
Abstract
Antisense oligonucleotides (ASOs) have shown potential as therapeutic molecules for the treatment of inner ear dysfunction. The peripheral sensory organs responsible for both hearing and equilibrium are housed within the inner ear. Hearing loss and vestibular balance problems affect a large portion of the population and limited treatment options exist. Targeting ASOs to the inner ear as a therapeutic strategy has unique pharmacokinetic and drug delivery opportunities and challenges. Here, we review ASO technology, delivery, disease targets, and other key considerations for development of this therapeutic approach.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The inner ear is a membrane-lined cavity in the temporal bone that contains the cochlea and peripheral vestibular apparatus, the sensory organs responsible for hearing and equilibrium, respectively. Loss of these senses is common and debilitating making the development of therapies to treat their dysfunction important for improving human health and well-being.
Hearing Loss
Hearing loss is the most common neurological disorder in humans. More than 500 million people (nearly 7% of the world’s population) have disabling hearing loss, a number expected to rise to nearly 1 billion by 2050 (http://www.who.int/mediacentre/factsheets/fs300/en/) [1]. Significant hearing loss is found in 2 to 3 of every 1000 newborns and approximately one third of people over 65 years of age. Despite the enormous burden on human health, relatively few drugs have been approved to prevent or treat hearing disorders. Commonly, the goal is to manage symptoms using steroids and antibiotics, for example, or to use assistive devices that rely on sound amplification (e.g., hearing aids) or surgical placement of electrodes (e.g., cochlear implants) that stimulate the auditory nerve. Although such devices have been invaluable in improving the quality of life in those with hearing deficits, they have shortcomings that limit their utility and make the need for more effective treatments an important goal.
Hearing loss caused by damage or dysfunction in the inner ear sensory apparatus, termed sensorineural hearing loss (SNHL), accounts for 90% of all human hearing loss [2]. Approximately 75% of all SNHL is nonsyndromic [3], meaning losses limited to the auditory system and not involving other organ systems, and 30% is syndromic [2, 3], which indicates hearing loss accompanied by other organ abnormalities [4, 5]. There are many types and causes of hearing impairment including age-related hearing loss (presbycusis), noise-induced hearing loss, autoimmune disease, drug-induced ototoxicity, Meniere’s disease, tinnitus, and infection, and forms caused by gene mutations. Genetic factors are estimated to cause more than 50% of SNHL cases, most involving a single gene or gene region. Genetic mutations can also increase risk for age-related hearing loss and other causes of hearing impairment. To date, more than 6000 causative gene variants in more than 150 genes have been associated with nonsyndromic hearing loss [6, 7] and nearly 40 genes are implicated in syndromic hearing loss, with more than 400 syndromes having a hearing loss component [3]. Most genetic variants (80%) associated with hearing loss are autosomal recessive and 20% are autosomal dominant.
Vestibular Dysfunction
Vestibular dysfunction manifests as dizziness and spatial disorientation and often is accompanied by impaired balance [8]. Affecting as many as 90 million people in the USA alone, the prevalence of vestibular dysfunction increases with age, with 35% of adults in their 40s suffering from the condition and as many as 70% of people over the age of 70 [9,10,11]. Vestibular problems increase the risk of falls and fall-related injuries and death, and thereby have a major impact on health and socioeconomic well-being [8, 9, 12]. Balance disorders may result from peripheral vestibular impairment or central nervous system deficits. Vestibular dysfunction can have genetic causes or can result from any number of insults, such as ototoxins or viral infection, or conditions, such as benign paroxysmal positional vertigo, or Meniere’s disease, though many cases of impairment are degenerative and idiopathic [8]. Considerably less is known about genetic causes of vestibular dysfunction compared to hearing loss. There are few known gene mutations associated with vestibular problems with the exception of those also associated with hearing loss, wherein vestibular dysfunction has been found to affect 34% of children with genetic forms of hearing impairment [13]. Vestibular rehabilitation and a compensatory process likely mediated by the central nervous system can lessen acute symptoms, and there are currently no effective drugs for the treatment of persistent, or recurring vestibular dysfunction.
The Ear Anatomy and Function
The ear is an interesting target for treatments in that there are several unique options available for the delivery of therapeutic agents because of its location and anatomical features. Like any tissue, dysfunction can result from defects in different specialized cell types, which must be considered when designing delivery methods and tools [14]. The ear is comprised of the outer, middle, and inner ear compartments. The inner ear houses 2 sensory organs, i.e., the vestibular system, which provides positional information crucial for balance, and the cochlea, which detects sound and is responsible for hearing. In some, but not all cases, vestibular problems accompany hearing loss.
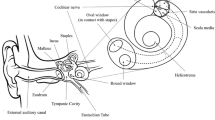
The ear is designed to detect sound by coupling the entry of sound waves into the outer ear with the vibration of the tympanic membrane and mechanical movement of a chain of 3 bones (i.e., ossicles) in the middle ear, followed by the transduction of the mechanical signals into electrical signals in the inner ear (Fig. 1). The inner ear is divided into the cochlea, the primary sensory organ for hearing, and the otoliths and semi-circular canals, which comprise the peripheral vestibular system. The movement of the ossicles puts pressure on a membrane-covered opening of the inner ear called the oval window. This pressure displaces the perilymph of the inner ear, creating a wave that travels through the cochlea, depressing the basilar membrane lining the cochlea, and this movement is finally compensated for at the round window. The round window itself is a membrane at the base of the cochlea consisting of an outer epithelial, middle fibrous, and inner epithelial layer. The basilar membrane forms the base of the organ of Corti, which contains 3 types of cells, i.e., inner hair cells (IHCs), outer hair cells (OHCs), and supporting cells (SC) (Fig. 2). Hair cells are highly organized mechanosensory cells with hair-like projections on their surface called stereocilia that, when deflected by the movement of perilymph following a sound vibration, create receptor potentials that induce neurotransmitter release onto the synaptic endings of neurons of the cochlear (i.e., auditory) nerve. This sensory signal is then transmitted through the spiral ganglion and relayed across nuclei in the pons, midbrain, and thalamus to the auditory cortex in the temporal lobe. In this way, sound waves are transmitted from the outer ear to the middle ear to the inner ear and the cochlea and are transduced to electrical signals via the IHCs. These signals are then transmitted to the brain via afferent neurons and perceived as sound. Loss or absence of hair cells results in the inability of neurons to relay information to the central auditory pathways, causing hearing loss. Hair cell loss is permanent as regeneration does not occur in humans.

