
Abstract
Coronavirus disease 2019 is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). We have used bioinformatics to investigate seventeen mutations in the spike protein of SARS-CoV-2, as this mediates infection of human cells and is the target of most vaccine strategies and antibody-based therapies. Two mutations, H146Y and S221W, were identified as being most pathogenic. Mutations at positions D614G, A829T, and P1263L might also have deleterious effects on protein function. We hypothesized that candidate small molecules may be repurposed to combat viral infection. We investigated changes in binding energies of the ligands and the mutant proteins by assessing molecular docking. For an understanding of cellular function and organization, protein–protein interactions are also critical. Protein–protein docking for naïve and mutated structures of SARS-CoV-2 S protein was evaluated for their binding energy with the angiotensin-converting enzyme 2 (ACE2). These interactions might limit the binding of the SARS-CoV-2 spike protein to the ACE2 receptor or may have a deleterious effect on protein function that may limit infection. These results may have important implications for the transmission of SARS-CoV-2, its pathogenesis, and the potential for drug repurposing and immune therapies.
Similar content being viewed by others


Introduction
Coronaviruses are a large family of enveloped RNA viruses that were named for the crown-like spikes on their surface (Su et al. 2016). There are seven types of human coronaviruses including HCoV-229E (229E), HCoV-OC43 (OC43), severe acute respiratory syndrome coronavirus (SARS-CoV), HCoV-NL63 (NL63), HCoV-HKU1 (HKU1), Middle East respiratory syndrome coronavirus (MERS-CoV) (Su et al. 2016), and the coronavirus known as SARS-CoV-2 that may result in severe respiratory tract disease (Kaur and Gupta 2020). It is thought that COVID-19, the disease caused by SARS-CoV-2, began in China and has spread in the world (Wu et al. 2020a). The common symptoms in COVID-19 patients are a dry cough, fever, breathing difficulties (dyspnea), and pneumonia (Wu et al. 2020b). The development of inflammation-induced lung injury can lead to acute respiratory distress syndrome (ARDS) (Shi et al. 2020), respiratory failure, and death (Wu et al. 2020b).
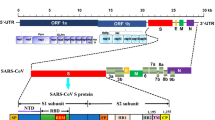
SARS-CoV-2 contains two categories of proteins: structural proteins that include Spike (S), Nucleocapsid (N), Matrix (M), and Envelope (E) and non-structural proteins that include proteases (nsp3 and nsp5) and RdRp (nsp12) (Ibrahim et al. 2020). To enter the host cells, coronaviruses fuse to the cell membrane using the viral surface spike protein which is a crucial step in the viral entry into cells (Ibrahim et al. 2020; Prompetchara et al. 2020). The spike protein contains two subunits, S1 and S2, which are mediators of attachment and membrane fusion, respectively. S1 is also divided into proportions, including N-terminal domain (NTD) and C-terminal domain (C-domain). Either NTD or C-domain may function as the receptor-binding domain (RBD), depending on the virus (Ou et al. 2020). The spike protein binds to the human angiotensin-converting enzyme 2 (ACE2) receptor, which is expressed in the lung, heart, intestinal epithelium, vascular endothelium, and kidneys and is considered to be the dominant entry portal (Clerkin et al. 2020; Dhar and Mohanty 2020). The efficiency of viral infection depends on this process, and several factors are associated with protein–protein interactions, including the nature of residues and the type of chemical interactions between ligand and receptor. Therefore, the presence of residues that have the lower free energy are more favored and might drive binding kinetics and result in the fusion event (Ortega et al. 2020).
The Spike protein has become a focus for vaccine development for protection against coronaviruses (Du et al. 2009). Besides the important role of this protein in viral entry, it is also able to stimulate the immune response during the viral infection which was the key in both SARS-CoV and MERS-CoV vaccination studies (Prompetchara et al. 2020), and it has therefore been utilized for SARS-CoV-2 vaccine design (Ahmed et al. 2020). The spike protein was found to target T cells after natural infection which results in the induction of CD8+ T-cell responses against the membrane (M) and N proteins. T-cell response may also be involved in long-term defense and memory T cell activities may remain for many years after infection (Vabret et al. 2020). Since antiviral T-cell responses specific to RBD have been detected in people after the COVID-19 recovery, it has been reported to be a promising vaccine target. Also, the antibodies that target the RBD of the spike protein or other segments of the spike protein have been investigated to maintain immunity as target epitopes for T cells (Vabret et al. 2020). Moreover, memory B cells are responsible for the rapid production of plasma cells with high affinity in response to the re-infection and the long-term protection will be accomplished by inducing these long-lived plasma cells and memory B cells (Vabret et al. 2020). Furthermore, a study on SARS-CoV-2 pseudotype lentiviruses claims that there are two immune-dominant linear B-cell epitopes known as S14P5 and S21P2 on the SARS-CoV-2 spike protein, which are critical in neutralizing and controlling COVID-19 infection. Epitope S14P5 is located near the RBD, so it is possible that antibodies binding to this site may inhibit binding to ACE2 receptor. In addition, there is a possibility of an allosteric effect upon ACE2 binding. There is a portion of the fusion peptide sequence in the S21P2 epitope likely to play a potential role in the virus cell fusion (Poh et al. 2020). According to another study, only 5–6 amino acids can be detected by antibodies; so there will be 983 antibody binding sites if all epitopes in the 1255 amino acid long SARS-CoV-2 Spike protein can be used by the antibodies; they will also bind to human protein epitopes (Sørensen et al. 2020). A similar increase in antibody-dependent enhancement (ADE) for coronaviruses can be seen in animal models. This increase allows them to enter cells that express FcγR. Some have suggested that amino acid diversity and antigen drift due to mutations may lead to antibody-dependent progression (Negro 2020; Ricke and Malone 2020).
The mutation rate for RNA viruses is high, and this is associated with changes in virulence (Duffy 2018). As the virus spreads, the SARS-CoV-2 genome becomes potentially capable of gaining rapid mutations as the virus spreads and tries to adapt to a new environment. These variations allow the virus to survive and propagate in host cell. Because the spike protein can mutate so readily, it is important to obtain a broad mutation profile of this protein from extensive genome sequencing. Recent advances in sequencing have provided a wealth of information on the genetic mutations present in various organisms, including SARS-CoV-2. But the major challenge is recognizing and characterizing certain genetic mutations which have functional effects. Missense mutations are of particular concern. These can interfere with the functions of the transcribed proteins by affecting their stability and modulating interactions with other biological molecules. Predicting the effects of mutations on protein stability and interactions is therefore important for understanding different biological processes including resistance to illness and to the medications because they might also impact severity of disease. It is worth mentioning that according to current information, there are several ways in which mutations of the Spike protein can affect pathogenicity. For instance, it can enhance receptor binding, fusion activation, or antibodies to mediate antibody-dependent enhancement (ADE) elicitation against this peptide (Korber et al. 2020a). So, development of computational techniques is strongly required to predict the effects of mutations on protein stability to support the quick and routine analysis of the sequencing data needed for personalized medicine. To predict the impact of mutations on protein stability, we report our revised knowledge-based SDM (Site-Directed Mutator) and the webserver SDM2 in this study. SDM pioneered the use of conformational constrained environment-specific substitution tables (ESSTs) for measuring thermal stability differences between naive and mutant protein (Blundell 2017). Generally, we show that a mutation of S protein that results in more transmissible SARS-CoV-2 inhibits the S1 domain shedding and enhances the incorporation of S protein in the virion. As we need further research to assess the effect of this shift on the development and extent of COVID-19, in this study, the pathogenicity and stability of non-mutant and mutant SARS-CoV-2 Spike proteins were predicted and compared using different bioinformatics tools in order to investigate the impact of mutations on the structure and function of the protein, so we evaluate the drug efficiency and the interactions between the SARS-CoV-2 spike proteins and the human cell receptor ACE2 due to the mutations as well.
