
Abstract
Spontaneous intracerebral hemorrhage (ICH) accounts for 10–30% of all strokes and affects more than one million people every year worldwide, and it is the stroke subtype associated with the highest rates of mortality and residual disability. So far, clinical trials have mainly targeted primary cerebral injury and have substantially failed to improve clinical outcomes. The understanding of the pathophysiology of early and delayed injury after ICH is, hence, of paramount importance to identify potential targets of intervention and develop effective therapeutic strategies. Matrix metalloproteinases (MMPs) represent a ubiquitous superfamily of structurally related zinc-dependent endopeptidases able to degrade any component of the extracellular matrix. They are upregulated after ICH, in which different cell types, including leukocytes, activated microglia, neurons, and endothelial cells, are involved in their synthesis and secretion. The aim of this review is to summarize the available experimental and clinical evidence about the role of MMPs in brain injury following spontaneous ICH and provide critical insights into the underlying mechanisms.
Similar content being viewed by others


Introduction
Spontaneous intracerebral hemorrhage (ICH) accounts for 10–30% of all strokes and affects more than one million people every year worldwide [1, 2]. ICH is associated with the highest rate of mortality and morbidity. Around 40% of ICH patients die within 1 month after onset and residual disability is common among survivors, with only one-third living independently at 1 year [3, 4].
Primary injury induced by ICH is due to the mass effect produced by extravasated blood that can develop from minutes to hours after the bleeding onset. The rapid accumulation of blood within cerebral parenchyma results in physical disruption of brain anatomy, mechanical compression of adjacent tissue, and an increase in intracranial pressure [5]. Secondary damage develops in the late acute and subacute phases and relates to the local and systemic responses to hematoma formation and its toxic effect on adjacent brain tissue. The secondary damage occurs through a variety of pathological pathways including iron-mediated oxidative stress, inflammatory response, excitotoxicity, cytotoxicity, spreading depression, and hypermetabolism [6,7,8,9,10,11,12,13,14], which eventually result in the blood–brain barrier (BBB) dysfunction, perihematomal edema, and brain cell death [5].
So far, clinical trials have mainly targeted primary injury focusing on blood pressure control, hemostasis, and surgical approaches [15,16,17,18,19,20,21,22], in which these approaches have failed to substantially improve clinical outcome, and the management of patients with ICH remains supportive. Therefore, understanding of the pathophysiology of early and delayed injury after ICH is, hence, of paramount importance to identify potential targets of intervention and develop effective therapeutic strategies [23,24,25,26]. Increasing evidence shows that inflammation plays an important role in the progression of ICH [5]. For that reason, therapeutic targeting of post-ICH inflammation in acute stroke has gained interest as a potential neuroprotective strategy. Matrix metalloproteinases (MMPs) are part of the neuro-inflammatory process that occurs during ICH and are, thus, also ideal as biomarkers and therapeutic targets in ICH treatment.
The role of MMPs as a potential target for the treatment of ICH has been widely discussed in the last decade. The impact of MMPs on extracellular matrix destruction and BBB disruption in patients suffering from ICH has been of interest. Hence, we primarily aimed to summarize the available published experimental and clinical data about the role of MMPs in brain injury following spontaneous ICH and provide critical insights into the underlying mechanisms. We identified relevant studies on this topic through a MEDLINE search (June 2019), using the following terms: “matrix metalloproteinase” or “matrix metallopeptidase” and “stroke,” “cerebral hemorrhage,” “cerebral hematoma,” “intracerebral hemorrhage,” “intracerebral hematoma,” “intracranial hemorrhage,” or “intracranial hematoma.” There were no date limitations or language restrictions. The reference lists of retrieved articles were reviewed to search for additional reports of relevant data. Studies addressing the role of MMPs in spontaneous ICH both in animal models and human patients were considered. We did not discuss ICH associated with aneurysms, cerebral vascular malformation or brain trauma, subdural hematoma, intracranial hemorrhage in premature infants, subarachnoid hemorrhage, and hemorrhagic transformation of cerebral ischemia.
MMPs: Structure and Functioning
MMPs represent a ubiquitous superfamily of structurally related zinc-dependent endopeptidases able to degrade any component of the extracellular matrix [27]. MMPs are synthesized by several cell types and either secreted from the cell or anchored to its surface [28]. Under physiological conditions, the expression of MMPs is negligible and it is found in small amounts in tissues and fluids [29]. Thus far, more than 20 types of MMPs have been identified in human and classified into six groups according to their substrate. 1) Collagenases, including MMP-1, MMP-8, MMP-13, and MMP-18, that cleave to collagens I, II, and III and other ECM and non-ECM molecules like laminin, fibronectin, and elastin, and can also activate MMP-2 and MMP-9. 2) Gelatinases, including gelatinase A (MMP-2) and gelatinase B (MMP-9), which mainly digest denatured collagens (gelatins). 3) Stromelysin, including stromelysin-1 (MMP-3) and stromelysin-2 (MMP-10). 4) Matrilysins, including MMP-7, which degrade collagen IV and X, elastin, aggrecan, and proteoglycan. 5) Membrane-type MMPs (MT-MMPs), which can degrade collagen I, II, III, and activate pro-MMP to MMP. 6) Other MMPs, like MMP-11 (stromelysin-3), which differs from stromelysin-1 in structure and the specificity of the substrate, and MMP-12 (metalloelastase), which is expressed by macrophages and degrade collagen IV, elastin, fibronectin, and laminin [28]. The structure of the MMPs generally consists of a prodomain, a catalytic domain with a zinc-binding site, a hinge region, and a hemopexin domain, which confers much of the substrate specificity. MT-MMPs have an additional transmembrane binding domain [28].
MMPs are synthesized in zymogenic inactive form and latency is preserved by the cysteine switch motif in the prodomain, which chelates the active zinc site [30]. The activation of the latent forms usually occurs extracellularly, but it has been also demonstrated inside cells [31, 32]. The cleavage of the cysteine-zinc bridge is the first step for the conversion of pro-MMPs into MMPs, and it is followed by the autocatalysis of the prodomain. Prodomain cleavage can result from different mechanisms, namely direct proteolysis mediated by plasmin, thrombin, and other serine proteases or MMPs, allosteric activation with distortion of the catalytic site, and disruption of the cysteine-zinc interaction by reactive oxygen or nitrogen-free radicals.
The levels and the functioning of MMPs are further regulated at the transcription level and by inhibition of the proteolytic activity. Different signals by inflammatory cytokines (mainly tumor necrosis factor-α [TNF-α], interleukin [IL]-1β and IL-6), chemokines, nuclear factors, oxygen-free radicals, and mechanical stress can induce MMPs transcription [29, 33]. The activity of MMPs can be inhibited in the plasma by α2-macroglobulin, and inside tissues by tissue inhibitors of metalloproteinases (TIMPs), which comprise a family of four protease inhibitors [30, 32]. TIMPs are 21–28 kDa proteins that bind and reversibly block MMPs activity. They are either anchored to the ECM or secreted in a soluble form. TIMPs consist of a C-terminal domain and an N-terminal domain that each contains three conserved disulfide bonds. The N-terminal domain folds within itself and binds to the active site of MMPs to inhibit their activity.
MMPs and Secondary Brain Injury in Experimental Cerebral Hemorrhage
The modulation of MMPs is complex because they have a pleiotropic and biphasic nature with multiple roles that grossly depend on the stage of the hematoma development.
Early after ICH, some MMPs are upregulated and different cell types are involved in their synthesis and secretion including either leukocytes (such as neutrophils, lymphocytes, and macrophages) or resident cells of the central nervous system (such as activated microglia, astrocytes, oligodendrocytes, neurons, endothelial cells, and pericytes) [34,35,36,37,38,39].
