
Abstract
Historically, the brain has been considered an immune-privileged organ separated from the peripheral immune system by the blood–brain barrier. However, immune responses do occur in the brain in neurological conditions in which the integrity of the blood–brain barrier is compromised, exposing the brain to peripheral antigens and endogenous danger signals. While most of the associated pathological processes occur in the central nervous system, it is now clear that peripheral immune cells, especially mononuclear phagocytes, that infiltrate into the injury site play a key role in modulating the progression of primary brain injury development. As inflammation is a necessary and critical component for the subsequent injury resolution process, understanding the contribution of mononuclear phagocytes on the regulation of inflammatory responses may provide novel approaches for potential therapies. Furthermore, predisposed comorbid conditions at the time of stroke cause the alteration of stroke-induced immune and inflammatory responses and subsequently influence stroke outcome. In this review, we summarize a role for microglia and monocytes/macrophages in acute ischemic stroke in the context of normal and metabolically compromised conditions.
Similar content being viewed by others

Introduction
Stroke, a focal reduction of cerebral blood flow, causes energy depletion, excitotoxicity, and ion imbalances across the plasma membrane. This ischemic cascade results in loss of cellular integrity and subsequent cellular demise by necrosis or apoptosis [1]. The dying cells and debris elicit sterile inflammation, an inflammatory response in the absence of microbes, in the ischemic brain [2]. Damage-associated molecular patterns (DAMPs), which include modified or oxidized lipid species, cytoplasmic proteins, DNA, RNA, and modified extracellular matrix components, are released from the intracellular compartment at the time of cell demise, triggering stroke-induced inflammation [3, 4]. Regardless of the specific trigger (i.e., microbial or endogenous), innate immunity via pattern recognition receptors (PRRs) is believed to elicit inflammatory responses in stroke. DAMPs activate families of Toll-like receptors and scavenger receptors on microglia, perivascular macrophages, and endothelial cells [5]. The cytokines, chemokines, adhesion molecules, and matrix metalloproteinases these cells produce facilitate the infiltration of circulating immune cells into the injured brain [6].
Owing to a tight correlation between the presence of inflammation and injury in the stroked brain, postischemic inflammation has been considered a negative contributing factor to stroke outcomes. However, a growing body of evidence indicates the importance of immune-mediated responses for central nervous system (CNS) injury repair and recovery [7], revealing the controversial complexity of inflammatory responses in ischemic stroke. Besides microglia, a host of immune cells, including neutrophils, monocytes/macrophages, natural killer cells, and lymphocytes, infiltrate into the stroked hemisphere. Not only are the types of infiltrating immune cells temporally and spatially regulated [8], but the ischemic environment drives the function of infiltrating cells depending on the poststroke stage-specific milieu [9]. Among peripheral immune cells, monocytes/macrophages play a pivotal role in postischemic inflammation. Furthermore, the recognition of their distinct pro- and anti-inflammatory subsets and of mononuclear phagocyte phenotypic switching have provided further insight into the interactions between the immune system and CNS disease progression.
Impact of Stroke on Immunity
Excessive immune responses in the poststroke brain lead to disturbances in peripheral immunity. The literature has consistently described stroke-induced early peripheral immune activation, which is defined by increased inflammatory cytokines in both experimental animal models and humans [10–13]. This early immune activation often progresses to immune suppression in patients with stroke. Some studies have suggested that lymphopenia and immunosuppression occur as a compensatory mechanism against brain damage by attenuating autoreactive T-cell targeting of CNS self-antigens [14, 15]. Other studies have demonstrated that stroke-induced immune suppression increases the risk of secondary infections and increases mortality rates in patients [16, 17]. Up to 65 % of patients with stroke suffer from pneumonia and urinary tract infections [18, 19], and approximately 30 % of them die from stroke-related infections [20–22]. Studies focusing on the innate immune system have shown that stroke impairs the respiratory burst and subsequent generation of inflammatory mediators in granulocytes and monocytes [23, 24]. Alterations of peripheral immunity are also associated with atrophy of lymphatic organs, such as the spleen and thymus. This type of atrophy was accompanied by a reduced number of leukocytes in the spleen and an early transient increased number of leukocytes in the blood followed by a sharp reduction, demonstrating the deployment of immune cells into the circulatory system in response to stroke [25, 26]. The reduced number of leukocytes in the periphery was attributed to immune cell migration to injured sites as the reduction of peripheral immune cells in the spleen and blood temporally coincided with the increased appearance of immune cells in the stroked brain [8, 25].
