwindner S, Grebner C, Hermansson NO, Jazayeri A, Johansson P, Leong L, Prihandoko R, Rappas M, Soutter H, Snijder A, Sundström L, Tehan B, Thornton P, Troast D, Wiggin G, Zhukov A, Marshall FH, Dekker N. Structural insight into allosteric modulation of protease-activated receptor 2. Nature. 2017;545:112–5. https://doi.org/10.1038/nature22309.
Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–8. https://doi.org/10.1038/nature00786.
Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res. 2000;87:E1-9. https://doi.org/10.1161/01.res.87.5.e1.
Gallagher TM, Buchmeier MJ. Coronavirus spike proteins in viral entry and pathogenesis. Virology. 2001;279:371–4. https://doi.org/10.1006/viro.2000.0757.
Gaunt ER, Hardie A, Claas EC, Simmonds P, Templeton KE. Epidemiology and clinical presentations of the four human coronaviruses 229E, HKU1, NL63, and OC43 detected over 3 years using a novel multiplex real-time PCR method. J Clin Microbiol. 2010;48:2940–7. https://doi.org/10.1128/JCM.00636-10.
Hamming I, Cooper ME, Haagmans BL, Hooper NM, Korstanje R, Osterhaus AD, Timens W, Turner AJ, Navis G, van Goor H. The emerging role of ACE2 in physiology and disease. J Pathol. 2007;212:1–11. https://doi.org/10.1002/path.2162.
Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271-280.e8. https://doi.org/10.1016/j.cell.2020.02.052.
Kirchdoerfer RN, Wang N, Pallesen J, et al. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci Rep. 2018;8:17823. https://doi.org/10.1038/s41598-018-36918-8.
Kleta R, Romeo E, Ristic Z, Ohura T, Stuart C, et al. Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder. Nat Genet. 2004;36:999–1002. https://doi.org/10.1038/ng1405.
Koltun ES, Tsuhako AL, Brown DS, Aay N, Arcalas A, Chan V, Du H, Engst S, Ferguson K, Franzini M, Galan A, Holst CR, Huang P, Kane B, Kim MH, Li J, Markby D, Mohan M, Noson K, Plonowski A, Richards SJ, Robertson S, Shaw K, Stott G, Stout TJ, Young J, Yu P, Zaharia CA, Zhang W, Zhou P, Nuss JM, Xu W, Kearney PC. Discovery of XL413, a potent and selective CDC7 inhibitor. Bioorg Med Chem Lett. 2012;22:3727–31. https://doi.org/10.1016/j.bmcl.2012.04.024.
Kowalczuk S, Bröer A, Tietze N, Vanslambrouck JM, Rasko JE, Bröer S. A protein complex in the brush-border membrane explains a Hartnup disorder allele. FASEB J. 2008;22:2880–7. https://doi.org/10.1096/fj.08-107300.
Kuba K, Imai Y, Rao S, Gao H, Guo F, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–9. https://doi.org/10.1038/nm1267.
Lauer GM, Walker BD. Hepatitis C virus infection. N Engl J Med. 2001;345:41–52. https://doi.org/10.1056/NEJM200107053450107.
Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–4. https://doi.org/10.1038/nature02145.
Liu Z, Xiao X, Wei X, Li J, Yang J, Tan H, Zhu J, Zhang Q, Wu J, Liu L. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J Med Virol. 2020;92:595–601. https://doi.org/10.1002/jmv.25726.
Ma M, Lin XH, Liu HH, Zhang R, Chen RX. Suppression of DRP1-mediated mitophagy increases the apoptosis of hepatocellular carcinoma cells in the setting of chemotherapy. Oncol Rep. 2020;43:1010–8. https://doi.org/10.3892/or.2020.7476.
Malik YS, Sircar S, Bhat S, Sharun K, Dhama K, Dadar M, Tiwari R, Chaicumpa W. Emerging novel coronavirus (2019-nCoV)-current scenario, evolutionary perspective based on genome analysis and recent developments. Vet Q. 2020;40:68–76. https://doi.org/10.1080/01652176.2020.1727993.
Masłyk M, Janeczko M, Martyna A, Kubiński K. CX-4945: the protein kinase CK2 inhibitor and anti-cancer drug shows anti-fungal activity. Mol Cell Biochem. 2017;435:193–6. https://doi.org/10.1007/s11010-017-3068-z.
Millet JK, Whittaker GR. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015;202:120–34. https://doi.org/10.1016/j.virusres.2014.11.021.