Several experimental studies demonstrated increased levels of MMPs in brain tissue after cerebral hemorrhage (Table 1). Power et al. [36] showed increased mRNA levels of MMP-2, MMP-3, MMP-7, MMP-9, and MMP-12 at 24 h in collagenase-induced ICH in rats. Wells and colleagues [35] observed increased mRNA levels of MMP-3 and MMP-12 in a collagenase-induced mouse model of ICH. In the same mouse model, an early increase in mRNA activity of MMP-9 has been further confirmed [37, 40]. Increased gelatinolytic activity, likely of MMP-2 and MMP-9, has been shown at 6 and 24 h in the autologous blood-induced mouse model of ICH [41]. Furthermore, increased activity of the same MMPs was found in collagenase-induced ICH in rats [42]. Exploring the time course of stroke-related inflammatory mediators over 5 days after ICH modeled by infusing autologous blood into the striatum in rats, Wu et al. [43] found that the mRNA and/or protein expression of MMP-9 was upregulated between 2 h and 5 days after ICH, and peaked at day 2, in parallel with brain water content. Despite difficulties in comparing data from different species, the expression of MMPs looks to be bimodal, with an early peak incidence occurring at days 1–3, followed by a decrease in the subacute phase and a further increase by day 7, before trending down to normal levels in the chronic stage [36, 37, 44,45,46].
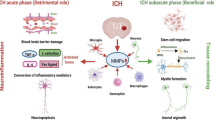
The enhanced activities of MMPs play a key role in the pathophysiology of secondary brain injury following ICH. Indeed, by favoring the loss of vascular integrity, increasing the permeability of vascular walls, enhancing the inflammatory response through the conversion of several inflammatory mediators into their activated forms and by the mechanism of anoikis, MMPs are involved in BBB breakdown, edema formation, hematoma expansion and neuronal loss that follow the hematoma development (Fig. 1). The activation of MMPs (mainly MMP-3 and MMP-9) is responsible for the degradation of the neurovascular matrix through the digestion of the principal components of the basal lamina (collagen type IV, laminin, and fibronectin) surrounding blood vessels [47] and the proteins (such as occludins and claudin-5) forming the tight junctions [31, 48]. The excessive proteolytic activity results in damage to vascular walls, loss of vascular integrity and increased permeability, which promotes the disruption of BBB and, in turn, the development of edema and extravasation of leukocytes into the brain parenchyma [49, 50]. Reactive oxygen species released during the conversion from oxyhemoglobin to methemoglobin, nitric oxide produced at the lesion sites, and both proteases and chemokines released by the brain infiltrating cells contribute to the activation and/or the induction of MMPs, amplify the reactions and, thereby, exacerbate cerebral injury in a vicious circle [30, 38, 51]. In addition, MMPs contribute to enhancing the inflammatory cascade that follows the injury to the brain by converting inflammatory mediators, like TNF-α, transforming growth factor-α, tumor necrosis factor receptors, IL-6, L-selectins, and Fas ligand into their activated forms [38, 51,52,53]. Consistently with all the aforementioned pathways, increased levels of MMPs early after stroke have been associated with BBB damage and perihematomal edema in several animal models of ICH [39, 45]. The expression of MMP-9 induced by the vascular endothelial growth factor (VEGF) has been also associated with hematoma growth through the damage of the basal lamina and walls of brain vessels [54]; MMP-9 has been also suggested to increase the amount of cerebral hemorrhage by inducing abnormal blood vessel proliferation through the activation of insulin-like and tumor growth factors [55].

Matrix metalloproteinases roles in pathophysiology of intracerebral cerebral hemorrhage. Upper bracket represents the roles of MMPs in pathways related to secondary damage, whereas the lower bracket represents the roles of MMPs in pathways related to recovery after ICH (see text for details). MMPs matrix metalloproteinases
Interestingly, the increase in MMP-2 and MMP-9 levels was found not only in areas adjacent to the ICH but also in distant territories, in which they correlated with depressed cortical excitability in contralateral cortex, suggesting the spread of inflammatory and degenerative processes in remote regions functionally connected to the site of hematoma [56].
The role of MMPs in causing cell death after ICH can be also hypothesized because the direct application of MMPs was able to cause neuronal death in cellular studies. This phenomenon of programmed cell death is known as anoikis and occurs in anchorage-dependent cells when degradation of extracellular matrix interferes with cell-matrix interactions, cell attachment, and integrin signaling [57, 58].
Interactions and interdependency between proteases like MMPs and thrombin also exist in the development of neurotoxicity; remarkably, MMP-3 can convert the MMP-9 zymogen into its active form, and MMP-9 and thrombin co-work to promote neurotoxicity. Cultured neurons are killed by MMP-3 and death is most marked when all three proteases, MMP-3, MMP-9, and thrombin, are combined [59]. Brain damage and neuronal death in vivo were significantly reduced in MMP-3 or MMP-9 null mice compared with wild-type counterparts, and MMP-3 and MMP-9 double null mice had even less cerebral injury [59]. These findings also suggested that multiple proteases may need to be targeted simultaneously in order to mitigate ICH-related brain injury.
Further evidence on the contribute of MMPs to the development of secondary injury after ICH came from knockout mice experiments, which demonstrated reduced brain edema, microglia/macrophage activation, neutrophil accumulation, and neuronal death and improved sensorimotor function in MMP-9 and MMP-12 null mice [35, 39, 41]. However, opposite results have been reported in other experimental models and deleterious effects as facilitation of cellular death and an increase in hemorrhage and brain edema have been observed by inhibiting MMPs or using MMP-knock out mice. Such inconsistent results may be partially explained by the enhanced expression of other MMPs isoforms than the silenced one because of the effects of long-term inhibition or deficiency of the knockout MMP activity on normal collagen or matrix homeostasis. [44, 60].
Remarkably, MMPs also have pro-reparative functions. Far from being the simple replacement of damaged neurons, neuro-repair is a much more complex phenomenon that includes endogenous mechanisms of angio-vasculogenesis, gliogenesis, neurogenesis and re-myelination. As ECM is widely present in the brain and contributes to the integration of stimuli from tissue microenvironment and provision of remodeling responses, MMPs are key players of neuro-reparative pathways with actions including neural cell stem migration, release and activation of pro-angiogenic and neurotrophic factors, and vessel remodeling (Fig. 1) [61]. Acute insults to the brain can prompt neurogenesis in the subventricular zone and hippocampal subgranular zone, in which newly formed blood vessels favor the migration of neuronal cells to damaged tissue [62]. Although the mechanisms and molecular steps underlying brain injury-induced angiogenesis and neurogenesis are not completely understood, it is worth to notice that MMPs increase in the subventricular zone in the subacute phase, and there is evidence suggesting their role in either neurogenic migration or neurovascular remodeling at the site of injured areas [63]. The induction of ICH by collagenase injection in rats resulted in significant increases in the levels of MMP-9, VEGF, and nerve growth factor, as well as in the numbers of 5-bromo-2-deoxyuridine- and doublecortin-positive cells in the ipsilateral brain [64]. The inhibition of MMP-9 through the intracerebroventricular injection of MMP-9 small interfering RNA (siRNA) on day 7 and 10 after hemorrhage decreased markers of remodeling through reductions in bioavailable growth factors and was associated with worse functional outcomes. In contrast to the deleterious actions in the acute stage of ICH, MMPs may, hence, play beneficial roles during stroke recovery [63].