The trafficking of immune cells in the poststroke brain is both temporally and spatially coordinated [8, 25, 27, 28]. With an early rise of resident microglia after stroke, peripheral immune cells, including monocytes/macrophages, myeloid dendritic cells, and neutrophils, appear in the brain within 1 day poststroke, peak in number at 3 days poststroke, and remain sustained until 7 days poststroke. These events are followed by relatively small increases of T and B lymphocytes. Stroke-induced disruption of the blood–brain barrier (BBB) facilitates the migration of immune cells into the brain [29–31]. Circulating monocytes gain access to brain parenchyma via endothelial migration and differentiate into tissue macrophages. Although many studies have found that microglia and monocytes/macrophages contribute to brain inflammation and injury in stroke, both cell types have overlapping functions and the ability to polarize toward pro- (M1) or anti-inflammatory (M2) phenotypes. As such, their expression of common antigens and the morphological similarity between them have resulted in equivocal findings on their roles in stroke [9, 32–34]. The unique temporal and spatial presence of microglia and macrophages in the context of stroke suggest distinctive and complementary roles of each cell type in response to stroke injury.
Microglia
Microglia, resident macrophages in the CNS, are part of the mononuclear phagocyte system. They arise in the brain from early development and persist into adulthood. A long-standing controversy regarding the ontology of microglia led to a view that a subset of primitive myeloid precursors localized in the yolk sac give rise to early microglia [35–37]. Microglial precursors express CX3CR1 and CD45 and travel to the neuroectoderm in a matrix metalloproteinase-8- and matrix metalloproteinase-9-dependent manner [36, 38]. Importantly, these are not exchanged with fetal liver or bone marrow-derived hematopoietic stem cells and they possess self-renewing capability under physiologic conditions [35, 36, 39, 40]. The morphology and protein expression of microglia are not uniform, and this heterogeneity could explain differential microglial responses in different surroundings [41, 42]. In the resting state, resident microglia constantly monitor the surroundings. The microglial surveillance maintains hemostasis in the developing and adult CNS by controlling the number of synapse and neuronal firings and remove debris [43, 44].
Upon an ischemic event, microglia rapidly react to the danger signal and become activated [28]. The activated microglia peak at 2 to 3 days poststroke and remain sustained for several weeks [8]. Recent evidence suggests that activated pericytes, located on the abluminal side of endothelial cells lining the microvasculature, leave the vessel wall, and give rise to activated microglia following stroke [45, 46]. However, this requires further investigation. Studies indicate that microglia are susceptible to energy deficits and their activation and recruitment depend on the metabolic status of the lesion environment. In a permanent occlusion model, microglia reside in the penumbra but not in the infarct core. However, animals with transient ischemic stroke have microglia in both the infarct core and penumbra [47]. Adenosine triphosphate (ATP) released from damaged cells contributes to microglial activation and migration in the injured site [48].