In addition to the importance of the Spike protein for vaccine design (Ahmed et al. 2020), its crucial role in drug development is axiomatic as well (Wu et al. 2020b). In the combat against COVID-19 infection, many researchers have come up with different strategies in drug development. Currently, there is no available specific medication for COVID-19 and procedure a new drug is a long-term process, so repurposing FDA-approved drugs creates opportunities to advance potential treatments. There are several candidate drugs that might be able to inhibit the infection and replication of SARS-CoV-2. In current studies, the drugs are being tested in order to target 3CL protein, block the ACE2, the host cell receptor for the S protein of SARS-CoV-2, and inhibit TMPRSS2, which is required for S protein priming may prevent cell entry of SARS-CoV-2 (Huang et al. 2020; McKee et al. 2020). However, in this study we focused on targeting the S protein.
So, our fundamental strategy was to use existing molecular databases to screen for molecules that may have therapeutic effect on coronavirus, especially those that might affect the spike protein including Imatinib, Remdesivir, Telaprevir, Arbidol, Zafirlucast, a flavanone called Hesperidin, Pemirolast, Isoniazid pyruvate, Nitrofurantoin, Cefoperazone, and Ivermectin which were chosen to be investigated in this study. The method is based on the pathological characteristics and genomic information of different mutated forms of coronavirus in order to develop new targeted drugs from scratch. The selected drugs found would show better anti-coronavirus effects, but the research procedure of discovering new drug might take several years. We conducted SARS-CoV-2 spike proteins using the Swiss model to be the ligand targets, so that we could predict a variety of analysis of therapeutic targets for SARS-CoV-2.
Materials and Methods
Datasets
The protein sequence of the spike glycoprotein was extracted in FASTA format from the UniProt database (UniProt ID: P0DTC2) (http://www.uniprot.org/). The PDB structure was retrieved from the Protein Data Bank for structural analysis (PDB ID: 6xr8).
Predicting Functional Impact of Missense Mutation
Recently, numerous methods of in silico predictions have been developed to determine the effects of mutations in amino acids on proteins that may change the function. Many methods of prediction focus on the physicochemical properties of amino acids and the structure of their side chains, and others use annotations that are accessible, such as Gene Ontology. Classification methods such as those that are based on machine learning techniques including neural networks, support vector machines, Bayesian methods, and mathematical operations are also available. The computationally derived information about the structure and function of the protein and the properties of both the residues of native and substituted amino acids is combined and eventually characterizes the mutation as being either disease linked or neutral (Mueller et al. 2015). Top 17 abundant non-synonymous mutations (the most frequent the world) observed in S protein of SARS-CoV-2 obtained from the National Genomics Data Center (https://bigd.big.ac.cn/ncov/protein) (Table 1).
Pathogenic Prediction of nsSNPs (Non-Synonymous Single-Nucleotide Polymorphisms)
The seventeen protein mutations (S221w, H146y, A829T, D614g, H49y, S247r, L5f, V483a, Y28n, P1263L, D839n, A879t, R21k, D936y, V320g, S477n, and L54f) were subject to computational prediction using Meta-SNP server (http://snps.biofold.org/meta-snp/index.html), MutPred2, and PROVEAN. Meta-SNP evaluates the results to distinguish between disease-related and polymorphic non-synonymous SNPs, based on the random forest binary classifier. This prediction tool comprises other algorithms such as PANTHER (Mi et al. 2007), PhD-SNP (Capriotti et al. 2006), SIFT (Ng and Henikoff 2003), SNAP (Johnson et al. 2008), and Meta-SNP to predict the pathogenicity of the mutations. The scores range between 0 and 1, and the score > 0.5 for the mutation is predicted to be disease.
PhD-SNP (Predictor of Human Deleterious Single-Nucleotide Polymorphisms)
Predictor of human Deleterious Single Nucleotide (PhD-SNP) (https://snps.biofold.org/phd-snp/phd-snp.html) is a support vector machine (SVM) based on an online classifier. It is used for the pathogenicity prediction that is dependent on a single support vector machine by training/testing datasets of protein sequence and profile.
The range of scores reported to be somewhere between 0 and 1, and the score > 0.5 for the transformation is anticipated to be infection.
SIFT (Sorting Intolerant from Tolerant)
SIFT (https://sift.bii.a-star.edu.sg/) classifies whether the protein feature is affected by a mutation based on sequence homology and the physical properties of the amino acids. This method can be used to characterize the mutations that occur naturally and the mutations caused by the laboratory. The values are positive and the mutation score > 0.05 is predicted to be neutral.
SNAP (Screening for Non-Acceptable Polymorphisms)
SNAP is a method based on neural networks that applies an advanced machine learning approach to study the effects of nsSNPs (Bromberg et al. 2008). The estimation of the loss or gain of a protein’s function due to mutation is depicted based on sequence data and structural components, such as secondary structure, solvent accessibility, and the conservation of residue within sequence families. The scores range between 0 and 1, and the score > 0.5 for the mutation is predicted to be disease.
PROVEAN
Protein Variation Effect Analyzer (PROVEAN is a software) (PROVEAN v1.1.3.) is used for prediction of the effect of an amino acid substitution or indel on protein biological function. It is useful for sequence variants filtration to distinguish non-synonymous or indel variants that are anticipated to be significant in terms of function.
MutPred2
MutPred2 (http://mutpred.mutdb.org/) is a machine-based system and software that combines genetic and molecular data to provide probabilistic reasoning about amino acid substitution pathogenicity (Pejaver et al. 2020). This is done by offering a general prediction of pathogenicity and a graded list of particular molecular modifications that could potentially influence the phenotype. It is trained on a set of 53,180 pathogenic and 206,946 unlabeled (putatively neutral) variants obtained from the Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/ac/index.php), dbSNP, SwissVar (https://web.expasy.org/swissvar.html), and inter-species pairwise alignment. The MutPred2 model is a bagged ensemble of 30 feed-forward neural networks, each trained on a balanced subset of pathogenic and unlabeled variants (Pejaver et al. 2020).
Stability Prediction of nsSNPs
Prediction of protein stability changes resulting from single amino acid variations helps in understanding the structure of the protein. The stability analysis was performed using I-Mutant 3.0 (Capriotti et al. 2008), MUpro (Cheng et al. 2006), and SDM (Topham et al. 1997) to analyze the impact of deleterious variants on the Spike glycoprotein.
I-Mutant 3.0
The I-Mutant 3.0 (http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi) is based on a support vector machine (SVM) algorithm that uses single-point mutations to predict protein stability changes. This tool is a classifier that is used after mutation and regression estimation to estimate the sign of change in protein stability and predicts deltaDeltaG values. The final file represents the DDG (predicted energy change). This is calculated based on the Gibbs free energy change of the mutant protein by subtracting the native protein’s apparent free energy value (Kcal/mol). DDG > 0 value represents an increase of stability while DDG < 0 value represents a decrease of stability (Capriotti et al. 2008).