MMPs and Cerebral Hemorrhage: Gathering Evidence from Clinical Studies
Different studies assessed the temporal profile and expression of MMPs in patients with acute ICH and the correlation with clinical and radiological outcomes, providing further support to their involvement in the pathophysiology of hemorrhagic brain injury (Table 2). Abilleira and colleagues [65] were the first to investigate the plasma concentration of MMP-9 in patients with acute ICH within 24 h after symptom onset. They found increased expression of MMP-9 after acute spontaneous ICH in comparison to healthy controls. Furthermore, a significant association between higher MMP-9 plasma levels and perihematomal edema volume or the development of neurological worsening within the acute stage in deep hematomas was observed [65].
The temporal profile of MMPs and their natural inhibitors after ICH (as well as their influence on hematoma, perihematomal edema, and clinical outcomes) have been also explored [60]. MMP-2, MMP-9, MMP-3, TIMP-1, and TIMP-2 were serially determined within 12 h from admission (baseline), followed by 24 h, 48 h, 7 days, and 3 months in 21 patients. The highest levels of MMP-2 and TIMP-2 were found at baseline, MMP-9 and TIMP-1 at 24 h, and MMP-3 at 24 to 48 h. There was no interaction between baseline ICH volume and MMP-TIMP level. Although MMP-9 expression at baseline was positively correlated to perihematomal edema, its inhibitor (TIMP-1) had a negative correlation. MMP-3 was the strongest predictor of mortality and both MMP-3 and MMP-9 were associated with the 3-month residual scar volume [66].
Dynamic changes in MMP-9 expression both in blood and cerebrospinal fluid (CSF) were investigated in ICH patients who underwent hematoma evacuation [67]. The MMP-9 levels at 1, 4, and 7 days after the intervention were higher in patients than in age-matched controls and, notably, MMP-9 was not detected in CSF samples of the control group. Plasma and CSF levels of MMP-9 were increased at day 1, peaked at day 4, and remained upregulated at day 7, with levels corresponding to the CSF samples higher than those found in plasma [67]. A positive correlation emerged between MMP-9 levels, stroke severity, and perihematomal edema volume. Serum levels of MMP-9 within the first week after ICH onset were also analyzed in another prospective cohort of 62 patients [68], in which the highest levels were observed on the third day following ICH onset and were positively correlated with National Institutes of Health Stroke Scale score and brain edema. Castellazzi et al. [69] evaluated the temporal changes of serum MMP-9 and MMP-2 and their inhibitors TIMP-1 and TIMP-2 levels in acute and subacute phases of spontaneous ICH. Compared to values obtained at 24 h, mean serum concentrations of MMP-9 was increased at 48 h and peaked at 7 days, and serum active mean MMP-2 mean levels progressively declined at 2 and 7 days. Perihematomal edema volume was positively correlated with levels of MMP-9 and MMP-2 at 24 h and MMP-9 at 48 h, whereas was inversely associated with MMP-2 at 7 days, suggesting a distinct role of the different isoforms of MMPs in the inflammatory response associated with perihematomal edema from acute to subacute stages.
The analyses of brain parenchyma obtained from patients with ICH confirmed the upregulation of MMP-2 [70,71,72], MMP-9 [64,65,66,67,68, 73, 74], and MMP-19 in perihematomal areas [75]. In two prospective studies, serum levels of MMP-9 at admission in patients with ICH resulted significantly associated with hematoma expansion detected at follow-up neuroimaging performed at 48 ± 6 h from onset [76] and within 42–54 h from the first head CT scan or immediately after clinical neurological deterioration [77]. Li et al. [78] assessed the concentrations of serum MMP-3 and MMP-9 within 24 h of symptom onset in 59 patients with spontaneous ICH. They found that increased MMP-3 levels were independently associated with perihematomal edema volume and identified predicting thresholds for the poor clinical outcome assessed by the modified Rankin scale (mRS) at 90 days (MMP-3 ≥ 12.4 ng/mL and MMP-9 ≥ 192.4 ng/mL), independent of baseline stroke severity and hematoma volume. Recently, the levels of seven isoforms of MMPs at five pre-specified time points (within 24 h, followed by 1–2, 3–5, 6–8, and 10 days post-injury) were assessed and their association with radiological outcomes investigated [79]. In univariate analyses, MMP-10 within 2 days from stroke was significantly associated with hematoma expansion and MMP-3 within 24 h from injury was the only variable associated with early neurological deterioration. Interestingly, “delayed” endpoints as perihematomal edema and delayed neurological deterioration were only associated with markers obtained at later time points (MMP-1 at day 3–5 and MMP-8 at day 6–8). MMP-1, MMP-8, and MMP-10 also correlated with discharge status and mRS at 90 days [79]. The same research group explored sex-specific associations of MMPs with secondary injury and outcomes after ICH [80]. By examining the serum samples from a prospective biobank, they observed MMP-1, MMP-2, MMP-3, and MMP-9 levels gradually increased over time in males until 10 days post-ICH. In female patients, a different pattern of upregulation was found: MMP-8 was the only isoform to significantly change over time, reaching the peak at 3–5 days post-injury, although only a near significance was found for MMP-10 level. Broad positive associations of MMP-1, MMP-2, MMP-3, and MMP-9 with initial and peak perihematomal edema were found in females. Additionally, serum levels of MMP-3 in males and MMP-10 in females were independent predictors of functional outcomes at 3 months.
Cerebral Hemorrhage Associated with Cerebral Amyloid Angiopathy
Spontaneous ICH is mainly associated with arterial hypertension that leads to bleeding in deep brain tissue. However, the second cause of ICH among the elderly is cerebral amyloid angiopathy (CAA), which is characterized by the deposition of fibrillar beta-amyloid protein in the leptomeninges and mid-sized cerebral arteries that is typically located in the lobar region. Evidence emerged from in vitro studies has demonstrated that MMPs containing gelatinase activity from vascular cells are upregulated by beta-amyloid protein stimulation [70]. This suggests that their overexpression could contribute to the development of spontaneous ICH due to CAA. Although no differences were found in plasma levels between ICH patients and controls, MMP-2 and MMP-9 were upregulated in the perihematomal area of brains in CAA patients with an increase of MMP-2 expression in reactive astrocytes around beta-amyloid-affected vessels and in infiltrated histiocytes in vascular lesions, whereas MMP-9 was only found acutely in inflammatory cells in distant areas from ICH [70]. Zhao et al. evaluated MMP-9 expression in postmortem brain tissue of human CAA. They found that MMP-9 colocalized with CAA, primarily in the tunica media of compromised vessels, in a CAA-severity dependent fashion [81]. Moreover, the topical application of recombinant MMP-9 induced ICH in a time- and dose-dependent manner; transgenic mice with CAA resulted more susceptible to MMP-9 induced bleeding and developed a more extensive ICH, which also appeared sooner after the exposure [81]. These findings suggest a significant contribution of MMPs in the development of ICH in the setting of CAA.
Inhibition of MMPs as a Potential Therapeutic Target for ICH
The inhibition of MMPs in the acute stage of ICH represents an attractive target to mitigate the secondary brain injury and, hence, improve clinical outcomes. Minocycline represents one of the most investigated agents. It is a semi-synthetic, second-generation tetracycline that can pass through BBB due to its high lipophilicity. Minocycline has anti-inflammatory, neuroprotective, and antiapoptotic properties, which can be, at least partially, attributed to the innate MMP inhibitory capacity [82].
Minocycline had beneficial effects in experimental models of ICH. The intraperitoneal injection of minocycline decreased the expression of MMP-12 and TNF-α, protected BBB, reduced the extravasation of plasma proteins and development of edema, and decreased the number of neutrophils in the brain of rats with collagenase-induced ICH [83]. Power et al. [36] demonstrated that post-ICH in vivo treatment with minocycline significantly reduced brain level of MMP-12, glial activation and apoptosis, and improved neurobehavioral outcomes in rats. Several experimental studies confirmed the role of minocycline administration in reducing BBB damage [84, 85], inhibition of microglia activation [86], attenuation of perihematomal edema, neurological deficits, and brain atrophy [87,88,89]. In rat collagenase-induced ICH models, delayed minocycline administration significantly reduced MMP-12 levels, but it is worth to notice that its efficacy was lost and no effects on neuronal loss were observed when treatment was continued for 7 days [90]. Similarly, neither reduction in brain lesion nor functional benefits (as assessed with sensory-motor tests) were observed when minocycline was given for either 5 or 14 days [91].