Microglia functions have been associated with both detriments and benefits in stroke outcomes. The toxicity originates from in vitro studies that demonstrated neuronal survival is attenuated by exposure to both microglia treated with proinflammatory stimuli and the supernatant of the treated microglia [49, 50]. These activated microglia release cytotoxic factors such as superoxide, nitric oxide, and tumor necrosis factor (TNF)-α [51–55]. Similarly, the detrimental role of activated microglia in vivo has been reported. Ritzel et al. [56] reported that microglia are vulnerable to severe ischemia. These cells display proinflammatory properties with elevated levels of reactive oxygen species and TNF, supporting their role in poststroke inflammation. Rats treated with minocycline for 4 weeks after middle cerebral artery occlusion had reduced microglial activation with improved neurogenesis and neurological function [57, 58]. Additionally, indomethacin, an anti-inflammatory agent that inhibits microglia activation, enhances neurogenesis in focal brain ischemia [59]. There are several contradicting reports that argue for beneficial roles for microglia in stroke. A report by Kim et al. [60] showed reduced neurogenesis with minocycline treatment. The depletion and replenishment of microglia were respectively augmented and reduced by N-methyl-D-aspartate- or oxygen glucose deprivation-induced neuronal cell death [61, 62]. Faustino et al. [63] also reported that microglia depletion enhances inflammation and injury in acute neonatal focal stroke. Selective ablation of proliferating microglia after focal ischemia in a transgenic mouse also demonstrated exacerbation of stroke injury with an altered inflammatory response [64]. A similar beneficial effect of microglia in stroke was reported in a study where there was a 60 % increase in infarct size by selective elimination of microglia, which the effect was reversed by repopulating the cells [65]. In contrast, microglia ablation was either protective or had no effect in models of other neurodegenerative diseases, including Alzheimer’s disease [66], amyotrophic lateral sclerosis [67], and experimental autoimmune encephalomyelitis [68].
Monocytes and Monocyte-Derived Macrophages
Monocytes and macrophages belong to the mononuclear phagocyte system and are derived from macrophage/dendritic cell progenitors in the bone marrow [69]. While monocytes are also formed in the liver and spleen [70–72], the view that monocytes are derived from the bone marrow has been established since the early 1960s and still dominates [73]. The monocytes produced from the bone marrow exit to the circulatory system in a C-C chemokine receptor 2 (CCR2)-dependent manner and enter tissues to give rise to tissue macrophages [74]. Circulating monocytes possess 2 distinct phenotypes with unique functional properties in human and mice. Based on the expression of specific surface markers, these cells are classified into proinflammatory (classically activated) or anti-inflammatory (alternatively activated) subsets. In humans, monocytes differentially express CD14 and CD16 (FcγRIII). The proinflammatory subset consists of 95 % of monocytes that express CD14hiCD16– and the rest expressing CD14+CD16+ are classified as an anti-inflammatory subset [75, 76]. Rodent monocytes share similarities to human monocytes, although the subset composition is different. Mouse monocytes are divided approximately equally between pro- and anti-inflammatory subsets as identified by Ly-6C (Gr-1) expression. Monocytes expressing a high level of Ly-6C (Gr-1+, Ly-6Chi) are classified as a proinflammatory subset, and monocytes expressing a low level of Ly-6C (Gr-1–, Ly-6Clo) are classified as an anti-inflammatory subset [77, 78]. In rats, 10 % to 20 % of monocytes that express low CD43 are classified as a proinflammatory subset and the remaining monocytes that expresses higher levels of CD43 are classified as an anti-inflammatory subset [79]. Recently, CD43 has also been used to distinguish monocyte subsets in mice [80]. Classically activated proinflammatory monocytes express CCR2 with low or no expression of CX3CR1 across different species. CCR2 expression is critical for the trafficking of circulating monocytes into inflamed tissue where they give rise to macrophages. The anti-inflammatory monocytes, which do not express CCR2 but do express higher levels of CX3CR1, patrol blood vessels in a steady state, performing in situ phagocytosis [78, 81].