MUpro
The web server for predictions of protein stability changes upon mutations (MUpro) for single amino acid mutations, available at http://mupro.proteomics.ics.uci.edu/, was used based on SVM and Neural Networks (two machine learning programs). The output of the program is in the form of a plus or minus energy change sign. Positive energy change indicate that the mutation leads to increased stability by the mutation (marked as neutral), while negative value of ΔΔG energy change indicate that the mutation has destabilized (marked as destructive) (Cheng et al. 2006).
SDM (Site-Directed Mutator)
SDM2 has been regularly tested on a large variety of datasets. The newly revised environment-specific substitution tables (ESSTs), based on residue packing density, have improved the method’s overall efficiency. Analysis of the use of residue packing density has demonstrated an improved ability to identify disease and non-disease mutations and thus SDM2 with the new Modified ESST is expected to be a helpful method for understanding disease mutations and directing protein engineering based on packaging densities. SDM, http://mordred.bioc.cam.ac.uk/~sdm/sdm.php, is a statistical approach for prediction of SNPs effect on the proteins stability developed by Topham et al. (1997). This useful tool can be used for Mutagenesis studies design or prediction of the effect of mutations on proteins structure. SDM uses substitution frequencies of environment-specific amino acid in the families of homologous protein to calculate a stability score similar to the free energy difference between the native and mutant proteins. The performance of this method has been better or comparable than other methods presented in the classification of mutations as stabilizing or destabilizing (Worth et al. 2011).
Evolutionary Conservation Analysis
To measure conservation degree at each aligned position, the ConSurf server (http://consurf.tau.ac.il) (Glaser et al. 2003) was used for calculation of the Spike glycoprote conservation pattern. From the very beginning, it uses various grouping arrangements and detects monitored positions, and then, at that point, gauges the evolutionary survival rate utilizing empirical Bayesian inference. Finally, it gives the evolutionary survival profiles of the structure or protein sequence. The range of ConSurf score varies from 1 to 9, in which 1 addressing quickly developing sites, 5 addressing the average ones, and 9 addressing gradually evolving sites. This technique additionally assesses the structural or functional effect of amino acids within the protein.
Physicochemical Property Analysis
NCBI Amino Acid Explorer (https://www.ncbi.nlm.nih.gov/Class/Structure/aa/aa_explorer.cgi) gives an itemized clarification of properties like size, charge, side-chain adaptability, hydrogen bonds, and hydrophobicity to evaluate changes in the biophysical and chemical properties of native and mutant amino acids (Bulka and Freeland 2006).
Molecular Docking
For the molecular docking experiment, the non-mutant structure of the template SARS-CoV-2 spike protein (PDB: 6xr8) and its seventeen mutant structures were chosen to be the target proteins which were obtained from the RCSB (http://www.rscb.org) and Protein Data Bank and Swiss model (http://swissmodel.expasy.org) respectively, in (PDB) format.
The 3D structures of different compounds were tested with the non-mutant and mutant SARS-CoV-2 Spike proteins, including Imatinib, Remdesivir, Telaprevir, Zafirlukast, Hesperidin, Pemirolast, Isoniazid pyruvate, Nitrofurantoin, Cefoperazone, and Ivermectin, using PubChem database (https://pubchem.ncbi.nlm.nih.gov) in Structure-data file (SDF) format.
To simulate protein and ligands binding affinity, ligands converted to PDB format by SMILES (https://cactus.nci.nih.gov/translate) and inserted into AutoDock Tools (version 1.5.6) (http://autodock.scripps.edu).
From that point onward, the downloaded proteins embedded into the work place are made ready. At first, the water particles were omitted and then software tools were used to add polar hydrogen and Kollman charges. After the arrangements, simulations of bioactive adaptations were made via AutoDock Vina (http://autodock.scripps.edu). A complete parameter that controls the search extent was selected as 8, and for each ligand, 9 models were provided. Best docked complexes were obtained by absolute clustering of root-mean-square deviation (RMSD) factor (with value of 0.0.). Based on the obtained results, lower energy scores showed the best interactions of protein–ligand. Various forms of the ligands influence the docking score estimation. In order to obtain an accurate estimation, their most active forms were utilized. RMSD values of 3 or greater than 3 show no docking occurrence. There was only one valid docking position with the root-mean-square deviations of atomic positions (RMSD = 0).
For protein–protein docking, we searched the docking results of ACE2 (PDB: 1r42) with the non-mutant structure of the template SARS-CoV-2 spike protein and its seventeen mutant structures (S221w, H146y, A829T, D614g, H49y, S247r, L5f, V483a, Y28n, P1263L, D839n, A879t, R21k, D936y, V320g, S477n, and L54f). Data were obtained by ClusPro online server (https://cluspro.bu.edu) in which the association of the two proteins was assumed to be driven by their electrostatic interactions, so results were obtained by the electrostatic-favored weights. ClusPro uses the scoring feature of PIPER that contains terms of shape complementarity, electrostatics, and pairwise potentials applied on the top 1000 conformations generated and ranked according to cluster size (Kozakov et al. 2013, 2017b; Vajda et al. 2017).
After completion, docking results were imported to Discovery Studio 4.5 and the best position of each ligand was loaded as outputs. The different poses of the ligands affect the estimation of the docking score, so the most active forms were used in order to obtain an accurate estimation. The interactions of amino acids and ligands were also examined by Discovery Studio 4.5.
Results
Pathogenicity and Stability Predictions
Different tools for predicting the impact of mutations on the structure and function of the protein were used to predict the pathogenicity and stability. Seventeen mutated variants were exposed to tools of pathogenic prediction (Meta-SNP) and stability prediction (SDM, MUpro, I-Mutant 3.0).
Accordingly, 6 variants were identified as pathogenic in at least one of the databases, and in most databases, S221W and H146Y had the highest pathogenicity (Tables 2, 3).
Conservation Analysis
The conservation pattern exhibits the significance of a residue that assists with keeping up the structure and function of protein. ConSurf surveys the conservation degree at each aligned position, showing the localized development (Glaser et al. 2003).
First, Multiple Sequence Alignment is used to detect the conserved position and then an empirical Bayesian interface is used to measure the rate of developmental preservation. The ConSurf tool was used to assess the amino acids conservation levels of 17 variant positions. A more conserved position of mutation may influence the protein function. The effects of the highly conserved area on the species are shown in Fig. 1. Thus, mutations of D614G, A829T, and P1263L positions may have destructive effects on the protein.

Conservation analysis of the protein sequence of Spike glycoprotein using ConSurf. The positions P1263L is highly conserved with a score of 9 and present exposed region of the protein
Analysis of Physicochemical Properties
Substitution of amino acids causes physicochemical effects, which can in turn cause local and general changes in the protein. These changes are based on changes in charge, size, hydrophobicity, hydrogen bonds, lateral chain flexibility, etc. In this study, a comparison between native and mutant proteins in terms of physicochemical properties was performed using NCBI-amino acid explorer (Table 3).