The feasibility and safety of minocycline in ICH patients have been explored in a pilot, double-blinded, placebo-controlled randomized clinical trial [92]. Twenty patients with ICH presenting within 12 h of symptom onset with hematoma volume less than 30 ml were randomly assigned to high-dose (10 mg/kg) intravenous minocycline or placebo. High-dose minocycline resulted in the reduction of MMP-9 levels, even though hematoma volume and functional outcomes between the two groups were similar. This was the first study exploring minocycline in human patients, and larger randomized clinical trials are required before drawing definitive conclusions about its therapeutic potential in clinical practice [92].
The effects of GM6001, a synthetic, broad-spectrum MMPs inhibitor, have been explored in different animal studies. Wang et al. [37] demonstrated that intraperitoneal administration of GM6001 (100 mg/kg) to a mouse model of collagenase-induced ICH decreased gelatinase activity, neutrophil infiltration, oxidative stress, brain edema, injury volume, and improved functional outcome. The favorable effects of GM6001 in reducing brain edema formation [45] and brain damage [93] have been further confirmed in other rodent studies. Recently, it has been shown that the intraperitoneal injection of GM6001 (40 mg/kg) improved the BBB damage and reduced cerebral edema and inflammatory cellular infiltration by inhibiting the cleavage of β-dystroglycan triggered by MMP-2 and MMP-9 in rats [94].
The early treatment with CM352 that is a short half-life MMPs inhibitor prevented hematoma expansion, reduced lesion size, attenuated neurological deficits and improved functional and neurological recovery at 24 h and 14 days in a rat model of collagenase-induced ICH [95]. In rats undergoing intracerebral injection of type IV collagenase, TIMP-2 was able to reduce extracellular matrix proteolysis and protect the proteolytic opening of BBB [96].
A broad spectrum of MMPs inhibitors such as FN-349, BB-94, BB-1101, and 10% dimethyl sulfoxide (a selective MMP-2 and MMP-9 inhibitor) were effective in reducing BBB permeability and blocking edema formation in rodent models of ICH, whereas BB-2983, with a profile of inhibitory activity closed to BB-1101, IW499 (a selective MMP-2 inhibitor), and methylprednisolone had no effect on BBB opening [42, 85, 97].
The potential benefit to inhibit the MMPs activity is also suggested by several studies revealing attenuation of inflammatory injury, BBB permeability and cerebral edema, and increasing neuroprotective effects or improvement of neurological deficit after the administration of compounds that are so far not considered as directly inhibiting the MMPs, but that can result in the reduction of MMPs expression or upregulation of TIMPs [98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129]. In a rat collagenase model of ICH, atorvastatin significantly relieved brain edema and protect neurons from apoptosis by decreasing MMP-9 levels [98]. In a similar ICH model using CD-1 mice, simvastatin attenuated brain edema and reduced cellular apoptosis through downregulation of nuclear factor (NF)-kB expression and resulted in reduced expression levels of MMP-9, caspase-3, and aquaporin-4 (AQP4) [99]. Glibenclamide protected BBB and improved neurological outcome in a rat model of ICH by inhibiting the Sur1-Trpm4 channel, which reduced the expression of MMPs and, thereby, increased tight-junction protein levels [100]. A decreased in perihematomal levels of MMP-9, BBB permeability and brain edema, and improved neurological outcomes were also observed after rosiglitazone infusion therapy in rabbits undergoing minimally invasive surgery for ICH evacuation [101]. Memantine resulted in a decrease of hematoma expansion, coupled with an inhibitory effect on the tissue plasminogen activator/urokinase plasminogen activator and MMP-9 levels in the brain reduced inflammatory infiltration and apoptosis and promoted functional recovery after ICH [102]. Treatment with valproic acid (VPA) reduced cerebral bleeding, perihematomal cell death, caspase activities, and inflammatory cell infiltration, and improved recovery following ICH in rats. These neuroprotective effects resulted from the gene expression regulation and transcriptional changes induced by VPA, including downregulation of the mRNA of MMP-9 [103]. In collagenase-induced ICH in mice, progesterone injections reduced VEGF expression and MMP-9 activity, attenuated cellular and molecular inflammatory responses, and reduced lesion volume, brain swelling, myelin loss, and improved long-term neurologic function [104]. Estradiol treatment prevented BBB breakdown and improved neurological deficits after ICH in hyperglycemic ICH mice by decreasing the expression of silent information regulator 1 (Sirt1) and inhibiting the activity of MMP-9 [105]. In a rat ICH model, the transplantation of mesenchymal stem cells reduced the levels of pro-inflammatory cytokines, MMP-9, inducible nitric oxide synthase, and the density of microglia/macrophages and neutrophil infiltration at the site of brain injury. It also increased the levels of zonula occludens-1 and claudin-5 and was, therefore, associated with attenuation of BBB leakage [106]. Dexamethasone, given immediately and 3 days after ICH in rats was successful to decrease brain edema by lowering the MMP-9 and intercellular adhesion molecule-1 levels, partially through the IκB/nuclear factor (NF)-κB signaling pathway [107]. In adult male 129S2/sv mice subjected to autologous blood-induced ICH, recombinant human erythropoietin decreased MMP-2 expression and increased TIMP-2 expression, resulting in preserved expression of occludin and collagen IV, and reduced BBB breakdown-associated inflammation [108]. Hypothermia has been also demonstrated to reduce BBB leakage, edema development, cell apoptosis, and neurological deficits by inhibiting the expression of protease-activated receptor-1, MMP-9 and AQP4 [109,110,111].
Signally, the reduction of MMPs activity obtained through treatment in most experiments occurred within the frame of a more general suppression of the inflammatory response, without a clear demonstration of the mediating effect of MMPs inhibition. The low specificity of many inhibitors does not allow to definitively attribute the response to the intervention to the differential MMP activity and a close cause-effect relationship cannot be clearly proven. It is also worth to mention that, although some therapeutic approaches resulted effective in experimental settings, they do not necessarily translate into clinical benefit when used to treat patients with ICH. For example, the administration of dexamethasone for 3 days after ICH showed beneficial effects in animal models, whereas it increased rates of complications in human trials [130].
The risk of off-target effects should also be considered. Given the role of MMPs in neuro-repair, their inhibition may potentially attenuate the neuro-regeneration and, hence, result in impairment of recovery and functional outcome. Moreover, as MMPs have a ubiquitous role in the human body, the side effects related to the suppression of physiological processes need to be elucidated. To date, a substantial gap still exists in the knowledge of tolerability and safety issues of MMPs inhibition.
Clinical Implications and Future Perspectives
In summary, there is enough evidence from both experimental and clinical studies that MMPs are upregulated after acute ICH. They are released into the area of brain injury by a variety of cells, including activated microglia, leukocytes, astrocytes, neurons, and endothelial cells.
The expression of MMPs increases early after ICH: the brain and peripheral levels of MMPs peak within the first days and remain high across the first week after hematoma development. Only a few studies, however, have provided a comprehensive longitudinal assessment of the temporal expression profile of the different members of the MMP family and their correlations with brain changes and functional outcomes. Notably, the levels of some MMP isoforms increase whereas others decrease over time and their role and effects change from hyper-acute to subacute and chronic ICH phases.