A substantial number of monocytes reside in the spleen. Swirski et al. [82] have reported that the number of monocytes in the heart following myocardial infarction exceeds the number in circulation, identifying the spleen as a reservoir of undifferentiated monocytes which are rapidly deployed after myocardial infarction [82]. The role of the spleen in cerebral ischemia has also been investigated. Both clinical and animal studies have demonstrated transient reduction in spleen size following stroke, which has been attributed to the poststroke deployment of splenocytes, including monocytes [25, 83–85]. The extent of spleen atrophy is associated with the severity of stroke-induced brain injury, leading to the view that the spleen plays a contributory role in stroke pathology [26, 86]. Increased apoptosis in the spleen, albeit controversial (see Kim et al. [25]), and cell release from the spleen are related to reduced spleen size [26]. Others have also reported that splenectomy or splenic irradiation reduces ischemic injury in animal models of stroke [87–89]. While these data provided a rationale for targeting the spleen as a therapeutic strategy for patients with stroke, there are reports that argue against this approach. Kim et al. [25] addressed the role of splenic monocytes in ischemic stroke. In this study, pro- and anti-inflammatory monocyte subsets were longitudinally determined at acute and subacute phases of ischemic stroke from 1 h to 7 days poststroke in the spleen, blood, bone marrow, and brain. The study showed that both monocyte subsets were decreased in the spleen and blood with compensatory production of monocytes in the bone marrow at later time points, while the number of monocytes in both subsets was significantly increased in the brain after stroke. However, the removal of spleen at the time of stroke did not provide neuroprotection despite splenectomized mice having fewer monocytes in their brains [25]. While the time of splenectomy and nonselective ablation of both monocyte subsets by splenectomy may confound the interpretation of the spleen’s precise role in stroke pathology, the increased incidence of hemorrhagic and ischemic stroke in patients with splenic injury or splenectomy [90], and the protective role of the proinflammatory Ly6Chi monocyte subset in acute stroke [91], suggests that the role of individual monocyte subsets in the spleen should be carefully investigated.
Monocyte Trafficking
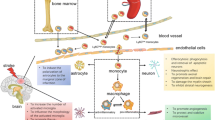
There are reported discrepancies in the order of infiltration of neutrophils, monocytes, and lymphocytes into the stroked hemisphere [8, 92]. Despite the controversy, the presence of these immune cells in the postischemic brain clearly indicates the mobilization of peripheral immune cells into the site of CNS injury [93, 94]. Among these cell types, the accumulation of monocytes/macrophages in the stroked hemisphere has been consistently associated with injury development in acute stroke. Chemokine gradients generated in the injury sites are the major driving force behind trafficking of monocyte subsets from the periphery to injury sites (Fig. 1). Studies on experimental autoimmune encephalitis (EAE) in mice demonstrated the importance of the monocyte chemoattractant protein-1 (MCP-1)/CCR2 axis in monocyte recruitment [95]. In this model, the trafficking of monocytes, but not T cells, was coupled to the severity of EAE progression.

Trafficking of peripheral monocytes to the brain in ischemic stroke. Stroke produces danger signals [e.g., damage-associated molecular patterns (DAMPs)] and activates microglia. Excess cytokines and chemokines produced by activated microglia, endothelial cells, and astrocytes in the injury site allow for transendothelial migration of monocytes/macrophages through the compromised blood–brain barrier. The spleen, an immediate reservoir for monocytes/macrophages, deploys pro- [Ly-6Chi/C-C chemokine receptor 2 (CCR2)+/CX3CR1lo) and anti-inflammatory (Ly-6Clo/CCR2-/CX3CR1hi) monocytes into circulation upon stroke. The deployment of monocytes from the bone marrow is delayed until later phases of acute stroke [25]. The circulating proinflammatory monocytes enter into the stroked hemisphere via the monocyte chemoattractant protein (MCP)/CCR2 axis during the acute phase, giving rise to tissue macrophages. IFN = interferon; TNF = tumor necrosis factor; MMP = matrix metalloproteinase