Anatomy of the ear. Diagram of the major structures of the outer, middle and inner ear

Cross-section schematic of the outer and middle ear and the cochlea with the major structures indicated and with delivery routes commonly used for drug delivery shown. (inset) A depiction of the portion of the cochlea magnified to show the hair cells within the organ of Corti with spiral ganglion innervations
Hearing loss is typically assessed by testing hearing sensitivity, performed by measuring hearing thresholds, which are the lowest level of sound that can be heard 50% of the time. Hearing thresholds are measured in decibels (dB) and the threshold is assessed at different frequencies (Hertz, Hz) in each ear to determine the degree of hearing loss as a function of how high a hearing threshold is above normal. For example, thresholds that are 20 to 40 dB higher than normal indicate mild hearing loss, whereas thresholds of 90 dB are an indication of profound loss. A widely used test used to determine a hearing response and thereby assess thresholds is the auditory brainstem response (ABR), which is an auditory-evoked potential generated in response to sound stimulation that is produced by electrical impulses of the cochlear nerve and brainstem. These neural impulses are detected as waves and are indicative of the response in the peripheral system (i.e., cochlear nerve) followed by waves of activity in central nervous system auditory circuits [15]. Delays, or absence of ABR responses, indicate auditory dysfunction. Additional measurements of hearing that are often used in evaluating loss include the assessment of otoacoustic emissions (OAEs), which are sounds generated from the inner ear as a result of sound amplification by the outer hair cells. OAEs are absent following damage of OHCs, and thus measurement of these sounds allows the direct evaluation of outer hair cell function.
The brain requires the vestibular system to understand its position and movement. The peripheral vestibular apparatus is comprised of the otoliths and semi-circular canals, which report to the brain on linear and rotational head movement, respectively. Similar to the cochlea, the vestibular organs also function through the movement of stereocilia on the surface of sensory hair cells; though here, unlike cochlear hair cells, the mechanical activation of the stereocilia is largely engaged through changes in head position and angle with respect to Earth’s gravitational pull. Like hearing, balance can be evaluated by behavioral and physiological methods [13]. One direct measure of vestibular function is the vestibular sensory evoked potential (VsEP), which involves an evaluation of the action potentials generated by the vestibular nerve in response to head movement as recorded from the surface of the skull, similar to the ABR.
Treatment of Inner Ear Dysfunction
In order to develop effective drugs to treat hearing loss, careful consideration must be given to the specific type of hearing loss and the cause of the deficit. Estimates indicate that 50% of congenital hearing loss, that which is present at or soon after birth, has a genetic etiology, which presents opportunity for correction by gene therapy approaches. However, hearing loss at birth likely indicates a problem with development of the hearing apparatus which would necessitate intervention at early stages of fetal development in humans, where differentiation of hair cells begins during the third month of pregnancy (10 to 12 weeks postconception) [16, 17]. Unlike humans, in mammals such as the mouse, which is the most common animal model for genetic hearing loss, the cochlea is not fully developed or functional at birth, allowing for effective early interventions by treating shortly after birth. Thus, translating results from mouse models of hearing loss to humans must consider the differences in hearing development between species.
Though many forms of hearing and vestibular dysfunction are developmental and suggest that treatment would require early intervention to restore appropriate gene expression at the required time point, there are many cases of late-onset or slowly progressing hearing loss caused by genetic mutations that could respond to therapies applied later in life. In addition, age-related and noise-induced hearing loss and other degenerative forms of inner ear dysfunction could be treated in a responsive or prophylactic manner in children and adults. While many approaches that have demonstrated therapeutic benefit to hearing loss and vestibular function have treated mice at early postnatal time points during the developmental period of the inner ear, there have been some studies demonstrating therapeutic efficacy in juvenile and adult inner ears, suggesting that the organ will be responsive to therapies that are engaged at later stages and that timing will depend on the specific target and deficit rather than any intrinsic barriers to therapy [18, 19].
For genetic forms of hearing loss, information about the effect of a mutation is important for designing therapeutics. For example, whether a mutation is autosomal dominant or recessive offers insight into whether an approach to knockdown toxic gain-of-function gene products or correct aberrant gene expression resulting from loss-of-function mutations would be beneficial. Both of these outcomes can be achieved by appropriate ASO design as described below.
Delivery of therapeutic molecules to the ear is another issue in drug development that must be addressed. Though in some ways the inner ear is an ideal organ for drug delivery due to its isolation within the bony capsule of the temporal bone and the relatively easy access via the middle ear, there are unique barriers to drug entry into the system that must be overcome [20, 21]. There are a number of different routes for drug delivery to the inner ear that have been explored that can be broadly classified as systemic or local. For ease of delivery, a therapeutic administered systemically, via intravenous, intramuscular, or oral delivery, for example, may be ideal if the molecule can gain access to the relevant cells of the inner ear. However, endothelial cells of blood vessels create a blood-perilymph and blood-strial barrier that restrict the entry of systemically applied drugs into the inner ear and also control elimination of locally administered reagents. Thus, local delivery could be advantageous in that it would provide drug presentation directly and exclusively to the target organ, thereby maximizing local concentration and minimizing systemic effects while also bypassing many of the physical barriers present following systemic delivery. Local delivery routes that have been explored include (1) intratympanic (IT) administration via injection through the tympanic membrane and into the middle ear, (2) intracochlear through the round window or cochleostomy, and (3) intralabyrinthine to the posterior semi-circular canal via canalostomy (Fig. 2a) [22,23,24,25,26,27,28]. IT application is limited by passage through the middle ear epithelium, which covers the round window and the oval window, and elimination through the Eustachian tube. IT administration of therapeutic molecules in hydrogel systems, in some cases, has been shown to address some of these problems [29, 30]. The latter 2 methods have an advantage in that they deliver directly to the perilymphatic system, bypassing adsorption barriers, though, at the same time, these may be considered more invasive and with more potential risks than systemic or intratympanic delivery approaches. Indeed, round window injection has variable efficacy, possibly due to the increased risk of middle ear effusion [31]. Canalostomy may have some advantages for drug delivery directly to the inner ear without the risk for hearing loss [22, 32]. One important consideration for either route of delivery is the limited volume of drug that can be introduced into the inner ear labyrinth, a space that is restricted by the surrounding bony labyrinth. One way that this volume limitation can be addressed is to inject through the round window membrane following fenestration of the semi-circular canal, which adds an egress for fluid and effectively doubles the allowable injection volume [23]. Ultimately, the type of drug, the target cells, the protein function/dysfunction, mutation type, and developmental requirements will all need careful consideration when designing treatments for the inner ear [33].
Current therapies that are in the clinic for inner ear conditions are primarily pharmaceuticals such as aminoglycosides and steroids; however, many other treatment reagents are in development including small-molecule drugs, gene editing, viral vectors for exogenous gene expression, and antisense oligonucleotides [34].