The mutation of serine to arginine/tryptophan at position S247R resulted in an alteration of the side-chain flexibility from low to high/moderate. The mode of interaction in serine was found to consist of H-bonds and van der Waals interactions, whereas arginine/tryptophan contributed to Ionic, H-bonds, van der Waals, and aromatic stacking interaction. There was an increase in hydrophobicity, molecular weight, and change in polarity from polar to Positive/non-polar. Histidine’s mutation to Tyrosine resulted in increased hydrophobicity, polarity changes from Positive to Polar, and molecular weight. The side-chain flexibility was changed from high/moderate to reduce in the case of the mutation Tyrosine/Aspartic acid to alanine. The interaction mode in Glutamate/Aspartate consisted of interactions between Ionics, hydrogen bonds, and van der Waals. There was a reduction in hydrogen bonds, an increase in hydrophobicity and a decrease in molecular weight, and a polarity change from negative to non-polar as well. The mutation of Alanine to Threonine resulted in decrease in hydrophobicity and the number of side-chain H-bonds, but molecular weight increased and polarity changes from non-polar to Polar. Also, the side-chain flexibility was modified from limited to low. In the case of the mutation Leucine to Phenylalanine, there was an increase in molecular weight and hydrophobicity. There was a decrease in hydrophobicity and molecular weight and a change in polarity from positive to polar in the mutation Histidine to Tyrosine. With regards to the mutation Proline to Leucine, there can be seen that the side-chain flexibility changed from moderate to restricted mood and hydrophobicity and molecular weight increased due to the mutation. Also, general property changed from Imino into Aliphatic. The mutation of Valine to Alanine and Glycine decreased the hydrophobicity and the molecular weight and changed the side-chain flexibility from low to limited and none, respectively. The mutation of Aspartic acid to Tyrosine resulted in increase in hydrophobicity, molecular weight, and isoelectric point. The polarity changed from negative to polar. An increase in the number of side-chain H-bonds, isoelectric point, and hydrophobicity can be seen in the mutation of Aspartic acid to Asparagine. Arginine’s mutation to Lysine resulted in an increase in the number of side-chain H-bonds, molecular weight, isoelectric point, and hydrophobicity, while all these factors saw a decline in the mutation of Serine to Asparagine. In addition, side-chain flexibility changes into moderate from low.
Protein–Ligand Docking
In AutoDock Vina, Imatinib, Remdesivir, Telaprevir, Zafirlukast, Hesperidin, Pemirolast, Isoniazid pyruvate, Nitrofurantoin, Cefoperazone, and Ivermectin were docked with the non-mutant and seventeen structures of mutant SARS-CoV-2 spike protein (Table 4, Fig. 2). The results showed significant binding of the ligands with the target proteins. Proteins and the best pose of each ligand were visualized in Discovery Studio and the residue interactions were visualized as well. As the template, all ligands docked with the non-mutated structure of SARS-CoV-2. The lowest energy conformations of all the ligands were selected for proteins as outputs.

Docking results of seventeen mutation variants of SARS-CoV-2 spike protein with candidate drugs (best mode: RMSD = 0.000)
Imatinib
Imatinib is an Abl kinase inhibitor used for treating Philadelphia chromosome-positive chronic myelogenous leukemia (CML) and acute lymphocytic leukemia (ALL) (Kerkelä et al. 2006). First, we docked Imatinib (ligand) with the non-mutant structure of SARS-CoV-2 spike protein and it showed the binding affinity of − 9.6 kcal/mol (Fig. 3a). Then the ligand was docked with all mutant structures and D936y mutation of SARS-CoV-2 spike protein and showed the most significant binding energy with Imatinib with the binding affinity of − 10.6 kcal/mol which has the most affinity in comparison with the other mutated structures (Fig. 4aʹ).

Molecular interactions between a Imatinib and naive spike protein, b Remdesevir and naive spike protein, c Telaprevir and naive spike protein, d Zafirlukast and naive spike protein, e Hesperidin and naive spike protein, f Pemirolast and naive spike protein, g Isoniazid pyruvat and naive spike protein, h Nitrofurantoin and naive spike protein, i Cefoperazone and naive spike protein, and j Ivermectin and naive spike protein

Molecular interactions between aʹ Imatinib and D936y-mutated variant of SARS-CoV-2 spike protein, bʹ Remdesevir and H49Y and bʺ S247R-mutated variants of SARS-CoV-2 spike protein, cʹ Telaprevir and V483A-mutated variant of SARS-CoV-2 spike protein, dʹ Molecular interactions between Zafirlukast and D839N-mutated variant of SARS-CoV-2 spike protein, eʹ Hesperidin and Y28n-mutated variant of SARS-CoV-2 spike protein, fʹ Pemirolast and L54f-mutated variant of SARS-CoV-2 spike protein, gʹ Isoniazid pyruvat and V483A-mutated variant of SARS-CoV-2 spike protein, hʹ Nitrofurantoin and H146Y-mutated variant of SARS-CoV-2 spike protein, iʹ Cefoperazone and V483A-mutated variant of SARS-CoV-2 spike protein. jʹ Ivermectin and S477n-mutated variant of SARS-CoV-2 spike protein
Remdesevir
Remdesivir is a nucleotide analog pro-drug that is metabolized intracellular to an analog of adenosine triphosphate inhibiting viral RNA polymerases. Remdesivir with its broad-spectrum activity against members of many families of viruses such as coronaviruses (e.g., SARS-CoV and MERS-CoV) demonstrated therapeutic and prophylactic efficiency in non-clinical models of these viruses. According to docking results, the binding affinity of Remdesivir with non-mutant SARS-CoV-2 spike protein (Fig. 3b) and the most effective mutant structure, h49y and s247r, were − 9.3 kcal/mol and − 9.9 kcal/mol, respectively (Fig. 4bʹ, bʺ).
Telaprevir
Telaprevir is an anti-HCV drugs and known to be a potential inhibitor against coronaviruses. In this study we tested the binding affinity of Telaprevir with non-mutant and mutant SARS-CoV-2 spike protein structures. The docking of Telaprevir with non-mutant structure as well as the other mutant structures of SARS-CoV-2 spike protein (Fig. 3c) revealed that the drug shows high-affinity interaction to the protein with the total affinity of more than − 12.0 kcal/mol and so, the mutation did not significantly affect the binding affinities except in V483a-, R21k-, and S477n-mutated structures with the higher binding energies of − 11.0, − 11.2, and − 11.7, respectively (Fig. 4cʹ).
Zafirlukast
Zafirlukast is a leukotriene receptor antagonist used for the chronic treatment of asthma has been considered as one of the efficient drugs for controlling Coronaviruses the same as the SARS-CoV-2 (Xu et al. 2020). In this study, we analyzed the drug’s binding affinities with non-mutant and mutant SARS-CoV-2 spike protein structures. Zafirlukast had a high binding affinity that might inhibit RBD–receptor interaction (Fig. 3d).The drug binding affinity of the non-mutant structure and the most effective mutant structure, D839N, were − 10.6 kcal/mol and so, there were no recorded mutations that might have increased the binding affinity (Fig. 4dʹ).
Hesperidin
Hesperidin is a flavanone that can be efficient in controlling coronaviruses with its Anti-inflammatory and anti-oxidant effect especially for SARS-CoV-2. Accordingly, hesperidin is able to target the binding between Spike RBD of SARS-CoV-2 and human ACE2 (Wu et al. 2020a). In this study, the docking of Hesperidin with non-mutant structure revealed that the compound shows high affinity interaction with affinity of − 10.8 kcal/mol which was the highest affinity compared to the mutant structures (Fig. 4eʹ), so none of the mutations were able to show lower binding energies (Fig. 3e).