Overexpression of specific isoforms of MMPs plays a role in the development of secondary brain damage. By promoting the loss of vascular integrity, the permeability of vascular walls, inflammatory response, cellular death, MMPs contribute to the disruption of BBB, edema propagation, hematoma growth, and neuronal loss. It is absolutely remarkable that MMPs are also involved in neuro-repair: by taking part in neural cell stem migration, release and activation of pro-angiogenic and neurotrophic factors, vessel remodeling, myelin formation, and axonal growth [52], they are key mediators of neural network remodeling and recovery after ICH.
Overall, there is substantially converging evidence from experimental studies to suggest that early and short-term inhibition of MMPs after ICH can be an effective strategy to reduce cerebral damage and improve the outcome, whereas long-term treatment may be associated with more harm than benefit. It is, however, worth to notice that, so far, we do not have a clear understanding of the time-specific role that the different MMPs assume within the pathophysiology of secondary brain injury and recovery after ICH. In addition, most of the studies exploring pharmacological strategies to modulate MMPs can only provide indirect evidence of the benefit to target MMP activity.
The prospects for effective therapeutic targeting of MMPs require the establishment of conditions to specifically modulate a given MMP isoform, or a subset of MMPs, in a given spatio-temporal context [131]. Further research is warranted to better understand the interactions between MMPs and their molecular and cellular environments, determine the optimal timing of MMPs inhibition for achieving a favorable therapeutic outcome, and implement the discovery of innovative selective agents to spare harmful effects before therapeutic strategies targeting MMPs can be successfully incorporated into routine practice [132, 133].
References
Feigin VL, Lawes CM, Bennett DA, Anderson CS. Stroke epidemiology: a review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. 2003;2:43–53.
Lattanzi S, Brigo F, Silvestrini M. Blood pressure and stroke: From incidence to outcome. J Clin Hypertens (Greenwich). 2019;21:605–607.
van Asch CJ, Luitse MJ, Rinkel GJ, van der Tweel I, Algra A, Klijn CJ. Incidence, case fatality, and functional outcome of intracerebral haemorrhage over time, according to age, sex, and ethnic origin: a systematic review and meta-analysis. Lancet Neurol 2010;9:167–176.
Lattanzi S, Silvestrini M. Stroke outcome prediction: what do we know and where are we going? Eur J Neurol. 2018;25:1398–1399.
Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. 2011;42:1781–1786.
Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab. 2003;23:629–652.
Gong C, Hoff JT, Keep RF. Acute inflammatory reaction following experimental intracerebral hemorrhage in rat. Brain Res. 2000;871:57–65.
Di Napoli M, Slevin M, Popa-Wagner A, Singh P, Lattanzi S, Divani AA. Monomeric C-Reactive Protein and Cerebral Hemorrhage: From Bench to Bedside. Front Immunol. 2018;9:1921.
Lattanzi S, Brigo F, Trinka E, Cagnetti C, Di Napoli M, Silvestrini M. Neutrophil-to-Lymphocyte Ratio in Acute Cerebral Hemorrhage: a System Review. Transl Stroke Res. 2019;10:137–145.
Qureshi AI, Ali Z, Suri MF, et al. Extracellular glutamate and other amino acids in experimental intracerebral hemorrhage: an in vivo microdialysis study. Crit Care Med. 2003;31:1482–1489.
Mun-Bryce S, Wilkerson AC, Papuashvili N, Okada YC. Recurring episodes of spreading depression are spontaneously elicited by an intracerebral hemorrhage in the swine. Brain Res. 2001;888:248–255.
Ardizzone TD, Lu A, Wagner KR, Tang Y, Ran R, Sharp FR. Glutamate receptor blockade attenuates glucose hypermetabolism in perihematomal brain after experimental intracerebral hemorrhage in rat. Stroke. 2004;35:2587–2591.
Jafari M, Di Napoli M, Lattanzi S, et al. Serum magnesium level and hematoma expansion in patients with intracerebral hemorrhage. J Neurol Sci. 2019;398:39–44.
Jafari M, Di Napoli M, Datta YH, Bershad EM, Divani AA. The Role of Serum Calcium Level in Intracerebral Hemorrhage Hematoma Expansion: Is There Any? Neurocrit Care. 2019;31:188–195.
Lattanzi S, Cagnetti C, Provinciali L, Silvestrini M. How Should We Lower Blood Pressure after Cerebral Hemorrhage? A Systematic Review and Meta-Analysis. Cerebrovasc Dis. 2017;43:207–213.
Lattanzi S, Silvestrini M. Optimal achieved blood pressure in acute intracerebral hemorrhage: INTERACT2. Neurology. 2015;85:557–558.
Divani AA, Liu X, Di Napoli M, Lattanzi S, Ziai W, James ML, Jafarli A, Jafari M, Saver JL, Hemphill JC, Vespa PM, Mayer SA, Petersen A. Blood Pressure Variability Predicts Poor In-Hospital Outcome in Spontaneous Intracerebral Hemorrhage. Stroke. 2019;50:2023–2029.
Lattanzi S, Silvestrini M. Blood pressure in acute intra-cerebral hemorrhage. Ann Transl Med. 2016;4:320.
Yuan ZH, Jiang JK, Huang WD, Pan J, Zhu JY, Wang JZ. A meta-analysis of the efficacy and safety of recombinant activated factor VII for patients with acute intracerebral hemorrhage without hemophilia. J Clin Neurosci. 2010;17:685–693.
Mayer SA, Brun NC, Begtrup K, Broderick J, Davis S, Diringer MN, Skolnick BE, Steiner T; FAST Trial Investigators. Efficacy and safety of recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med. 2008;358:2127–2137.
Hanley DF, Lane K, McBee N, et al.; CLEAR III Investigators. Thrombolytic removal of intraventricular haemorrhage in treatment of severe stroke: results of the randomised, multicentre, multiregion, placebo-controlled CLEAR III trial. Lancet. 2017;389:603–611.
Hanley DF, Thompson RE, Rosenblum M, et al; MISTIE III Investigators. Efficacy and safety of minimally invasive surgery with thrombolysis in intracerebral haemorrhage evacuation (MISTIE III): a randomised, controlled, open-label, blinded endpoint phase 3 trial. Lancet. 2019;393:1021–1032.
Mahmoudi J, Majdi A, Lattanzi S, Di Napoli M, Bershad EM, Rodrigues CMP, Divani AA. Imidazoline Receptor Agonists for Managing Hypertension May Hold Promise for Treatment of Intracerebral Hemorrhage. Curr Mol Med. 2018;18:241–251.
Hevesi M, Bershad EM, Jafari M, Mayer SA, Selim M, Suarez JI, Divani AA. Untreated hypertension as predictor of in-hospital mortality in intracerebral hemorrhage: A multi-center study. J Crit Care. 2018;43:235–239.
Divani AA, Hevesi M, Pulivarthi S, Luo X, Souslian F, Suarez JI, Bershad EM. Predictors of nosocomial pneumonia in intracerebral hemorrhage patients: a multi-center observational study. Neurocrit Care. 2015;22:234–242.
Salazar P, Di Napoli M, Jafari M, Jafarli A, Ziai W, Petersen A, Mayer SA, Bershad EM, Damani R, Divani AA. Exploration of Multiparameter Hematoma 3D Image Analysis for Predicting Outcome After Intracerebral Hemorrhage. Neurocrit Care. 2019 Jul 29. doi: https://doi.org/10.1007/s12028-019-00783-8. [Epub ahead of print]
Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res 2003;92:827–839.
Raffetto JD, Khalil RA. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol. 2008;75:346–359.
Florczak-Rzepka M, Grond-Ginsbach C, Montaner J, Steiner T. Matrix metalloproteinases in human spontaneous intracerebral hemorrhage: an update. Cerebrovasc Dis. 2012;34:249–262.