Antisense Oligonucleotide Therapeutics
Targeting and modifying gene expression with short nucleic acid sequences that function, in part, by complementary base-pairing to a target RNA has shown promise as a therapeutic strategy. There are a number of different types of antisense molecules that have been explored as therapeutics, differing primarily in their mechanism of action. These include antisense oligonucleotides (ASOs) that alter their target by creating a steric block upon binding, or those that degrade the targeted RNA upon binding, such as ribozymes, siRNA/miRNAs, or RNase H-dependent ASOs. In general, unmodified RNA or DNA molecules are unstable and degrade rapidly in vivo. Most ASOs are chemically modified to improve their pharmacological profile. The specific chemical modifications that are being made to ASOs are dependent on the type of ASO. Modifications to the ASO have been crucial to stabilize the ASO in vivo, increase binding affinity towards its target and improve cellular uptake and release, and, more recently, provide selective tissue and cell type-specific targeting features, all of which have been extensively reviewed elsewhere [35,36,37,38].
Two types of oligonucleotides that have been explored as therapeutics for hearing loss include splice-switching ASOs and RNA interference (RNAi) with siRNAs or miRNA sequences.
RNA Interference
ASOs that target RNA for degradation by the RNA-induced silencing complex (RISC) are designed as small interfering RNAs (siRNAs) or microRNAs (miRNAs) that trigger RNA degradation via the RNA interference (RNAi) pathway [39, 40]. RNAi refers to the phenomenon by which double-stranded RNAs (dsRNA) induce sequence-specific posttranscriptional silencing of a gene with a sequence homologous to the dsRNA by targeting its RNA transcript (Fig. 3a). This process involves the cleavage of a region of dsRNA into a shorter 21- to 23-nucleotide dsRNA fragment by the dsRNA-specific nuclease, Dicer. The resulting dsRNA is bound by an Argonaut protein, forming the RISC complex where 1 strand of the dsRNA is removed and the other strand is delivered to the target RNA sequence for base-pairing. After binding, the targeted RNA is either cleaved, degraded, or otherwise translationally repressed [40].

Examples of antisense approaches for the treatment of hearing loss and vestibular dysfunction. (a) A miRNA-mediated knockdown of the semi-dominant gain-of-function allele TMC1/Bth by RNAi. An artificial miRNA gene targeting TMC/Bth expressed from a cassette within the adeno-associated virus (AAV2/9) vector following viral transduction is processed to generate a short 21- to 23-nt double-stranded RNA that is bound by the RNA-induced silencing complex (RISC). One strand of the duplex is removed and the remaining single-stranded antisense RNA base pairs to the TMC1/Bth mRNA, which initiates cleavage or degradation, resulting in a decrease in overall mRNA abundance. (b) Scheme of a splice-switching ASO for the correction of splicing. A mutation (216G>A) in exon 3 of USH1C creates a cryptic splice site that results in mis-splicing and the creation of an open reading frameshift and the loss of full-length Harmonin, thereby causing Usher syndrome type 1C. The ASO (shown in blue) base pairs across the cryptic splice site blocks the aberrant splicing, thereby redirecting to the wild-type splice site and restoring the USH1C open reading frame and full-length Harmonin. Boxes represent exons and lines are introns. Diagonal lines indicate the splicing pathway
Utilizing the cells’ natural RNAi mechanism to specifically downregulate gene expression by introducing siRNAs to the cell has been an intensely studied tool for gene-based therapy, and the first siRNA drug, patisiran (ONPATTRO™) [41, 42], which targets and downregulates TTR gene expression in the liver, has recently been approved by the FDA for the treatment of hereditary transthyretin amyloidosis. Patisiran is a 21-nucleotide double-stranded nucleic acid with a portion of the sequence modified with 2′-O-methylcytidines and 2′-O-methyluridines. The modified nucleic acid is formulated as a lipid complex for delivery to hepatocytes. A key development leading to the successful advancement of this siRNA treatment was the formulation of lipid nanoparticles as agents to deliver the siRNAs.
For the treatment of or protection against hearing loss, a number of different RNAi strategies differing in RNA targets, delivery modalities, and types of siRNA/miRNA molecules have shown efficacy in animal models. These include but are not limited to RNA approaches involving the knockdown of mRNA encoding toxic, gain-of-function proteins [26, 43] or modulation of gene pathways that limit regeneration or protect against noise-induced hearing loss [44,45,46] or cisplatin-induced ototoxicity [47,48,49]. In most cases reported, unformulated siRNAs were delivered via trans-tympanic injection to the middle ear prior to the induction of hearing and hair cell damage, which results in modest protection from hearing loss. This mode of delivery appears to be effective for siRNAs, though improvements in delivery could elevate protection [50]. Promising results with AAV-mediated transduction of artificial miRNAs or nanoparticle-formulated siRNAs indicate alternative effective approaches for RNAi-based knockdown of gene expression. Here, we expand on examples of RNAi approaches for the treatment of 2 different types of hearing loss.
DFNA36/TMC1
The use of RNAi to suppress autosomal-dominant mutant alleles associated with hearing loss has been identified as an effective approach in an animal model of human autosomal-dominant NSHL, wherein a missense mutation in a single gene allele is responsible for the deficit. For this, a mouse carrying the semi-dominant Tmc1 c.1235T>A (p.M412K) allele, also known as the Beethoven (Bth) missense mutation, was treated with an artificial miRNA expressed from an adeno-associated virus (AAV) vector administered by injection through the round window membrane (Fig. 3a) [26, 51]. The orthologous human mutation, TMC1 c.1253T>A (p.M418K), is associated with progressive postlingual sensorineural hearing loss with an age of onset that varies from 5 to 25 years of age [52, 53]. Similarly in mice, the Bth allele causes progressive hearing loss. A single injection of AAV-TMC1 miRNA into postnatal day 0 to postnatal day 2 (P0-P2) mice resulted in a nearly 90% reduction in the targeted Tmc1 c.1235T>A allele 4 weeks after injection. This knockdown of the disease-associated allele improved hair cell survival compared to control mice. Expression of the Tmc1-targeted miRNA also slowed progression of hearing loss, as measured by ABR, for up to 21 weeks with some mice exhibiting stable ABR thresholds for up to 35 weeks, the latest time point assessed. Hearing preservation was significant at 8 and 16 kHz but not at 32 kHz, similar to reports from others on the lack of efficacy of hearing therapeutics in the high-frequency range in mice and may be related to the amount of exposure of treatment to the cells in the region of the cochlea that is responsible for detection of high-frequency tones [27, 54].
In a follow-up study, in which delivery was performed by injection through the round window membrane in combination with semi-circular canal fenestration, treatment of adult mice with the TMC1-targeted miRNA was found to slow the progression of hearing loss and stereocilia bundle degeneration, and improve hair cell survival [51]. This study demonstrated for the first time that antisense-based genetic therapies are effective when administered to an adult mouse model of human deafness, providing exciting proof of concept of the efficacy of treatment later in life. Also important to note, despite having only a single-nucleotide difference in the targeted sequence, expression of the miRNA reduced expression of the mutated allele only without affecting wild-type allele expression. This allele-specific knockdown is particularly important in cases of targeting a dominant negative target as the wild-type protein product likely is required and must be maintained in order to support normal hearing after the dominant negative protein is knocked down. Overall, these results demonstrate the feasibility of RNAi for the suppression of deafness-associated gene expression to treat hearing loss. Although this approach utilizes an AAV-mediated expression of the siRNA molecule, which has the benefit of long-term expression of the interfering RNA, the option of nonviral delivery similar to patisiran is a possibility if appropriate formulation could be developed for efficient inner ear delivery.