Pemirolast
Pemirolast is as an anti-allergic drug and has also been studied for the treatment of asthma (Fujitaka et al. 1999). Pemirolast has emerged as a potential candidate for spike protein disruption and interaction with SARS-CoV-2, so it could potentially disrupt the SARS-CoV-2 interface with ACE2 receptors (Smith and Smith 2020). Hence, we analyzed the drug’s binding affinity with non-mutant and mutant SARS-CoV-2 spike protein structures (Fig. 3f). The drug binding affinity of the non-mutant structure and mutant structures were remained quite steady between − 7.1 and − 7.6 kcal/mol which were positioned in the RBD of the protein (Fig. 4fʹ).
Isoniazid Pyruvate
Another candidate drug is an antibiotic used for the treatment of tuberculosis called Isoniazid pyruvate and has been introduced to combat against SARS-CoV-2 (Smith and Smith 2020). Docking results for mutant and non-mutant structures (Fig. 3g) were − 6.3 kcal/mol that reveals that mutations did not change the binding affinities compared to the naive structure of the spike protein except S247r, L5f, and V483A with their increasing effect in binding affinity with the docking scores of − 6.5, − 6.6, and − 6.8 kcal/mol, respectively (Fig. 4gʹ).
Nitrofurantoin
Nitrofurantoin or Macrobid an antibiotic used in bladder infections treatment suggested that this small molecule could mitigate SARS-CoV-2 infection (Smith and Smith 2020). In this study, top-scoring ligands for RBD–ACE2 receptor interface are contributed to A829T-, D614g-, H49y-, S247r-, L5f-, V483a-, Y28n-, P1263L-, D839n-, A879t-, R21k-, and V320g-mutated structures with the lower energies compared to the naive spike protein with docking score of − 7.7 kcal/mol (Figs. 3h, 4hʹ). In comparison, only three structures revealed highest energies of − 6.7 to − 6.9 kcal/mol.
Cefoperazone
Cefoperazone is an FDA-approved beta-lactam antibiotic used to treat multiple bacterial infections especially infections of the respiratory tract. Recent studies have suggested that Cefoperazone can be considered as a receptor-binding domain (RBD)–ACE2 interaction inhibitor (Senathilake et al. 2020). According to molecular docking, first binding pose of the drug showed hydrogen bonding with receptor-binding domain (RBD) residues with the lowest energy of − 10.5 kcal/mol in mutant structure V483a which was − 8.8 in the naive structure (Figs. 3i, 4iʹ).
Ivermectin
Another drug that we investigated through molecular docking is Ivermectin. It is used to treat several forms of parasite infestations, but antiviral effects against multiple SARS-CoV-2 infections have recently been reported (Caly et al. 2020). Here, Ivermectin docked with the naïve and mutated variants of spike protein RBD. The binding energy of Ivermectin to the naïve spike protein was − 10.1 kcal/mol. Although there was a slight increase in binding affinity up to 10.3 with some of the mutated variants, the affinity with H49y, L5f, V483a, D839n, V320g, S477n, and L54F decreased. The highest binding energy was accounted for S477n variant (Figs. 3j, 4jʹ).
Protein–Protein Docking
In this study, ClusPro, a web-based server, was used for the docking of two interacting proteins. There, Root-Mean-Square Deviation (RMSD)-based clustering of the lowest energy structures were created to identify the largest clusters that will represent the most possible complex models and refinement of the selected structures using energy minimization. Also, ClusPro selects the centers of highly populated clusters of the low-energy structures. Each cluster size shows the corresponding energy width which provides the data on entropic contributions to the free energy. Accordingly, cluster size is the best way to rank models, which is how the models are ranked from Cluspro (Kozakov et al. 2017a).
Accordingly, protein–protein docking between non-mutant and 17 mutant SARS-CoV-2 Spike protein structures was performed to evaluate their binding energy with Angiotensin-converting enzyme 2 (ACE2). ClusPro analysis provided 30 complexes with respective energy scores. The lowest energy complex was accounted for H146y mutant structure of SARS-CoV-2 Spike protein with the score of − 1130.2 kJ mol−1. Moreover, L5f and P1263l mutations showed an equal Weighted Score (Lowest Energy = − 1054.0 kJ mol−1) with the non-mutant spike protein. Some mutations also might attenuate the Spike protein and ACE2 interactions with the recorded highest energies including S247r, D614g, D839n, L54f, and A879t. The table that lists the clusters of docked structures in the order of cluster size is available (Table 5).
Discussion
COVID-19 has become a major health problem globally. SARS-CoV-2 spike protein binds to its receptor, the ACE2 (hACE2) via its receptor-binding domain (RBD) after which priming cleavage occurs at the interface of the S1 and S2 domain (S1/S2) by the proteases for the viral fusion (Duan et al. 2020). With the rapid spread of the virus, there is an opportunity for the virus to undergo a process of natural selection. This is the reason for some natural mutations in S protein (Wang et al. 2020). The S protein mutation is too important because it is the first major step in initiating virus transmission. The role that protein S plays in infection/virus transmission is particularly important in the development and production of monoclonal vaccines/antibodies. In these studies, evidence has been presented to show how is the reaction of circulating natural species to neutralizing antibodies (Wang et al. 2020).
In this study, SARS-CoV-2 spike protein mutations have been studied using bioinformatics tools to investigate and explain the role of spike protein and natural mutations. Wet laboratory tests are not a substitute for in silicon applications. However, their supportive role in empirically confirming disease-related alleles and prioritizing the most likely new pathogenic species is undeniable and can therefore play a more helpful role in diagnostic strategies. There are many tools for assessing the functional importance of these variables, but it is still a challenging task to obtain the reliability of predicted results.
To evaluate the S protein pathogenicity of SARS-CoV-2 missense variants, this study used a combined approach of molecular modeling and intra-silicon mutation predictions. This combination method can also detect deleterious variants. Based on the obtained results tools such as MutPred, PMUT, and Provean were among the most accurate in predicting the pathogenicity of SARS-CoV-2 missense variant. The phenotypic consequences of nsSNPs were also predicted by silicon algorithms. This leads to a great understanding of genetic differences in disease susceptibility and drugs responses. As a result, according to what has been said, seventeen mutations can be observed in the spike protein, two of which are the most pathogenic (H146Y and S221W) (Tables 2, 3). In addition, mutations in the D614G, A829T, and P1263L positions can cause adverse effects on the protein. Because the L5F mutation is a signal peptide mutation, it is difficult to predict its effect on the virus. An interesting issue is the recurrence of this mutation in many phylogenetic SARS-CoV-2 tree lineages and among all people around the world. But the frequency of the virus has not increased and has remained the same. It is near the end of the cytoplasmic spike tail that the P1263L mutation occurs and this mutation does not enter the SARS-CoV-2 structure of protein (Korber et al. 2020a).
According to another study (Wang et al. 2020), a variety of human and animal cell lines were infected by 106 pseudotyped SARS-CoV-2 viruses. Based on the results, the D614G variants or combined variants such as D614G+V341I, D614G+K458R, D614G+I472V, D614G+D936Y, D614G+S939F, and D614G+S943T showed a four to 100-fold increase in pathogenicity compared to the reference Wuhan-1 strain (GenBank: MN908947). Potential associations of D614G with elevated viral loads have also been reported in COVID-19 patients. Accordingly, it has been studied that deletions of 22 putative glycosylation sites in addition to amino acid modifications in the S protein of SARS-CoV-2 are also effective, and six glycosylation mutants were considered to have low infectivity. Infectivity was significantly decreased by the ablation of both but not either, N331 and N343 glycosylation at the receptor-binding domain (RBD). They also used 13 neutralizing monoclonal antibodies to investigate the antigenicity of the infectious mutants. Ten mutations, such as N234Q, L452R, A475V, V483A, and F490L, were discovered and were surprisingly immune to some monoclonal antibodies (Wang et al. 2020).