As a cognate receptor for MCP-1, CCR2 expressed on monocytes is an essential contributor to monocyte recruitment. Similarly, the crucial role of the MCP-1/CCR2 axis on monocyte trafficking in the ischemic brain has been reported in several studies [96–98]. Ly-6Chi (CCR2+) proinflammatory monocytes are chemoattracted to MCP-1 and they are specifically recruited to injury sites, becoming classically activated M1 macrophages. Secreted by microvascular endothelial cells, monocytes/macrophages, and astrocytes upon injury, MCP-1 expression increases in the stroked hemisphere. The overexpression of MCP-1 led to increased infarct volume and enhanced the recruitment of monocytes to the injury site [99]. In contrast, the absence of CCR2 or MCP-1 led to reduced infarct size [96, 100]. Relevant to the MCP-1/CCR2 axis in monocyte trafficking, a study demonstrated involvement of the scavenger receptor CD36 in exacerbating stroke-induced injury in hyperlipidemic mice [101]. The exacerbation was linked to the role of CD36 in regulating MCP-1 expression in a host-specific manner and CCR2 expression on immune cells, showing the influence of peripheral immunity on the CNS via the MCP-1/CCR2 axis. Compared with the proinflammatory axis, the Ly-6Clo monocyte subset that expresses CX3CR1, a receptor for CX3CL1 (fractalkine), does not express CCR2. These anti-inflammatory Ly-6Clo (CCR2–/CX3CR1+) monocytes are recruited to normal tissues and develop into resident M2 macrophages that function in host defense and repair after injury [102]. Compared with the proinflammatory MCP-1/CCR2 axis, there was less involvement of the anti-inflammatory CX3CL1 (fractalkine)/CX3CR1 axis in the acute phase of stroke [101], which may be related to less recruitment of Ly-6Clo subsets in the ischemic brain at this time point [25].
Phenotype of Mononuclear Phagocytes
Inflammation is an orchestrated biological event in response to encountered pathogens or danger signals in the host tissue. Optimal inflammation includes destruction of host tissue if it is necessary. However, an equally important aspect of inflammation is its self-limiting capability, which results in resolution of inflammation to reestablish homeostasis. Mononuclear phagocytes, specifically microglia and monocyte-derived macrophages, are critical modulators in inflammatory and resolution phases of stroke. The polarization of mononuclear phagocytes toward either proinflammatory or anti-inflammatory subsets greatly affects their function. Classically activated (M1) mononuclear phagocytes produce proinflammatory factors, clear pathogens, and present antigens. This subset expresses CD16, CD32, CD86, major histocompatibility complex (MHC) II, and inducible nitric oxide synthase (iNOS). Alternatively activated (M2) mononuclear phagocytes function in wound healing and repair through phagocytosis and immune tolerance [103]. The M2 cells are further divided into M2a, M2b, and M2c subtypes [103–105]. The M2a subset expresses arginase-1, Ym1, and Fizz, and is involved in immunity against parasites, T helper 2 cell recruitment, tissue repair, and growth stimulation. M2b cells express high levels of interleukin (IL)-10, MHC II, and co-stimulatory CD86. This subset exhibits both pro- and anti-inflammatory features and is associated with adaptive immunity. Arginase-1, CD163, and CD206 are markers for M2c cells, which mainly function in scavenging cell debris during the repair process [9, 103]. Apart from M1 and M2 phenotypes, a novel Mox phenotype has been described in atherosclerotic lesions of a hyperlipidemic mouse model. Mox cells are characterized by high expression of heme oxygenase-1 and Nrf2-dependent redox-regulatory genes. This subset is proinflammatory with reduced phagocytic function, which may be related to their localization in atherosclerotic lesions [106]. In addition, an M3 phenotype, which can be induced by macrophage colony stimulating factor or IL-34, has been described without extensive published data [107]. While the diversity of mononuclear phagocyte phenotypes represents a significant challenge, determining the specific role of each phenotype in the context of the stroke injury milieu will be necessary to further our understanding in this area.