Other nucleic acid-based therapeutic approaches for inner ear dysfunction are being pursued and have shown good therapeutic benefit in mouse models [55,56,57]. The use of different approaches in the same mouse models of human deafness have allowed a comparison of treatments. For example, the DFNA36/TMC1 mice that were effectively treated with AAV-delivered artificial miRNAs to downregulate dominant negative TMC/Bth expression, discussed above, also have been shown to respond positively to a gene-editing strategy using the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 gene editing system to correct the causative TMC1 Bth mutation. In this case, the Cas9 protein was packaged with its guide RNA in a lipid droplet that can fuse with cells. Injection of the nanoparticles into the inner ear of newborn mice resulted in delivery to the cells and establishment of inner hair cells and temporary improvement of hearing despite genomic editing that was limited to a small number of cells [58].
Hes1 and Noise-Induced Hearing Loss
Another RNAi strategy that has been investigated as a therapeutic approach for hearing loss caused by damage is the downregulation of gene expression pathways that suppress regeneration. Regeneration of hair cells following damage is considered an important goal for the treatment of hearing loss. Induction or expression of the transcription factor Atoh-1, which plays a role in hair cell differentiation during development but is not expressed at high levels in adults, has been a target for activating cell regeneration in the inner ear. One approach has been to downregulate repressors of Atoh-1 gene expression, such as the transcriptional repressors in the Hes gene family, using siRNAs [59]. To this end, the knockdown of HES1 gene expression with siRNAs resulted in hair cell differentiation and some improvement in auditory thresholds in guinea pigs following noise injury [19]. In this case, 3 days after injury, siRNAs were delivered encapsulated in poly(lactide-co-glycolide acid) (PLGA) nanoparticles for sustained release following administration via a mini-osmotic pump infusions through a cochleostomy lateral to the round window.
Splice-Switching Antisense Oligonucleotides
Splice-switching ASOs are typically comprised of 12-30 nucleotides that are chemically modified to improve stability, deliverability, and binding affinity [60, 61]. This type of ASO functions by binding alone. ASOs that confer their effects by simply binding to a target RNA can be designed to block interactions of a specific single-stranded region of RNA with proteins or other RNA sequences, thereby altering RNA processing. For splice-switching activity, an ASO base-pairing to a target RNA can alter the recognition of splice sites by the spliceosome, which leads to an alteration of normal splicing of the targeted transcripts. Importantly, in order to avoid cleavage by the endogenous nuclease, RNase H, which cleaves RNA that is duplexed with DNA, the ASO must be chemically modified to escape enzymatic cleavage [62, 63]. Chemical modifications to the backbone, such as phosphorodiamidate morpholino linkages, and to the sugar, such as 2′-modified and conformationally constrained nucleotides, have improved nuclease resistance [64, 65]. Thus, with appropriate chemical modifications, splice-switching ASOs modify gene expression without altering the overall abundance of the target RNA. Splice-switching ASOs are the basis of the FDA-approved drugs for spinal muscular atrophy (SMA), nusinersen (SPINRAZA™) [66], and Duchenne muscular dystrophy (DMD), eteplirsen (EXONDYS 51™) [67].
Though not yet explored as a therapeutic approach for inner ear dysfunction, the use of RNase H-dependent ASOs could be an effective tool when downregulation of gene expression is the goal, much in the way siRNAs and the RNAi pathway have been utilized for downregulation of gene expression. ASOs can be designed to have a core of DNA nucleotides flanked by modified RNA nucleotides for stability. These so-called gapmers, when duplexed with RNA, create a DNA:RNA duplex in which the RNA becomes a substrate for cleavage by the endogenous nuclease, RNAse H [68]. Gapmers are currently in the clinic for the treatment of familial hypercholesterolemia, mipomersen (KYNAMRO™) [69, 70], and TTR amyloidosis, inotersen (TEGSEDI™) [71].
Usher Syndrome
Splice-switching ASOs have been used to correct gene expression and rescue hearing and vestibular function in mouse models of Usher syndrome. Usher syndrome (USH) is characterized by the combination of sensorineural hearing loss with retinitis pigmentosa and progressive vision loss. With a prevalence of as much as 1 in 6000, it is the most common hereditary cause of combined deafness–blindness [72]. By some counts, more than 10% of children with hearing loss have a mutation in an Usher syndrome gene [72]. The onset of symptoms is variable but typically occurs during childhood. Usher syndrome is clinically classified by severity [73]. Usher type 1, caused by a mutation in 1 of 6 genes or loci, accounts for ~ 40% of cases, and hearing loss is congenital, profound, and often associated with vestibular areflexia. Usher type 2, caused by mutations in 3 genes, accounts for about 60% of cases, which exhibit moderate to severe, slow progressing, prelingual hearing loss without vestibular dysfunction. Usher type 3 is associated with mutations in 1 gene which results in rapidly progressing hearing loss, variable vestibular involvement, and vision loss that progresses slowly to blindness in early to mid-adulthood. In some people with Usher syndrome, the genetic cause of the condition has not been identified and it is likely that additional genes are associated with the disorder. Most of the gene mutations associated with Usher syndrome result in a loss of hair cells in the inner ear and a loss of photoreceptors in the retina, which cause the hearing loss, balance problems, and vision loss [73].
USH1C/Harmonin
The first demonstration that ASOs could be used to treat hearing loss came in 2013 with the report of hearing and vestibular rescue in a mouse model of Usher syndrome type 1C following ASO treatment [54]. Usher type 1C is caused by mutations in USH1C which encodes the protein Harmonin, a scaffolding protein involved in interactions critical for establishing and maintaining stereocilia structures and organization of hair cells in the cochlea [74,75,76]. A splice-switching ASO was designed to base-pair to a donor 5′ splice site sequence that was created by a G>A mutation at position c.216 in exon 3 of the USH1C gene (Fig. 3b). When treated with a single intraperitoneal injection at postnatal day 5 (P5), hearing was rescued to wild-type levels at some frequencies, as assessed by auditory-evoked brainstem response analysis [54]. This rescue was complete at low frequencies, 8 and 16 kHz, and to a lesser degree at 32 kHz. Outer hair cells were also functional in ASO-treated mice as demonstrated by significant DPOAE recordings in treated mice, which were absent in mice treated with a control, nontargeted ASO [77]. ABR and DPOAE analysis revealed that treatment of mice at earlier time points at P1 compared to P5 resulted in better IHC and OHC function [54, 77]. Remarkably, hearing was maintained for at least 6 months after treatment, though an increase in thresholds was observed compared to measurements at earlier time points. Immunohistochemical analysis of ASO localization in the cochlear hair cells using an ASO-specific antibody showed widespread distribution of the ASO through the basilar membrane with greater intensity of signal in the inner hair cells relative to the outer hair cells [77]. The rescue of hearing was accompanied by a significant preservation of OHC number and organization of the stereocilia [54]. Surprisingly, though there was a robust increase in Harmonin protein levels in the cochlea of Usher mice treated with the ASO targeted to the 216G>A mutation, there was a relatively low level of corrected splicing compared to splicing at the aberrant site [54, 77, 78]. This result suggests that a low amount of Harmonin RNA is sufficient to support hearing in these mice.