Through the next step, we modeled all these mutations and ran simulations to study the effects of these mutations in binding with nine candidate drugs and the ACE2 receptor to study the effects and consequences of every mutation. The molecular docking results can show the drug’s possible targets of action although some of these targets may be false positives due to the model inaccuracy for small flexible protein or partial model. Our docking results showed kinds of binding interactions of the candidate drugs with protein structures among which some interactions are more favorable and/or significant according to the binding affinity and interactions (Figs. 3, 4). Although these selected compounds are also unable to bind with the contact surface of ACE2–Spike complex or are not specifically designed to target the Spike protein, they might be a good start point in degrading the protein and then inhibiting the virus. Any small molecule bound to Spike might inhibit the process of viral infection by interfering the refolding of Spike protein which is useful to design PROTAC-based therapy.
Imatinib, Remdesivir, Telaprevir, Zafirlukast, and the flavanone, Hesperidin, showed high affinity interactions with all the protein structures, including non-mutant and mutant structures. Furthermore, Pemirolast, Isoniazid pyruvate, Nitrofurantoin, Cefoperazone, and Ivermectin were the five top candidates that have high affinity for the ACE2 receptor–spike protein interface. Thus, these interactions might limit the binding of the SARS-CoV-2 Spike protein with the ACE2 receptor that results in infection restriction. Because the binding of the S protein to ACE2 is unfavorable, the interaction between the S protein and the ACE2 receptor is preferable to decrease.
Overall, from the ranking, 5 ligands (Pemirolast, Isoniazid pyruvate, Nitrofurantoin, Ivermectin, and Cefoperazone) were found to interact with receptor-binding domain (RBD)–ACE2 complexes with scores equal to or better than the score of naïve spike protein, in average. Moreover, the lowest binding energies for all the candidate drugs docked with seventeen mutated structures of SARS-CoV-2 spike protein may confirm the attenuating effects of some of the mutations on the Spike protein. In comparison to the naïve spike protein structure, it can be inferred from Table 4 and Fig. 2 that Hesperidin, Zafirlukast, and Telaprevir had no positive effect on affinity with selected mutations which means that they were not able to lower the binding energies. However, Imatinib, Remdesevir, Pemirolast, Isoniazid pyruvate, Nitrofurantoin, Cefoperazone, and Ivermectin might be able to increase the affinity with some mutated structures by decreasing the binding energy although binding energy changes related to Isoniazid pyruvate, Pemirolast, Nitrofurantoin, and Ivermectin were quite steady and not significant in comparison. Significant changes in binding affinity were mostly due to the mutation V483A. With regards to the most pathogenic mutations, H146Y and S221W, it can be inferred that the binding affinity between S protein and several ligands decreased due to the mutation. More specifically, Nitrofurantoin had the weakest binding affinity with H146Y and S221W compared to the non-mutated S protein. The affinity was decreased between H146Y and S221W and Hesperidin and Zafirlukast as well. These mutations also saw a slight decrease in affinity with Imatinib. Interestingly, H146Y did not change the affinity between Remdesevir and S protein, but the affinity decreased by the result of S221W mutation.
It is also worth mentioning that these compounds are found to be efficient in controlling COVID-19 infection based on previous studies that will be mentioned here. So, they were chosen as the targets for our study. Imatinib substantially decreases SARS-CoV and MERS-CoV viral titers blocking the entry of SARS-CoV or MERS-CoV S protein (Sisk et al. 2018). SARS-CoV-2 is highly homologous to the SARS-CoV (Li et al. 2020), so studying the effects of Abl kinase inhibitors on SARS-CoV and MERS-CoV may be useful for identifying the host cell pathways required for COVID-19 infection. Also, in vitro results of Remdesivir on SARS-CoV-2 were successful (Grein et al. 2020). Based on clinical trials, in adults who were hospitalized with Covid-19 and had signs of lower respiratory tract infection, Remdesivir was significantly effective in shortening the time for recovery (Beigel et al. 2020). The Food and Drug Administration released an Emergency Use Authorization in support of preliminary data on Remdesivir. Since that time, Remdesivir has also obtained approval in many other countries (Beigel et al. 2020). It is worth mentioning that for all patients, treatment with an antiviral drug alone is not likely to be effective. Present methods test Remdesivir in conjunction with immune response modifiers [e.g., the Janus kinase (JAK) inhibitor Baricitinib in ACTT-2, and interferon beta-1a in ACTT-3]. To continue to improve outcomes in patients, a diversity of therapeutic approaches, including novel antivirals, immune response modifiers, and combination approaches, are required (Beigel et al. 2020). Telaprevir is able to bind to the active site of the virus pain-like protease (PLpro), which is characterized in different coronaviruses, so it may contradict the replication of the virus (Elfiky and Ibrahim 2020). The SARS-CoV PLpro and SARS-CoV-2 PLpro protein sequences act in a similar way Thus, effective protease inhibitors for SARS-CoV can also be effective in inhibiting SARS-CoV-2 (Korber et al. 2020a). Another selected drug is ivermectin. It is an inhibitor of HIV-1 (IN) integrase protein and the α/β1 importin heterodimer (IMP) interaction that imports IN nuclear. Based on studies, the drug has been able to bind to the SARS-CoV-2 receptor-binding domain of ACE2 in other studies too. Based on the binding of Ivermectin, there is a potential interference with the binding of spike to the membrane of human cell (Lehrer and Rheinstein 2020).
Research on SARS-CoV proteins has also shown the potential role of this drug for IMPα/β1 during infection in the SARS-CoV nucleocapsid protein signal-dependent closure. Subsequent reports suggested that ivermectin nuclear transport inhibitory activity also affected SARS-CoV-2. They control viral replication in the body within 24 to 48 h. In fact, the binding between ivermectin and the Impa/b1 heterodimer occurs, which in turn prevents Impa/b1 from binding to the viral protein and thus prevents the virus from entering to nucleus.
In addition, it has been indicated that high dose of Ivermectin is as safe as the standard low-dose treatment (Caly et al. 2020).
Moreover, the kind of interaction with the active site amino acids number can likewise be considered important. The hydrogen bond interactions can be very important. This is because they play an important role in structures binding and free energies binding; although the van der Waals and Pi interactions helped to stabilize the binding structures. The interactions occurring in the active proteins positions become much better and more desirable. Figures 3 and 4 indicate types of interactions and amino acids in the proteins inhibition including a large number of Pi-sigma interactions involving charge transfer which helps drug intercalation in the receptor-binding site. In addition, Pi–alkyl bond plays a role in the improvement of hydrophobic interactions of the ligand in the receptor-binding pocket.