Mun-Bryce S, Rosenberg G: Matrix metalloproteinases in cerebrovascular disease. J Cereb Blood Flow Metab 1998;18:1163–1172.
Cauwe B, Opdenakker G. Intracellular substrate cleavage: a novel dimension in the biochemistry, biology and pathology of matrix metalloproteinases. Crit Rev Biochem Mol Biol 2010;45:351–423.
Mannello F, Medda V: Nuclear localization of matrix metalloproteinases. Prog Histochem Cytochem 2012;47:27–58.
Yong V. The potential use of MMP inhibitors to treat CNS diseases. Expert Opin Investig Drugs 1999;8:255–268.
Xue M, Yong VW. Matrix metalloproteinases in intracerebral hemorrhage. Neurol Res. 2008;30(8):775–82.
Wells JE, Biernaskie J, Szymanska A, Larsen PH, Yong VW, Corbett D. Matrix metalloproteinase (MMP)-12 expression has a negative impact on sensorimotor function following intracerebral haemorrhage in mice. Eur J Neurosci 2005;21:187–196.
Power C, Henry S, Del Bigio MR, et al. Intracerebral hemorrhage induces macrophage activation and matrix metalloproteinases. Ann Neurol 2003;53:731–742.
Wang J, Tsirka SE. Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain 2005; 128:1622–1633.
Yong VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci 2001;2:502–511.
Tejima E, Zhao BQ, Tsuji K, et al. Astrocytic induction of matrix metalloproteinase-9 and edema in brain hemorrhage. J Cereb Blood Flow Metab 2007;27:460–468.
Lu A, Tang Y, Ran R, Ardizzone TL, Wagner KR, Sharp FR. Brain genomics of intracerebral hemorrhage. J Cereb Blood Flow Metab 2006;26:230–252.
Xue M, Hollenberg MD, Yong VW. Combination of thrombin and matrix metalloproteinase-9 exacerbates neurotoxicity in cell culture and intracerebral hemorrhage in mice. J Neurosci 2006;26:10281–10291.
Rosenberg GA, Navratil M. Metalloproteinase inhibition blocks edema in intracerebral hemorrhage in the rat. Neurology 1997;48:921–926.
Wu H, Zhang Z, Li Y, et al. Time course of upregulation of inflammatory mediators in the hemorrhagic brain in rats: correlation with brain edema. Neurochem Int. 2010;57:248–53.
Grossetete M, Rosenberg GA. Matrix metalloproteinase inhibition facilitates cell death in intracerebral hemorrhage in mouse. J Cereb Blood Flow Metab 2008;28:752–763.
Kawakita K, Kawai N, Kuroda Y, Yasashita S, Nagao S. Expression of matrix metalloproteinase-9 in thrombin-induced brain edema formation in rats. J Stroke Cerebrovasc Dis 2006;15:88–95.
Chang JJ, Emanuel BA, Mack WJ, Tsivgoulis G, Alexandrov AV. Matrix metalloproteinase-9: dual role and temporal profile in intracerebral hemorrhage. J Stroke Cerebrovasc Dis. 2014;23:2498–505.
Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia 2002;39:279–291.
Zhao X, Zhang Y, Strong R, Grotta JC, Aronowski J. 15d-Prostaglandin J2 activates peroxisome proliferator-activated receptor-gamma, promotes expression of catalase, and reduces inflammation, behavioral dysfunction, and neuronal loss after intracerebral hemorrhage in rats. J Cereb Blood Flow Metab. 2006;26:811–20.
Gurney KJ, Estrada EY, Rosenberg GA. Blood–brain barrier disruption by stromelysin-1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol Dis 2006;23:87–96.
Hamann G, Okada Y, Fitridge R, del Zoppo G. Microvascular basal lamina antigens disappear during cerebral ischemia and reperfusion. Stroke 1995;26:2120–2126.
Yong VW. Metalloproteinases: Mediators of pathology and regeneration in the CNS. Nat Rev Neurosci 2005; 6: 931–944.
Black RA, Rauch CT, Kozlosky CJ, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997;385:729–733.
English WR, Puente XS, Freije JM, et al. Membrane type 4 matrix metalloproteinase (MMP17) has tumor necrosis factor-alpha convertase activity but does not activate pro-MMP2. J Biol Chem 2000;275:14046–14055.
Lee CZ, Xue Z, Zhu Y, Yang GY, Young WL. Matrix metalloproteinase-9 inhibition attenuates vascular endothelial growth factor-induced intracerebral hemorrhage. Stroke 2007;38:2563–68.
Rundhaug JE. Matrix metalloproteinases and angiogenesis. J Cell Mol Med 2005;9:267–85.
Mun-Bryce S, Wilkerson A, Pacheco B, et al. Depressed cortical excitability and elevated matrix metalloproteinases in remote brain regions following intracerebral hemorrhage. Brain Res. 2004;1026:227–34.
Giancotti FG, Ruoslahti E. Integrin signaling. Science 1999; 285:1028–1032.
Frisch SM, Screaton RA. Anoikis mechanisms". Current Opinion in Cell Biology. 2001;13:555–62.
Xue M, Fan Y, Liu S, Zygun DA, Demchuk A, Yong VW. Contributions of multiple proteases to neurotoxicity in a mouse model of intracerebral haemorrhage. Brain. 2009;132(Pt 1):26–36.
Tang J, Liu J, Zhou C, et al. Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutant mice. J Cereb Blood Flow Metab. 2004;24:1133–1145.
Montaner J, Ramiro L, Simats A, Hernández-Guillamon M, Delgado P, Bustamante A, Rosell A. Matrix metalloproteinases and ADAMs in stroke. Cell Mol Life Sci. 2019;76:3117–3140.
Masuda T, Isobe Y, Aihara N, Furuyama F, Misumi S, Kim TS, Nishino H, Hida H. Increase in neurogenesis and neuroblast migration after a small intracerebral hemorrhage in rats. Neurosci Lett. 2007;425:114–119.
Zhao BQ, Tejima E, Lo EH. Neurovascular proteases in brain injury, hemorrhage and remodeling after stroke. Stroke. 2007;38(2 Suppl):748–752.
Lei C, Lin S, Zhang C, et al. Activation of cerebral recovery by matrix metalloproteinase-9 after intracerebral hemorrhage. Neuroscience. 2013 ;230:86–93.
Abilleira S, Montaner J, Molina CA, Monasterio J, Castillo J, Alvarez-Sabín J. Matrix metalloproteinase-9 concentration after spontaneous intracerebral hemorrhage. J Neurosurg. 2003;99:65–70.
Alvarez-Sabín J, Delgado P, Abilleira S, et al. Temporal profile of matrix metalloproteinases and their inhibitors after spontaneous intracerebral hemorrhage: relationship to clinical and radiological outcome. Stroke. 2004;35:1316–1322.
Wu CH, Huang FY, Wang KY, et al. Expression of matrix metalloproteinase MMP-9 in the plasma and hematoma fluid of intracerebral hemorrhage patients. Zhonghua Yi Xue Za Zhi. 2008;88:174–176.
Petrovska-Cvetkovska D, Dolnenec-Baneva N, Nikodijevik D, Chepreganova-Changovska T. Correlative study between serum matrix metalloproteinase-9 values and neurologic deficit in acute, primary, supratentorial, intracerebral haemorrhage. Pril (Makedon Akad Nauk Umet Odd Med Nauki). 2014;35:39–44.
Castellazzi M, Tamborino C, De Santis G, et al. Timing of serum active MMP-9 and MMP-2 levels in acute and subacute phases after spontaneous intracerebral hemorrhage. Acta Neurochir Suppl. 2010;106:137–140.