ASO treatment of Usher mice also eliminated all behaviors suggestive of vestibular dysfunction in the mice including circling, head-tossing, and inability to swim [54]. A more detailed investigation of the treatment of vestibular dysfunction was performed by testing the Usher mice for vestibular sensory evoked potentials (VsEPs), which directly assesses vestibular function. Interestingly, it was found that if treatment was administered during a critical period between postnatal day 1 and postnatal day 5 (P1-P5), mice had normal vestibular function as indicated by VsEP, whereas mice treated at P15 did not display abnormal vestibular behavior (circling and swim dysfunction) but had abnormal VsEPs [78]. Exploratory behavior is also disrupted in Usher mice, a deficit not entirely related to the abnormal vestibular behavior. An investigation of the organization of exploratory movements in these mice was used to assess spatial organization in both dark and light settings. Typically, mice utilize a combination of environmental as well as self-movement cues as they explore and these activities were found to be disrupted in Usher mice and recovered in Usher mice treated with ASO to correct Ush1c splicing [79].
Hearing development in mice continues postnatally until approximately P15. This developmental period corresponds to the time frame in which ASO-Ush must be delivered to confer therapeutic benefit to hearing and balance phenotypes, suggesting that a window of opportunity for treatment exists during hearing development. However, in humans, the auditory system becomes functional at around 25 to 29 weeks gestational age with the earliest evidence of ABRs at 16 weeks gestational age [16, 55]. This developmental window suggests that a treatment for congenital hearing loss would likely require administration in utero. With this in mind, a recent study found that an intra-amniotic cavity delivery of ASO results in a significant effect on the target RNA in the inner ear. When injected into the amniotic cavity of E13-13.5 embryos, the ASO targeting the Ush1c c.216A aberrant splice site increased correctly spliced Ush1c mRNA to a level comparable to that observed when the ASO was delivered by IP injection at P5 [80]. These results suggest that intra-amniotic delivery of ASOs could be considered a therapeutically relevant route of delivery for a relatively noninvasive treatment approach for fetal and congenital diseases.
Other strategies for the treatment of Usher syndrome type 1C have also been reported. An AAV-mediated gene delivery approach has been shown to have similar efficacy in the Ush1c c.216AA mice as a splice-switching ASO [27]. In this case, the AAV-Ush1c virus was injected directly into the inner ear via the round window and similar to the outcomes with the ASO, vestibular function and auditory function were recovered for up to 6 months post-treatment. Similar to results with ASO treatment, therapeutic benefit was restricted to treatments that occurred before P10-P12 and to low-frequency hearing, with little recovery in the 32-kHz range. A more direct comparison of these 2 approaches will require a study to test the activity of the ASO delivered directly to the inner ear and a more long-term assessment of sustained auditory function. Together these studies demonstrate the utility of both ASOs and traditional gene therapy in the treatment of this form of Usher syndrome. Importantly, the 2 treatments are not expected to interfere with each other and, thus, could be used in combination, an advantage that could prove important, for instance in cases where AAV treatment/redosing may be limited by immunogenicity.
USH2A/Usherin
Mutations in the USH2A gene, encoding the Usherin protein, are the most frequent causes of Usher Type 2 (USHbases; http://www.lovd.nl/USH2A) [81]. One mutation, USH2A c.7595-2144A>G creates a new donor splice site within intron 40 of the gene that results in splicing of a 40 nucleotide pseudoexon (PE40) into the USH2A mRNA [82]. ASOs designed to base-pair to the 3′ splice acceptor site region of PE40 effectively block PE40 splicing and restore appropriate splicing of USH2A in patient-derived fibroblasts [83] as well as in a humanized zebrafish knockin model, though aberrant splicing of PE40 was minimal in these animals [84].
Further analysis of the USH2A gene in individuals with Usher type 2, as determined by the identification of a heterozygous mutation in the gene, identified 3 additional deep intronic mutations in the gene, all of which are predicted to create de novo splice sites and inclusion of pseudoexons [85]. The effectiveness of splice-switching ASOs in blocking pseudoexon splicing was demonstrated in vitro using a morpholino antisense oligonucleotide to block the aberrant splicing caused by 1 of the mutations c.9959-4159A>G.
USH2A exon 13 mutations, such as c. 2299delG, are found in a large proportion of Usher type 2 patients [86,87,88,89]. Many of these mutations introduce premature termination codons (PTCs) or frameshifts that result in loss of Usherin protein. The complete elimination of exon 13 does not disrupt the open reading frame of the protein and, thus, inducing skipping of the exon with splice-switching ASOs would eliminate stop mutations, essentially correcting the reading frame and recovering Usherin protein expression, albeit an isoform lacking the 214 amino acids encoded by exon 13. Analysis of patients revealed that those with 2 truncating USH2A mutations developed more severe hearing impairment than patients with only 1 truncating mutation or 2 nontruncating mutations [90], suggesting that using ASOs to induce skipping to essentially change a truncating mutation to a nontruncating mutation may be a viable therapeutic approach. A similar rationale is the basis of the ASO therapeutic for Duchenne’s Muscular Dystrophy, EXONDYS 51 [67]. This approach for USH2A is being pursued by ProQR as QR-421a, which recently received fast-track designation from the FDA, for treatment of vision loss in USH patients with USH2A exon 13 mutations (http://www.proqr.com; patent no. US 10,131,910 B2).
These ASO approaches for Usher type 2 caused by mutations in USH2A have not yet been shown to have therapeutic effects on hearing, due to the lack of appropriate animal models, but nonetheless demonstrate that ASOs can induce the desired effect on splicing. Given the demonstration of therapeutically relevant ASO delivery to the inner ear to prevent hearing impairment and vestibular dysfunction in Usher mice with the USH1C mutation, as described above, there is a strong precedence that ASOs with similar characteristics will have efficacy in vivo.