Protein–protein interaction may be important to gain an understanding of cell function and organization. Based on initial studies of molecular docking, there is an effect of SARS-CoV-2 protein mutation on ACE2 receptor-binding energy and RBD region of SARS-CoV-2 protein. The model scores were represented by ClusPro results in a set of equilibrium coefficients and actual weight coefficients of energy conditions when Spike protein structures were fused to ACE2. The display of energy values should be based on cluster size rather than energy, with an emphasis on selecting models. In fact, there is no direct connection with the binding affinity in the calculated energy. But it is noteworthy that there is a tendency to produce large clusters of docked structures in low-energy areas. A cluster’s size is roughly proportional to its probability, so the energy system represents the largest possible composition of the complex indirectly. In this study, it was concluded that compared to the non-mutant structure of S protein, H146Y has the lowest binding energy and D614G and S247R have the highest binding energy to ACE2 receptor. The intra-silicon study generally suggests that H146Y in the SARS-CoV-2 protein will have the most potent interaction with the ACE2 receptor. This increases the binding of the SARS-CoV-2 virus to human cells that preset the ACE2 receptor.
Based on molecular docking results in previous studies, D614G, V367F, and H49Y could enhance cell entry compared with wild-type S protein. These results are particularly crucial because the D614G mutation is rapidly spreading around the globe (Wang et al. 2020). D614 is located on the surface of the Spike protein protomer, where it contacts with the neighboring protomer. The change to G614 would eliminate the side-chain hydrogen bond, increase the flexibility of the main chain, and change interactions between protomers. However, although G614 is associated with higher viral loads in patients, it not related to the severity of disease. The D614G S protein is biologically and structurally different from the wild-type S protein, so it has been hypothesized that this mutation might affect the antigenicity of the S protein (Korber et al. 2020b; Ozono et al. 2020). Also, D614G mutation may impact the virus’ infectivity by improving receptor binding, fusion activation, or antibody-dependent enhancement (ADE) antibody elicitation (Korber et al. 2020a). Although higher infectiousness of the variants may account for the rapid viral spread and persistence, considering other factors is also important such as epidemiological factors that might cause changes in genotype frequency to mimic evolutionary pressures (Korber et al. 2020b).
According to the findings, a regional variation of spike protein mutations and its possible effects can be effective in the spread and exacerbation of the disease. Escape of antibody–variants may emerge as a result of these changes. As a result, studies on sequence changes should be conducted to help better in returning to therapeutic targeting. Mutations should be studied more extensively as the consequences of mutations in COVID-19 should be ascertained. This study will help us better understand the made variations and the design of the SARS-CoV-2 vaccine.
Conclusion
We have developed methods to assess and track SARS-CoV-2 spike protein 17 mutations, since the spike protein mediates human cell infection and is the target of most vaccine strategies and antibody-dependent therapies. Two mutations, H146Y and S221E, were identified to have the most pathogenic effect. We focused on the impacts of each spike protein mutation on binding energies with drugs. Our docking results showed the different kinds of binding interactions of the highlighted nine top candidate drugs with protein structures among which some interactions were more favorable according to the binding affinity and interactions. Compared to the non-mutated S protein, the most pathogenic mutations showed the lowest affinity with some of the candidate drugs, including Nitrofurantoin, Hesperidin, Zafirlukast, and Imatinib. We also used molecular simulations of structural models of both naive and mutant SARS-CoV-2 spike protein and bind them to the ACE2 receptor to generate an ensemble of configurations for docking which showed that due to some mutations, the spike protein and ACE2 interactions could possibly change in comparison with the non-mutant structure of spike protein. Accordingly, H146Y had the lowest energy which means that it can show the most enhancing interaction with ACE2 receptor. These results are critical in the repurposing of small molecules against the SARS-CoV-2 infection that is needed for future experimental studies.
Data Availability
The data supporting the findings of this study are available within the article.
Abbreviations
- ARDS:
-
Acute respiratory distress syndrome
- SARS-CoV:
-
Severe acute respiratory syndrome coronavirus
- MERS-CoV:
-
Middle East respiratory syndrome coronavirus
- ACE:
-
Angiotensin-converting enzyme
- nsSNPS:
-
Non-synonymous single nucleotide polymorphisms
- ADE:
-
Antibody-dependent enhancement
- PhD-SNP:
-
Predictor of human deleterious single nucleotide polymorphisms
- SIFT:
-
Sorting intolerant from tolerant
- SNAP:
-
Screening for non-acceptable polymorphisms
- SDM:
-
Site directed mutator
- S:
-
Spike
- N:
-
Nucleocapsid
- M:
-
Matrix
- E:
-
Envelope
- PDB:
-
Protein Data Bank
- PDBQT:
-
Protein Data Bank, Partial Charge (Q), & Atom Type (T)
- MVD:
-
Molecular docking software
- CML:
-
Chronic myelogenous leukemia
- ALL:
-
Acute lymphocytic leukemia
- TKI:
-
Tyrosine kinase inhibitor
- EGFR:
-
Epidermal growth factor receptor
- BTK:
-
Bruton’s tyrosine kinase
- CRS:
-
Cytokine release syndrome
- JAK inhibitor:
-
Janus kinase inhibitor
- ACE2:
-
Angiotensin-converting enzyme 2
- AT2R:
-
Angiotensin II receptor
- DPP4i:
-
Dipeptidyl peptidase 4 inhibitors
- RdRP:
-
RNA-dependent RNA polymerase
- RDV:
-
Remdesivir
References
Ahmed SF, Quadeer AA, McKay MR (2020) Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses 12:254
Beigel JH et al (2020) Remdesivir for the treatment of Covid-19—preliminary report. N Engl J Med 383(10):994–994
Blundell TL, Pandurangan AP, Ochoa-Montaño B, Ascher DB (2017) SDM: a server for predicting effects of mutations on protein stability. Nucleic Acids Res 45(W1):W229–W235
Bromberg Y, Yachdav G, Rost B (2008) SNAP predicts effect of mutations on protein function. Bioinformatics 24:2397–2398. https://doi.org/10.1093/bioinformatics/btn435
Bulka B, Freeland SJ (2006) an interactive visualization tool to explore the biophysical properties of amino acids and their contribution to substitution matrices. BMC Bioinform 7:329
Caly L, Druce JD, Catton MG, Jans DA, Wagstaff KM (2020) The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir Res 178:104787. https://doi.org/10.1016/j.antiviral.2020.104787
Capriotti E, Calabrese R, Casadio R (2006) Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 22:2729–2734
Capriotti E, Fariselli P, Rossi I, Casadio R (2008) A three-state prediction of single point mutations on protein stability changes. BMC Bioinform 9:S6
Cheng J, Randall A, Baldi P (2006) Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 62:1125–1132
Clerkin KJ et al (2020) COVID-19 and cardiovascular disease. Circulation 141:1648–1655
Dhar D, Mohanty A (2020) Gut microbiota and Covid-19-possible link and implications. Virus Res 285:198018. https://doi.org/10.1016/j.virusres.2020.198018
Du L, He Y, Zhou Y, Liu S, Zheng B-J, Jiang S (2009) The spike protein of SARS-CoV—a target for vaccine and therapeutic development. Nat Rev Microbiol 7:226–236. https://doi.org/10.1038/nrmicro2090