Hernandez-Guillamon M, Martinez-Saez E, Delgado P, et al. MMP-2/MMP-9 plasma level and brain expression in cerebral amyloid angiopathy-associated hemorrhagic stroke. Brain Pathol. 2012;22:133–141.
Rosell A, Ortega-Aznar A, Alvarez-Sabín J, et al. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke. 2006;37:1399–1406.
Ke SF, Jin XP, Zhang KH, et al. Study on expression of matrix metalloproteinase-2 and matrix metalloproteinase-9 in brain tissue adjacent to hemorrhage after brain hemorrhage in human. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2007;19:336–339.
Wu H, Zhang Z, Hu X, et al. Dynamic changes of inflammatory markers in brain after hemorrhagic stroke in humans: a postmortem study. Brain Res. 2010;1342:111–117.
Zhang XQ, Zhang ZM, Yin XL, Zhang K, Cai H, Ling F. Exploring the optimal operation time for patients with hypertensive intracerebral hemorrhage: tracking the expression and progress of cell apoptosis of prehematomal brain tissues. Chin Med J (Engl). 2010;123:1246–1250.
Tanskanen M, Myllykangas L, Saarialho-Kere U, Paetau A. Matrix metalloproteinase-β19 expressed in cerebral amyloid angiopathy.Amyloid. 2011;18:3–9.
Silva Y, Leira R, Tejada J, Lainez JM, Castillo J, Dávalos A; Stroke Project, Cerebrovascular Diseases Group of the Spanish Neurological Society. Molecular signatures of vascular injury are associated with early growth of intracerebral hemorrhage. Stroke. 2005;36:86–91.
Yang Q, Zhuang X, Peng F, Zheng W. Relationship of plasma matrix metalloproteinase-9 and hematoma expansion in acute hypertensive cerebral hemorrhage. Int J Neurosci. 2016;126:213–218.
Li N, Liu YF, Ma L, et al. Association of molecular markers with perihematomal edema and clinical outcome in intracerebral hemorrhage. Stroke. 2013;44:658–663.
Howe MD, Zhu L, Sansing LH, Gonzales NR, McCullough LD, Edwards NJ. Serum Markers of Blood-Brain Barrier Remodeling and Fibrosis as Predictors of Etiology and Clinico radiologic Outcome in Intracerebral Hemorrhage. Front Neurol. 2018;9:746.
Howe MD, Furr JW, Zhu L, Edwards NJ, McCullough LD, Gonzales NR. Sex-specific Association of Matrix Metalloproteinases with Secondary Injury and Outcomes after Intracerebral Hemorrhage. J Stroke Cerebrovasc Dis. 2019;28:1718–1725.
Zhao L, Arbel-Ornath M, Wang X, Betensky RA, Greenberg SM, Frosch MP, Bacskai BJ. Matrix metalloproteinase 9-mediated intracerebral hemorrhage induced by cerebral amyloid angiopathy. Neurobiol Aging. 2015;36:2963–2971.
Yong VW, Wells J, Giuliani F, Casha S, Power C, Metz LM. The promise of minocycline in neurology. Lancet Neurol. 2004;3:744–751.
Wasserman JK, Schlichter LC. Minocycline protects the blood-brain barrier and reduces edema following intracerebral hemorrhage in the rat. Exp Neurol. 2007;207:227–237.
Shi W, Wang Z, Pu J, et al. Changes of blood-brain barrier permeability following intracerebral hemorrhage and the therapeutic effect of minocycline in rats. Acta Neurochir Suppl. 2011;110(Pt 2):61–67.
Rosenberg GA, Estrada EY, Mobashery S. Effect of synthetic matrix metalloproteinase inhibitors on lipopolysaccharide-induced blood-brain barrier opening in rodents: Differences in response based on strains and solvents. Brain Res. 2007;1133:186–192.
Pu J, Shi W, Wang Z, et al. Effects of minocycline on the expression of NGF and HSP70 and its neuroprotection role following intracerebral hemorrhage in rats. J Biomed Res. 2011;25:292–298.
Wu J, Yang S, Xi G, Fu G, Keep RF, Hua Y. Minocycline reduces intracerebral hemorrhage-induced brain injury. Neurol Res. 2009;31:183–188.
Wu J, Yang S, Hua Y, Liu W, Keep RF, Xi G. Minocycline attenuates brain edema, brain atrophy and neurological deficits after intracerebral hemorrhage. Acta Neurochir Suppl. 2010;106:147–150.
Zhao F, Hua Y, He Y, Keep RF, Xi G. Minocycline-induced attenuation of iron overload and brain injury after experimental intracerebral hemorrhage. Stroke. 2011;42:3587–3593.
Wasserman JK, Zhu X, Schlichter LC. Evolution of the inflammatory response in the brain following intracerebral hemorrhage and effects of delayed minocycline treatment. Brain Res. 2007;1180:140–154.
Szymanska A, Biernaskie J, Laidley D, Granter-Button S, Corbett D. Minocycline and intracerebral hemorrhage: influence of injury severity and delay to treatment. Exp Neurol. 2006;197:189–196.
Chang JJ, Kim-Tenser M, Emanuel BA, et al. Minocycline and matrix metalloproteinase inhibition in acute intracerebral hemorrhage: a pilot study. Eur J Neurol. 2017;24:1384–1391.
Xue M, Hollenberg MD, Demchuk A, Yong VW. Relative importance of proteinase-activated receptor-1 versus matrix metalloproteinases in intracerebral hemorrhage-mediated neurotoxicity in mice. Stroke. 2009;40:2199–204.
Zhang X, Gu Y, Li P, et al. Matrix Metalloproteases-Mediated Cleavage on β-Dystroglycan May Play a Key Role in the Blood-Brain Barrier After Intracerebral Hemorrhage in Rats. Med Sci Monit. 2019;25:794–800.
Rodríguez JA, Sobrino T, López-Arias E, et al. CM352 Reduces Brain Damage and Improves Functional Recovery in a Rat Model of Intracerebral Hemorrhage. J Am Heart Assoc. 2017;6.
Rosenberg GA, Kornfeld M, Estrada E, Kelley RO, Liotta LA, Stetler-Stevenson WG. TIMP-2 reduces proteolytic opening of blood-brain barrier by type IV collagenase. Brain Res. 1992;576:203–207.
Katsu M, Niizuma K, Yoshioka H, Okami N, Sakata H, Chan PH. Hemoglobin-induced oxidative stress contributes to matrix metalloproteinase activation and blood-brain barrier dysfunction in vivo. J Cereb Blood Flow Metab. 2010;30:1939–1950.
Cui JJ, Wang D, Gao F, Li YR. Effects of atorvastatin on pathological changes in brain tissue and plasma MMP-9 in rats with intracerebral hemorrhage. Cell Biochem Biophys. 2012;62:87–90.
Gu C, Wu Y, Fan Z, Han W. Simvastatin improves intracerebral hemorrhage through NF-κB-mediated apoptosis via the MyD88/TRIF signaling pathway. Exp Ther Med. 2018;15:377–382.
Jiang B, Li L, Chen Q, et al. Role of Glibenclamide in Brain Injury After Intracerebral Hemorrhage. Transl Stroke Res. 2017;8:183–193.
Wu G, Wu J, Jiao Y, Wang L, Wang F, Zhang Y. Rosiglitazone infusion therapy following minimally invasive surgery for intracerebral hemorrhage evacuation decreases matrix metalloproteinase-9 and blood-brain barrier disruption in rabbits. BMC Neurol. 2015;15:37.
Lee ST, Chu K, Jung KH, et al. Memantine reduces hematoma expansion in experimental intracerebral hemorrhage, resulting in functional improvement. J Cereb Blood Flow Metab. 2006;26:536–544.