Conclusions and Future Directions
Many issues must be addressed when developing an inner ear therapy for hearing loss and/or vestibular dysfunction including the specific drug design, the age at treatment, target tissue and cell type, and the route of injection/delivery. There are many opportunities for the use of this technology for the treatment of inner ear conditions. For example, missense mutations are responsible for most cases of autosomal-dominant (AD) nonsyndromic hearing loss. Eliminating the mutant RNA transcript by RNAi or RNAse H-mediated cleavage using antisense molecules is a broadly applicable approach to eliminating RNAs associated with toxic gain-of-function protein products typical in AD NSHL. Furthermore, AD NSHL is typically postlingual and progressive, offering a relative long therapeutic window of opportunity for intervention. For other types of mutations, splice-switching ASOs, similar to the 1 described to target the Ush1c c.G216A mutation, are promising as therapeutic molecules. An example of another splicing mutations that results in a form of Usher syndrome is Usher 3A. Here, a CLRN1 (USH3A) mutation that creates a new splice site activates splicing of an aberrant pseudoexon, resulting in a frameshift and premature termination codon in the mRNA [91]. In addition to blocking the use of cryptic or de novo splice sites activated by mutations, splice-switching ASOs can also be designed to induce splicing events that would correct the reading frame in cases of frameshift and stop mutations. The demonstrations to date that antisense oligonucleotides and other forms of nucleic acid therapeutics can successfully access and affect targeted gene expression are a major achievement that sets a precedence for further development of this form of therapeutic for hearing loss.
Drug delivery to the inner ear has a number of advantages compared to other organs such as the potential for access to the cochlea and/or vestibular organ via a number of different direct injection routes [56]. Additionally, there is evidence that ASOs can access the cochlea when delivered systemically or even to the amniotic cavity [54, 80]. Future developments in ASO delivery will likely benefit treatment development for the inner ear. For example, delivery of ASOs in nanoparticles, such as PLGA used in delivery of siRNAs targeting HES1, described above, could aid in inner ear treatment, although in this case the nanoparticle was injected through the round window membrane [19]. There has been limited research to date on targeted nanoparticles systemically delivered to the inner ear or through the round window [29]. Additionally, though not yet explored for inner ear targeting, tissue-specific delivery of ASOs has been rapidly developing, predicting modifications to the antisense molecules themselves that will maximize delivery to the cells of interest [38]. Together, antisense approaches to treat hearing and vestibular dysfunction offer a number of different options for therapies, many of which have shown therapeutic promise in animal models of human deafness, setting a precedence for future work and more aggressive pursuit of antisense technology for inner ear therapy.
References
Wilson BS, Tucci DL, Merson MH, O'Donoghue GM. Global hearing health care: new findings and perspectives. Lancet. 2017;390(10111):2503–15.
Smith RJ, Bale JF, Jr., White KR. Sensorineural hearing loss in children. Lancet. 2005;365(9462):879–90.
Shearer AE, Hildebrand MS, Smith RJH. Hereditary Hearing Loss and Deafness Overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews((R)). Seattle (WA)1993.
Van Camp G, Willems PJ, Smith RJ. Nonsyndromic hearing impairment: unparalleled heterogeneity. Am J Hum Genet 1997;60(4):758–64.
Alford RL, Arnos KS, Fox M, Lin JW, Palmer CG, Pandya A, et al. American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genet Med : Off J Am Coll Med Genet 2014;16(4):347–55.
Azaiez H, Booth KT, Ephraim SS, Crone B, Black-Ziegelbein EA, Marini RJ, et al. Genomic Landscape and Mutational Signatures of Deafness-Associated Genes. Am J Hum Genet 2018;103(4):484–97.
Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet 2016;135(4):441–50.
Lopez C. The vestibular system: balancing more than just the body. Curr Opin Neurol 2016;29(1):74–83.
Agrawal Y, Carey JP, Della Santina CC, Schubert MC, Minor LB. Disorders of balance and vestibular function in US adults: data from the National Health and Nutrition Examination Survey, 2001-2004. Arch Intern Med 2009;169(10):938–44.
Ji L, Zhai S. Aging and the peripheral vestibular system. J Otolaryngol 2018;13(4):138–40.
Dillon CF, Gu Q, Hoffman HJ, Ko CW. Vision, hearing, balance, and sensory impairment in Americans aged 70 years and over: United States, 1999-2006. NCHS Data Brief 2010(31):1–8.
Agrawal Y, Pineault KG, Semenov YR. Health-related quality of life and economic burden of vestibular loss in older adults. Laryngoscope Investig Otolaryngol 2018;3(1):8–15.
Jones SM, Jones TA. Genetics of peripheral vestibular dysfunction: lessons from mutant mouse strains. J Am Acad Audiol 2014;25(3):289–301.
Ahmed H, Shubina-Oleinik O, Holt JR. Emerging Gene Therapies for Genetic Hearing Loss. J Assoc Res Otolaryngol: JARO 2017;18(5):649–70.
Akil O, Oursler AE, Fan K, Lustig LR. Mouse Auditory Brainstem Response Testing. Bio Protoc. 2016;6(6).
Hall JW, 3rd. Development of the ear and hearing. J Perinatol 2000;20(8 Pt 2):S12–20.
Magarinos M, Contreras J, Aburto MR, Varela-Nieto I. Early development of the vertebrate inner ear. Anat Rec (Hoboken) 2012;295(11):1775–90.
Alagramam KN, Gopal SR, Geng R, Chen DH, Nemet I, Lee R, et al. A small molecule mitigates hearing loss in a mouse model of Usher syndrome III. Nat Chem Biol 2016;12(6):444–51.
Du X, Cai Q, West MB, Youm I, Huang X, Li W, et al. Regeneration of Cochlear Hair Cells and Hearing Recovery through Hes1 Modulation with siRNA Nanoparticles in Adult Guinea Pigs. Mol Ther : J Am Soc Gene Ther 2018;26(5):1313–26.
Salt AN, Hirose K. Communication pathways to and from the inner ear and their contributions to drug delivery. Hear Res 2018;362:25–37.
Glueckert R, Johnson Chacko L, Rask-Andersen H, Liu W, Handschuh S, Schrott-Fischer A. Anatomical basis of drug delivery to the inner ear. Hear Res 2018;368:10–27.
Isgrig K, Chien WW. Posterior Semicircular Canal Approach for Inner Ear Gene Delivery in Neonatal Mouse. J Vis Exp 2018(133).
Yoshimura H, Shibata SB, Ranum PT, Smith RJH. Enhanced viral-mediated cochlear gene delivery in adult mice by combining canal fenestration with round window membrane inoculation. Sci Rep 2018;8(1):2980.
Isgrig K, Chien WW. Surgical Methods for Inner Ear Gene Delivery in Neonatal Mouse. Methods Mol Biol 2019;1937:221–6.
Akil O, Rouse SL, Chan DK, Lustig LR. Surgical method for virally mediated gene delivery to the mouse inner ear through the round window membrane. J Vis Exp 2015(97).