Duan L, Zheng Q, Zhang H, Niu Y, Lou Y, Wang H (2020) The SARS-CoV-2 spike glycoprotein biosynthesis, structure, function, and antigenicity: implications for the design of spike-based vaccine immunogens. Front Immunol. https://doi.org/10.3389/fimmu.2020.576622
Duffy S (2018) Why are RNA virus mutation rates so damn high? PLoS Biol 16:e3000003
Elfiky A, Ibrahim NS (2020) Anti-SARS and anti-HCV drugs repurposing against the Papain-like protease of the newly emerged coronavirus (2019-nCoV). Research Square Preprints
Fujitaka M, Ono H, Sebata N, Shirakawa K, Sakura N, Ueda K (1999) Therapeutic Drug Monitoring for pemirolast potassium in childhood asthma. Nihon Shoni Arerugi Gakkaishi Jpn J Pediatr Allergy Clin Immunol 13:32–37
Glaser F, Pupko T, Paz I, Bell RE, Bechor-Shental D, Martz E, Ben-Tal N (2003) ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19:163–164
Grein J, Ohmagari N, Shin D, Diaz G, Asperges E, Castagna A, Feldt T, Green G, Green ML, Lescure FX, Nicastri E (2020) Compassionate use of remdesivir for patients with severe Covid-19. N Engl J Med 382(24):2327–2336
Huang Y-F, Bai C, He F, Xie Y, Zhou H (2020) Review on the potential action mechanisms of Chinese medicines in treating coronavirus disease 2019 (COVID-19). Pharmacol Res 158:104939. https://doi.org/10.1016/j.phrs.2020.104939
Ibrahim IM, Abdelmalek DH, Elshahat ME, Elfiky AA (2020) COVID-19 spike-host cell receptor GRP78 binding site prediction. J Infect 80:554–562. https://doi.org/10.1016/j.jinf.2020.02.026
Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O’Donnell CJ, De Bakker PI (2008) SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 24:2938–2939
Kaur SP, Gupta V (2020) COVID-19 vaccine: a comprehensive status report. Virus Res 288:198114. https://doi.org/10.1016/j.virusres.2020.198114
Kerkelä R et al (2006) Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med 12:908–916
Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, Foley B, Giorgi EE, Bhattacharya T, Parker MD, Partridge DG (2020a) Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. bioRxiv
Korber B et al (2020b) Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182:812–827
Kozakov D, Beglov D, Bohnuud T, Mottarella SE, Xia B, Hall DR, Vajda S (2013) How good is automated protein docking? Proteins 81:2159–2166
Kozakov D et al (2017) The ClusPro web server for protein-protein docking. Nat Protoc 12:255–278. https://doi.org/10.1038/nprot.2016.169
Lehrer S, Rheinstein PH (2020) Ivermectin docks to the SARS-CoV-2 spike receptor-binding domain attached to ACE2. In Vivo 34:3023–3026
Li YC, Bai WZ, Hashikawa T (2020) The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J Med Virol 92:552–555
McKee DL, Sternberg A, Stange U, Laufer S, Naujokat C (2020) Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol Res 157:104859. https://doi.org/10.1016/j.phrs.2020.104859
Mi H, Guo N, Kejariwal A, Thomas PD (2007) PANTHER version 6: protein sequence and function evolution data with expanded representation of biological pathways. Nucleic Acids Res 35:D247–D252
Mueller S et al (2015) Protein degradation corrects for imbalanced subunit stoichiometry in OST complex assembly. Mol Biol Cell 26:2596–2608
Negro F (2020) Is antibody-dependent enhancement playing a role in COVID-19 pathogenesis? Swiss Med Wkly. https://doi.org/10.4414/smw.2020.20249
Ng PC, Henikoff S (2003) SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 31:3812–3814
Ortega JT, Serrano ML, Pujol FH, Rangel HR (2020) Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: an in silico analysis. EXCLI J 19:410
Ou X et al (2020) Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun 11:1620. https://doi.org/10.1038/s41467-020-15562-9
Ozono S et al (2020) Naturally mutated spike proteins of SARS-CoV-2 variants show differential levels of cell entry. bioRxiv
Pejaver V et al (2020) Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat Commun 11:5918. https://doi.org/10.1038/s41467-020-19669-x
Poh CM et al (2020) Potent neutralizing antibodies in the sera of convalescent COVID-19 patients are directed against conserved linear epitopes on the SARS-CoV-2 spike protein. bioRxiv. https://doi.org/10.1101/2020.03.30.015461
Prompetchara E, Ketloy C, Palaga T (2020) Immune responses in COVID-19 and potential vaccines: lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol 38:1–9
Ricke D, Malone RW (2020) Medical countermeasures analysis of 2019-nCoV and vaccine risks for antibody-dependent enhancement (ADE). Available at SSRN 3546070
Senathilake K, Samarakoon S, Tennekoon K (2020) Virtual screening of inhibitors against spike glycoprotein of 2019 novel corona virus: a drug repurposing approach. https://doi.org/10.20944/preprints202003.0042.v2
Shi Y et al (2020) COVID-19 infection: the perspectives on immune responses. Cell Death Differ 27:1451–1454. https://doi.org/10.1038/s41418-020-0530-3
Sisk JM, Frieman MB, Machamer CE (2018) Coronavirus S protein-induced fusion is blocked prior to hemifusion by Abl kinase inhibitors. J Gen Virol 99:619
Smith M, Smith JC (2020) Repurposing therapeutics for the wuhan coronavirus nCov-2019: supercomputer-based docking to the viral S protein and human ACE2 interface. ChemRxiv
Sørensen B, Susrud A, Dalgleish AG (2020) Biovacc-19: a candidate vaccine for Covid-19 (SARS-CoV-2) developed from analysis of its general method of action for infectivity. QRB Discov 1:e6. https://doi.org/10.1017/qrd.2020.8
Su S et al (2016) Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol 24:490–502. https://doi.org/10.1016/j.tim.2016.03.003
Topham CM, Srinivasan N, Blundell TL (1997) Prediction of the stability of protein mutants based on structural environment-dependent amino acid substitution and propensity tables. Protein Eng 10:7–21
Vabret N et al (2020) Immunology of COVID-19: current state of the science. Immunity 52(6):910–941
Vajda S et al (2017) New additions to the C lus P ro server motivated by CAPRI. Proteins 85:435–444
Wang L, Wang L, Zhuang H (2020) Profiling and characterization of SARS-CoV-2 mutants’ infectivity and antigenicity. Signal Transduct Target Ther 5:185. https://doi.org/10.1038/s41392-020-00302-8
Worth CL, Preissner R, Blundell TL (2011) SDM—a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res 39:W215–W222
Wu C et al (2020a) Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B 10:766–788. https://doi.org/10.1016/j.apsb.2020a.02.008
Wu C et al (2020b) Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. https://doi.org/10.1016/j.apsb.2020b.02.008
Xu C et al (2020) Systemic in silico screening in drug discovery for coronavirus disease (COVID-19) with an online interactive web server. J Chem Inf Model 60(12):5735–5745
Acknowledgements
None.
Funding
None.
Author information
Authors and Affiliations
Contributions
Conceptualization: ES and MM. Data curation: SSB. Investigation: AEN. Methodology: SN and NA. Writing of the original draft: NA and ES. Writing, reviewing, and editing of the manuscript: RN and RF. Analysis and/or interpretation of data: MM and SZ. Revising the manuscript critically for important intellectual content: GAF. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have declared no conflict of interest.
Ethical Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
For this type of study, formal consent is not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Alizadehmohajer, N., Zahedifar, S., Sohrabi, E. et al. Using In Silico Bioinformatics Algorithms for the Accurate Prediction of the Impact of Spike Protein Mutations on the Pathogenicity, Stability, and Functionality of the SARS-CoV-2 Virus and Analysis of Potential Therapeutic Targets. Biochem Genet 61, 778–808 (2023). https://doi.org/10.1007/s10528-022-10282-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10528-022-10282-9