Macrophage Polarization in Stroke
There is a relative paucity of in vivo evidence for monocyte trafficking from the periphery to the primary injury site. Earlier studies from myocardial infarction and Listeria monocytogenes infection demonstrated that proinflammatory Ly-6Chi (CCR2+) and anti-inflammatory Ly-6Clo (CCR2–) subsets are sequentially recruited to the injury site in a controlled manner for respective inflammation and repair/healing [102, 108]. However, accumulating evidence supports that Ly-6Chi monocytes recruited in inflamed tissue can differentiate into both M1 and M2 macrophages. In renal tubulointerstitial damage, bone marrow-derived Ly-6Chi monocytes polarized to M1 macrophages in the early phase of inflammation but polarized to M2 macrophages in the later phase when the inflammation was receding. In contrast, the Ly-6Clo monocyte subset recruited during inflammation only differentiated into M2 macrophages [109, 110]. The differentiated macrophages can convert to other phenotypes in the specific local microenvironment, and conversion of early differentiated M1 macrophages into inflammation-resolving M2 macrophages occurs in the periphery [111].
In the stroked hemisphere, there are multiple distinct macrophage phenotypes (Fig. 2), and macrophages can convert to other phenotypes depending on the ischemic milieu [112]. For example, Hu et al. [113] suggested that a conversion from M2 to M1 occurs in the ischemic brain based on the observation of early transient expression of M2 markers followed by persistent expression of M1 markers as the lesion progressed. Others have suggested the acquisition of the repair M2 phenotype from M1 macrophages in sterile wounds. Early recruited peripheral monocytes/macrophages are predominantly Ly-6Chi (CCR2+) cells that become M1 tissue macrophages in the stroked hemisphere [25, 80, 114, 115]. Once recruited, these proinflammatory cells lose LY6C and CCR2 expression and become capable of releasing vascular endothelial growth factor and transforming growth factor-β, which are prorepair mediators [116]. A recent study also supports the view of macrophage conversion at the injury site. Wattananit et al. [117] showed a beneficial role for early recruited CCR2+ monocyte-derived macrophages on functional recovery through enhancing resolution of inflammation. While there have been advances in understanding the role of mononuclear phagocytes in stroke, defining the phenotype of mononuclear phagocytes relevant to their function in the poststroke brain has been understudied. Many studies define macrophage phenotype based on gene expression. Despite changing expression patterns during disease progression, protein expression of the majority of M1 and M2 markers have not been extensively validated at different poststroke time points. There is also substantial overlap among signature markers for M1 and M2, representing a potential continuum of phenotypes. Additionally, as M1 and M2 signature markers are shared between microglia and macrophages in the stroked hemisphere, future studies should characterize the functions for each specific mononuclear subset.

Phenotypes and phenotypic conversion of mononuclear phagocytes. M1 and M2 microglia and macrophage subsets are distinguished by expression of specific markers and functions. M1 mononuclear phagocytes have cytotoxic effects involving antigen presentation and pathogen clearance. M2 cells are involved in clearing up debris, phagocytosis, and immune tolerance. Upregulation of CD36 and lysosomal acid lipase (LAL) is associated with M2 polarization. Depending on the ischemic milieu, mononuclear phagocytes polarize to the M1 or M2 subset. MHC = major histocompatibility complex; iNOS = inducible nitric oxide synthase; IL = interleukin; HO-1 = heme oxygenase-1; VEGF = vascular endothelial growth factor; COX = cyclooxygenase; NRF = nuclear factor - erythroid 2-related factor
Inflammation Versus Resolution
M1 macrophages release cytotoxic substances, eliciting inflammation and contributing to cell death, while M2 macrophages clear cellular debris through phagocytosis and release trophic factors [103, 105]. In the presence of microbes, the acute inflammatory response is often rapid, specific, and self-limiting, to avoid inflammation-mediated damage to neighboring tissues [118]. This primordial defense response involves initial recognition of pathogen-associated molecular patterns via the innate immune system. Sterile inflammation occurs in postischemic tissues in the absence of microbes [119, 120]. The triggers of sterile inflammation are elements of damaged tissue, collectively termed DAMPs. Regardless of the nature of triggers, pathogen-associated molecular patterns or DAMPs, PRRs, including Toll-like receptors and scavenger receptors, are thought to be involved in eliciting inflammatory responses [121–124]. There are also nonsurface-bound secreted PRRs, including triggers of the complement system. A circulating protein C1q, mannose-binding lectin, ficolins, and collectins initiate the complement cascade in myeloid cells and induce inflammatory response [125]. The complement system regulates the activation of mononuclear phagocytes. The binding of C1q or cleavage products of C3 with the complement receptors on mononuclear phagocytes produce inflammatory cytokines [126, 127], and promote phagocytosis of pathogens or apoptotic cells [128–131].