Shibata SB, Ranum PT, Moteki H, Pan B, Goodwin AT, Goodman SS, et al. RNA Interference Prevents Autosomal-Dominant Hearing Loss. Am J Hum Genet 2016;98(6):1101–13.
Pan B, Askew C, Galvin A, Heman-Ackah S, Asai Y, Indzhykulian AA, et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat Biotechnol 2017;35(3):264–72.
Salt AN, Plontke SK. Pharmacokinetic principles in the inner ear: Influence of drug properties on intratympanic applications. Hear Res 2018;368:28–40.
Li L, Chao T, Brant J, O'Malley B, Jr., Tsourkas A, Li D. Advances in nano-based inner ear delivery systems for the treatment of sensorineural hearing loss. Adv Drug Deliv Rev 2017;108:2–12.
Goycoolea MV. Clinical aspects of round window membrane permeability under normal and pathological conditions. Acta Otolaryngol 2001;121(4):437–47.
Zhu BZ, Saleh J, Isgrig KT, Cunningham LL, Chien WW. Hearing Loss after Round Window Surgery in Mice Is due to Middle Ear Effusion. Audiol Neurootol 2016;21(6):356–64.
Guo JY, He L, Qu TF, Liu YY, Liu K, Wang GP, et al. Canalostomy As a Surgical Approach to Local Drug Delivery into the Inner Ears of Adult and Neonatal Mice. J Vis Exp 2018(135).
Maeda Y, Sheffield AM, Smith RJ. Therapeutic regulation of gene expression in the inner ear using RNA interference. Adv Otorhinolaryngol 2009;66:13–36.
Zhang W, Kim SM, Wang W, Cai C, Feng Y, Kong W, et al. Cochlear Gene Therapy for Sensorineural Hearing Loss: Current Status and Major Remaining Hurdles for Translational Success. Front Mol Neurosci 2018;11:221.
Havens MA, Hastings ML. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res 2016;44(14):6549–63.
Khvorova A, Watts JK. The chemical evolution of oligonucleotide therapies of clinical utility. Nat Biotechnol 2017;35(3):238–48.
Bennett CF. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu Rev Med 2019;70:307–21.
Seth PP, Tanowitz M, Bennett CF. Selective tissue targeting of synthetic nucleic acid drugs. J Clin Invest 2019.
Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136(4):642–55.
Lam JK, Chow MY, Zhang Y, Leung SW. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol Ther Nucleic Acids. 2015;4:e252.
Adams D, Gonzalez-Duarte A, O'Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med 2018;379(1):11–21.
Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med 2013;369(9):819–29.
Maeda Y, Fukushima K, Nishizaki K, Smith RJ. In vitro and in vivo suppression of GJB2 expression by RNA interference. Hum Mol Genet 2005;14(12):1641–50.
Hill K, Yuan H, Wang X, Sha SH. Noise-Induced Loss of Hair Cells and Cochlear Synaptopathy Are Mediated by the Activation of AMPK. J Neurosci 2016;36(28):7497–510.
Xiong H, Long H, Pan S, Lai R, Wang X, Zhu Y, et al. Inhibition of Histone Methyltransferase G9a Attenuates Noise-Induced Cochlear Synaptopathy and Hearing Loss. Journal of the Association for Research in Otolaryngology : JARO. 2019.
Wang X, Zhu Y, Long H, Pan S, Xiong H, Fang Q, et al. Mitochondrial Calcium Transporters Mediate Sensitivity to Noise-Induced Losses of Hair Cells and Cochlear Synapses. Front Mol Neurosci 2018;11:469.
Mukherjea D, Jajoo S, Kaur T, Sheehan KE, Ramkumar V, Rybak LP. Transtympanic administration of short interfering (si)RNA for the NOX3 isoform of NADPH oxidase protects against cisplatin-induced hearing loss in the rat. Antioxid Redox Signal 2010;13(5):589–98.
Kaur T, Mukherjea D, Sheehan K, Jajoo S, Rybak LP, Ramkumar V. Short interfering RNA against STAT1 attenuates cisplatin-induced ototoxicity in the rat by suppressing inflammation. Cell Death Dis 2011;2:e180.
Kim YJ, Kim J, Tian C, Lim HJ, Kim YS, Chung JH, et al. Prevention of cisplatin-induced ototoxicity by the inhibition of gap junctional intercellular communication in auditory cells. Cell Mol Life Sci 2014;71(19):3859–71.
Oishi N, Chen FQ, Zheng HW, Sha SH. Intra-tympanic delivery of short interfering RNA into the adult mouse cochlea. Hear Res 2013;296:36–41.
Yoshimura H, Shibata SB, Ranum PT, Moteki H, Smith RJH. Targeted Allele Suppression Prevents Progressive Hearing Loss in the Mature Murine Model of Human TMC1 Deafness. Mol Ther : J Am Soc Gene Ther 2019.
Zhao Y, Wang D, Zong L, Zhao F, Guan L, Zhang P, et al. A novel DFNA36 mutation in TMC1 orthologous to the Beethoven (Bth) mouse associated with autosomal dominant hearing loss in a Chinese family. PLoS One 2014;9(5):e97064.
Kurima K, Peters LM, Yang Y, Riazuddin S, Ahmed ZM, Naz S, et al. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat Genet 2002;30(3):277–84.
Lentz JJ, Jodelka FM, Hinrich AJ, McCaffrey KE, Farris HE, Spalitta MJ, et al. Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat Med 2013;19(3):345–50.
Wang L, Kempton JB, Brigande JV. Gene Therapy in Mouse Models of Deafness and Balance Dysfunction. Front Mol Neurosci 2018;11:300.
Devare J, Gubbels S, Raphael Y. Outlook and future of inner ear therapy. Hear Res 2018;368:127–35.
Sayyid ZN, Kim GS, Cheng AG. Molecular therapy for genetic and degenerative vestibular disorders. Curr Opin Otolaryngol Head Neck Surg 2018;26(5):307–11.
Gao X, Tao Y, Lamas V, Huang M, Yeh WH, Pan B, et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature. 2018;553(7687):217–21.
Du X, Li W, Gao X, West MB, Saltzman WM, Cheng CJ, et al. Regeneration of mammalian cochlear and vestibular hair cells through Hes1/Hes5 modulation with siRNA. Hear Res 2013;304:91–110.
Bennett CF, Baker BF, Pham N, Swayze E, Geary RS. Pharmacology of Antisense Drugs. Annu Rev Pharmacol Toxicol 2017;57:81–105.
Ku SH, Jo SD, Lee YK, Kim K, Kim SH. Chemical and structural modifications of RNAi therapeutics. Adv Drug Deliv Rev 2016;104:16–28.
Rigo F, Chun SJ, Norris DA, Hung G, Lee S, Matson J, et al. Pharmacology of a central nervous system delivered 2'-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J Pharmacol Exp Ther 2014;350(1):46–55.