Sinn DI, Kim SJ, Chu K, et al. Valproic acid-mediated neuroprotection in intracerebral hemorrhage via histone deacetylase inhibition and transcriptional activation. Neurobiol Dis. 2007;26:464–472.
Jiang C, Zuo F, Wang Y, et al. Progesterone exerts neuroprotective effects and improves long-term neurologic outcome after intracerebral hemorrhage in middle-aged mice. Neurobiol Aging. 2016;42:13–24.
Zheng Y, Hu Q, Manaenko A, et al. 17β-Estradiol attenuates hematoma expansion through estrogen receptor α/silent information regulator 1/nuclear factor-kappa b pathway in hyperglycemic intracerebral hemorrhage mice. Stroke. 2015;46:485–91.
Chen M, Li X, Zhang X, et al. The inhibitory effect of mesenchymal stem cell on blood-brain barrier disruption following intracerebral hemorrhage in rats: contribution of TSG-6. J Neuroinflammation. 2015;12:61.
Yang JT, Lee TH, Lee IN, Chung CY, Kuo CH, Weng HH. Dexamethasone inhibits ICAM-1 and MMP-9 expression and reduces brain edema in intracerebral hemorrhagic rats. Acta Neurochir (Wien). 2011;153:2197–2203.
Li Y, Ogle ME, Wallace GC 4th, Lu ZY, Yu SP, Wei L. Erythropoietin attenuates intracerebral hemorrhage by diminishing matrix metalloproteinases and maintaining blood-brain barrier integrity in mice. Acta Neurochir Suppl. 2008;105:105–112.
Sun H, Tang Y, Li L, Guan X, Wang D. Effects of local hypothermia on neuronal cell apoptosis after intracerebral hemorrhage in rats. J Nutr Health Aging. 2015;19:291–298.
Gao D, Ding F, Lei G, et al. Effects of focal mild hypothermia on thrombin-induced brain edema formation and the expression of protease activated receptor-1, matrix metalloproteinase-9 and aquaporin 4 in rats. Mol Med Rep. 2015;11:3009–3014.
Wei S, Sun J, Li J, et al. Acute and delayed protective effects of pharmacologically induced hypothermia in an intracerebral hemorrhage stroke model of mice. Neuroscience. 2013;252:489–500.
Liu M, Guo MZ, Li PY, et al. Effect of new qingkailing injection on cerebral edema following intracerebral hemorrhage in rats. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2006;26:244–247.
Liao W, Zhong J, Yu J, et al. Therapeutic benefit of human umbilical cord derived mesenchymal stromal cells in intracerebral hemorrhage rat: implications of anti-inflammation and angiogenesis. Cell Physiol Biochem. 2009;24:307–316.
He CY, Huang JH, Wang WJ, et al. Effects of Liangxue Tongyu Formula on brain edema and expressions of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 in rats with intracerebral hemorrhage. Zhong Xi Yi Jie He Xue Bao. 2010;8:347–351.
Moxon-Emre I, Schlichter LC. Neutrophil depletion reduces blood-brain barrier breakdown, axon injury, and inflammation after intracerebral hemorrhage. J Neuropathol Exp Neurol. 2011;70:218–235.
Shin JW, Kang HC, Shim J, Sohn NW. Scutellaria baicalensis attenuates blood-brain barrier disruption after intracerebral hemorrhage in rats. Am J Chin Med. 2012;40:85–96.
Cai P, Luo H, Xu H, et al. Recombinant ADAMTS 13 Attenuates Brain Injury After Intracerebral Hemorrhage. Stroke. 2015;46:2647–2653.
Liang H, Guan D, Gao A, et al. Human amniotic epithelial stem cells inhibit microglia activation through downregulation of tumor necrosis factor-α, interleukin-1β and matrix metalloproteinase-12 in vitro and in a rat model of intracerebral hemorrhage. Cytotherapy. 2014;16:523–534.
Zhao F, Liu Z. Beneficial effects of edaravone on the expression of serum matrix metalloproteinase-9 after cerebral hemorrhage. Neurosciences (Riyadh). 2014;19:106–110.
Zhou QB, Jin YL, Jia Q, et al. Baicalin attenuates brain edema in a rat model of intracerebral hemorrhage. Inflammation. 2014;37:107–115.
Li D, Lei C, Zhang S, Zhang S, Liu M, Wu B. Blockade of high mobility group box-1 signaling via the receptor for advanced glycation end-products ameliorates inflammatory damage after acute intracerebral hemorrhage. Neurosci Lett. 2015;609:109–119.
Zhu Y, Liu C, Sun Z. Early Combined Therapy with Pharmacologically Induced Hypothermia and Edaravone Exerts Neuroprotective Effects in a Rat Model of Intracerebral Hemorrhage. Cell Biochem Biophys. 2015;73:581–587.
Suda S, Yang B, Schaar K, et al. Autologous Bone Marrow Mononuclear Cells Exert Broad Effects on Short- and Long-Term Biological and Functional Outcomes in Rodents with Intracerebral Hemorrhage. Stem Cells Dev. 2015;24:2756–2766.
Li HQ, Li Y, Chen ZX, et al. Electroacupuncture Exerts Neuroprotection through Caveolin-1 Mediated Molecular Pathway in Intracerebral Hemorrhage of Rats. Neural Plast. 2016;2016:7308261.
Shi H, Zheng K, Su Z, et al. Sinomenine enhances microglia M2 polarization and attenuates inflammatory injury in intracerebral hemorrhage. J Neuroimmunol. 2016;299:28–34.
Wu H, Wu T, Han X, et al. Cerebroprotection by the neuronal PGE2 receptor EP2 after intracerebral hemorrhage in middle-aged mice. J Cereb Blood Flow Metab. 2017;37:39–51.
Wu CH, Shyue SK, Hung TH, et al. Genetic deletion or pharmacological inhibition of soluble epoxide hydrolase reduces brain damage and attenuates neuroinflammation after intracerebral hemorrhage. J Neuroinflammation. 2017;14:230.
Li L, Yun D, Zhang Y, et al. A cannabinoid receptor 2 agonist reduces blood-brain barrier damage via induction of MKP-1 after intracerebral hemorrhage in rats. Brain Res. 2018;1697:113–123.
Song Y, Yang Y, Cui Y, Gao J, Wang K, Cui J. Lipoxin A4 Methyl Ester Reduces Early Brain Injury by Inhibition of the Nuclear Factor Kappa B (NF-κB)-Dependent Matrix Metallopeptidase 9 (MMP-9) Pathway in a Rat Model of Intracerebral Hemorrhage. Med Sci Monit. 2019;25:1838–1847.
Poungvarin N, Bhoopat W, Viriyavejakul A, Rodprasert P, Buranasiri P, Sukondhabhant S, Hensley MJ, Strom BL. Effects of dexamethasone in primary supratentorial intracerebral hemorrhage. N Engl J Med. 1987;316:1229–33.
Rivera S. Metalloproteinases in nervous system function and pathology: introduction. Cell Mol Life Sci. 2019;76:3051–3053.
Lattanzi S, Silvestrini M. Future challenges of stroke treatment. Neurol Neurochir Pol. 2018;52:415–416.
Hostettler IC, Seiffge DJ, Werring DJ. Intracerebral hemorrhage: an update on diagnosis and treatment. Expert Rev Neurother. 2019;19:679–694.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PDF 1224 kb)
Rights and permissions
About this article

Cite this article
Lattanzi, S., Di Napoli, M., Ricci, S. et al. Matrix Metalloproteinases in Acute Intracerebral Hemorrhage. Neurotherapeutics 17, 484–496 (2020). https://doi.org/10.1007/s13311-020-00839-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-020-00839-0