Raizada MK, Ferreira AJ. ACE2: a new target for cardiovascular disease therapeutics. J Cardiovasc Pharmacol. 2007;50:112–9. https://doi.org/10.1097/FJC.0b013e3180986219.
Seitzberg JG, Knapp AE, Lund BW, Mandrup Bertozzi S, Currier EA, Ma JN, Sherbukhin V, Burstein ES, Olsson R. Discovery of potent and selective small-molecule PAR-2 agonists. J Med Chem. 2008;51:5490–3. https://doi.org/10.1021/jm800754r.
Seow HF, Bröer S, Bröer A, Bailey CG, Potter SJ, Cavanaugh JA, Rasko JE. Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19. Nat Genet. 2004;9:1003–7. https://doi.org/10.1038/ng1406.
Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U S A. 2005;102:11876–81. https://doi.org/10.1073/pnas.0505577102.
Simmons G, Reeves JD, Rennekamp AJ, Amberg SM, Piefer AJ, Bates P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc Natl Acad Sci U S A. 2004;101:4240–5. https://doi.org/10.1073/pnas.0306446101.
Simmons G, Zmora P, Gierer S, Heurich A, Pöhlmann S. Proteolytic activation of the SARS-coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antivir Res. 2013;100:605–14. https://doi.org/10.1016/j.antiviral.2013.09.028.
Song W, Gui M, Wang X, Xiang Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018;14:e1007236. https://doi.org/10.1371/journal.ppat.1007236.
Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J Biol Chem. 2004;279:17996–8007. https://doi.org/10.1074/jbc.M311191200.
Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. 2020;94:e00127-e220. https://doi.org/10.1128/JVI.00127-20.
Wilder-Smith A, Freedman DO. Isolation, quarantine, social distancing, and community containment: pivotal role for old-style public health measures in the novel coronavirus (2019-nCoV) outbreak. J Travel Med. 2020;27:taaa20. https://doi.org/10.1093/jtm/taaa020.
World Health Organization. Public health surveillance for COVID-19: interim guidance. Who.Int; 2021. https://www.who.int/publications/i/item/who-2019-nCoV-surveillanceguidance-2020.8.
World Health Organization. Summary of probable SARS cases with onset of illness. Who.int; 2002. https://www.who.int/csr/sars/country/en/-2005.
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–3. https://doi.org/10.1126/science.abb2507.
Zhang H, Wada J, Hida K, Tsuchiyama Y, Hiragushi K, Shikata K, Wang H, Lin S, Kanwar YS, Makino H. Collectrin, a collecting duct-specific transmembrane glycoprotein, is a novel homolog of ACE2 and is developmentally regulated in embryonic kidneys. J Biol Chem. 2001;276:17132–9. https://doi.org/10.1074/jbc.M006723200.
Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. Am J Respir Crit Care Med. 2020;202:756–9. https://doi.org/10.1164/rccm.202001-0179LE.
Zhou S, Liu R, Baroudy BM, Malcolm BA, Reyes GR. The effect of ribavirin and IMPDH inhibitors on hepatitis C virus subgenomic replicon RNA. Virology. 2003;310:333–42. https://doi.org/10.1016/s0042-6822(03)00152-1.
Zhou P, Yang XL, Wang XG, Hu B, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–3. https://doi.org/10.1038/s41586-020-2012-7.
Zhu N, Zhang D, Wang W, Li X, Yang B, et al. China novel coronavirus investigating and research team. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–33. https://doi.org/10.1056/NEJMoa2001017.
Zisman LS, Keller RS, Weaver B, Lin Q, Speth R, Bristow MR, Canver CC. Increased angiotensin-(1–7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin-converting enzyme Homologue ACE2. Circulation. 2003;108:1707–12. https://doi.org/10.1161/01.CIR.0000094734.67990.99.
Acknowledgements
Authors thank DST-SERB and ICMR for providing financial assistance to Dr. V. Damodara Reddy under Ramanujan Fellowship (SB/S2/RJN-043/2014) and Mrs. Swetha Pulakuntla under ICMR-SRF (ISRM/11(20/2017) scheme. Authors also thank DST-SERB GPU optimized Super computer for calculations at Dr. D. Y. Patil Biotechnology and Bioinformatics Institute, Pune India.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pulakuntla, S., Lokhande, K.B., Padmavathi, P. et al. Mutational analysis in international isolates and drug repurposing against SARS-CoV-2 spike protein: molecular docking and simulation approach. VirusDis. 32, 690–702 (2021). https://doi.org/10.1007/s13337-021-00720-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13337-021-00720-4