Crooke ST. Molecular Mechanisms of Antisense Oligonucleotides. Nucleic acid therapeutics 2017;27(2):70–7.
Summerton J. Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim Biophys Acta 1999;1489(1):141–58.
Yu RZ, Grundy JS, Geary RS. Clinical pharmacokinetics of second generation antisense oligonucleotides. Expert Opin Drug Metab Toxicol 2013;9(2):169–82.
Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med 2017;377(18):1723–32.
Li D, Mastaglia FL, Fletcher S, Wilton SD. Precision Medicine through Antisense Oligonucleotide-Mediated Exon Skipping. Trends Pharmacol Sci 2018;39(11):982–94.
Cerritelli SM, Crouch RJ. Ribonuclease H: the enzymes in eukaryotes. FEBS J 2009;276(6):1494–505.
Geary RS, Baker BF, Crooke ST. Clinical and preclinical pharmacokinetics and pharmacodynamics of mipomersen (kynamro((R))): a second-generation antisense oligonucleotide inhibitor of apolipoprotein B. Clin Pharmacokinet 2015;54(2):133–46.
Parham JS, Goldberg AC. Mipomersen and its use in familial hypercholesterolemia. Expert Opin Pharmacother 2018:1–5.
Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med 2018;379(1):22–31.
Kimberling WJ, Hildebrand MS, Shearer AE, Jensen ML, Halder JA, Trzupek K, et al. Frequency of Usher syndrome in two pediatric populations: Implications for genetic screening of deaf and hard of hearing children. Genet Med : Off J Am Coll Med Genet 2010;12(8):512–6.
Mathur P, Yang J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim Biophys Acta 2015;1852(3):406–20.
El-Amraoui A, Petit C. Usher I syndrome: unravelling the mechanisms that underlie the cohesion of the growing hair bundle in inner ear sensory cells. J Cell Sci 2005;118(Pt 20):4593–603.
Reiners J, Nagel-Wolfrum K, Jurgens K, Marker T, Wolfrum U. Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp Eye Res 2006;83(1):97–119.
Kremer H, van Wijk E, Marker T, Wolfrum U, Roepman R. Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum Mol Genet. 2006;15 Spec No 2:R262–70.
Ponnath A, Depreux, F.F., Jodelka, F.M., Rigo, F., Farris, H., Hastings, M.L., Lentz, J.J. Rescue of outer hair cells with antisense oligonucleotides in Usher mice is dependent on age of treatment. . J Assoc Res Otolaryngol provisionally accepted.
Vijayakumar S, Depreux FF, Jodelka FM, Lentz JJ, Rigo F, Jones TA, et al. Rescue of peripheral vestibular function in Usher syndrome mice using a splice-switching antisense oligonucleotide. Hum Mol Genet. 2017;doi: https://doi.org/10.1093/hmg/ddx234.
Donaldson TN, Jennings KT, Cherep LA, McNeela AM, Depreux FF, Jodelka FM, et al. Antisense oligonucleotide therapy rescues disruptions in organization of exploratory movements associated with Usher syndrome type 1C in mice. Behav Brain Res 2018;338:76–87.
Depreux FF, Wang L, Jiang H, Jodelka FM, Rosencrans RF, Rigo F, et al. Antisense oligonucleotides delivered to the amniotic cavity in utero modulate gene expression in the postnatal mouse. Nucleic Acids Res 2016;44(20):9519–29.
Baux D, Blanchet C, Hamel C, Meunier I, Larrieu L, Faugere V, et al. Enrichment of LOVD-USHbases with 152 USH2A genotypes defines an extensive mutational spectrum and highlights missense hotspots. Hum Mutat 2014;35(10):1179–86.
Vache C, Besnard T, le Berre P, Garcia-Garcia G, Baux D, Larrieu L, et al. Usher syndrome type 2 caused by activation of an USH2A pseudoexon: implications for diagnosis and therapy. Hum Mutat 2012;33(1):104–8.
Slijkerman RW, Vache C, Dona M, Garcia-Garcia G, Claustres M, Hetterschijt L, et al. Antisense Oligonucleotide-based Splice Correction for USH2A-associated Retinal Degeneration Caused by a Frequent Deep-intronic Mutation. Mol Ther Nucleic Acids 2016;5(10):e381.
Slijkerman R, Goloborodko A, Broekman S, de Vrieze E, Hetterschijt L, Peters T, et al. Poor Splice-Site Recognition in a Humanized Zebrafish Knockin Model for the Recurrent Deep-Intronic c.7595-2144A>G Mutation in USH2A. Zebrafish. 2018;15(6):597–609.
Liquori A, Vache C, Baux D, Blanchet C, Hamel C, Malcolm S, et al. Whole USH2A Gene Sequencing Identifies Several New Deep Intronic Mutations. Hum Mutat 2016;37(2):184–93.
Leroy BP, Aragon-Martin JA, Weston MD, Bessant DA, Willis C, Webster AR, et al. Spectrum of mutations in USH2A in British patients with Usher syndrome type II. Exp Eye Res 2001;72(5):503–9.
Beneyto MM, Cuevas JM, Millan JM, Espinos C, Mateu E, Gonzalez-Cabo P, et al. Prevalence of 2314delG mutation in Spanish patients with Usher syndrome type II (USH2). Ophthalmic Genet 2000;21(2):123–8.
Yan D, Ouyang X, Patterson DM, Du LL, Jacobson SG, Liu XZ. Mutation analysis in the long isoform of USH2A in American patients with Usher Syndrome type II. J Hum Genet 2009;54(12):732–8.
Aller E, Jaijo T, Beneyto M, Najera C, Oltra S, Ayuso C, et al. Identification of 14 novel mutations in the long isoform of USH2A in Spanish patients with Usher syndrome type II. J Med Genet 2006;43(11):e55.
Hartel BP, Lofgren M, Huygen PL, Guchelaar I, Lo ANKN, Sadeghi AM, et al. A combination of two truncating mutations in USH2A causes more severe and progressive hearing impairment in Usher syndrome type IIa. Hear Res 2016;339:60–8.
Khan AO, Becirovic E, Betz C, Neuhaus C, Altmuller J, Maria Riedmayr L, et al. A deep intronic CLRN1 (USH3A) founder mutation generates an aberrant exon and underlies severe Usher syndrome on the Arabian Peninsula. Sci Rep 2017;7(1):1411.
Acknowledgments
We thank Jessica Centa, Anthony Hinrich, Francine Jodelka, and Wren Michaels for the critical comments. We regret the omission of relevant content that could not be included in the interest of space.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PDF 498 kb)
Rights and permissions
About this article

Cite this article
Hastings, M.L., Jones, T.A. Antisense Oligonucleotides for the Treatment of Inner Ear Dysfunction. Neurotherapeutics 16, 348–359 (2019). https://doi.org/10.1007/s13311-019-00729-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-019-00729-0