As inflammation is an important process that leads to subsequent resolution, clearing up debris in the injured brain has been considered an essential process for tissue repair and remodeling. In the resolution phase of acute inflammation, proresolving mediators prevent excessive inflammation and promote the removal of microbes and dying cells. Four distinct chemical families have been identified and named lipoxins, resolvins, protectins, and maresins. These families are biosynthesized in acute inflammation and control the duration and magnitude of inflammation via activation of mononuclear phagocyte recruitment, uptake of apoptotic neutrophils by the phagocytes, endogenous antimicrobial defense mechanisms, and clearance on mucosal surfaces [132–138]. An additional modulator for tissue repair and remodeling in the CNS injury is arginase-1. Studies indicated that arginase-1 expression is correlated with improved recovery from CNS injury and disease [80, 139–141]. A recent study using EAE and spinal cord injury models has shown that infiltrated macrophages mainly express arginase-1 in the inflamed CNS [142], suggesting the importance of infiltrated myeloid cells in the CNS repair after injury. Dying cells release signals such as ATP, uridine triphosphate, and CX3CL1. CX3CR1-expressing mononuclear phagocytes migrate toward the dying neurons that release CX3CL1 [143–145]. M2 macrophages that highly express CX3CR1 are considered to be a major subset that engages in phagocytosis as shown in in vitro neuron–microglia co-culture and organotypic slice culture [146, 147]. However, CD68, a marker of phagocytic activity, was shown to be inversely correlated with other M2 markers during stroke injury development [112], casting uncertainty on the relationship between M2 marker expression and phagocytic function.
Phagocytosis requires recognition of lipid membrane asymmetry of phosphatidylserine, modified or oxidized lipid moieties, and heat shock protein 60 in the surface of apoptotic cells [148, 149]. These signals from apoptotic cells include brain-specific angiogenesis inhibitor 1, milk-fat globule-epidermal growth factor-E8), and triggering receptor expressed on myeloid cells 2. A list of additional apoptotic cell-associated cellular patterns and their corresponding receptors on mononuclear phagocytes are well summarized by Sierra et al. [150]. It has been shown that CD36, a scavenger receptor, has high affinity toward apoptotic cell-associated cellular patterns and its interaction with oxidized phosphatidylserine is essential for macrophage-dependent phagocytosis of apoptotic cells [121]. CD36 is highly expressed in microglia, monocytes/macrophages, microvascular endothelial cells, platelets, and epithelial cells and it regulates inflammation, innate immunity, angiogenesis, and lipid metabolism through interactions with lipid and nonlipid-based ligands [151]. Several studies demonstrated that CD36 expression is upregulated in the ischemic brain [152], and that CD36 is involved in stroke-induced inflammation and injury [101, 152–154]. Despite the detrimental effects of CD36 in an acute ischemic setting, CD36-dependent phagocytosis was associated with functional benefit in neonatal stroke [155]. Additionally, the clearing of red blood cells after hemorrhagic stroke was associated with increased CD36 expression and improved functional recovery [156]. This might be related to the role of CD36 in M2 phenotypic switching of mononuclear cells. CD36 levels are significantly increased in M2 phenotype mononuclear phagocytes, mediating signals to pathways for M2 polarization through endoplasmic reticulum stress, JNK, and peroxisome proliferator-activated receptor-γ [157, 158]. M1 and M2 macrophages also have distinct metabolic phenotypes [159, 160]. While M1 macrophages depend on aerobic glycolysis, fatty acid oxidation is required to sustain M2 macrophages. Huang et al. [161] reported that CD36-mediated uptake of triglycerides and subsequent lipolysis by lysosomal acid lipase are important for sustaining the M2 phenotype [161]. Besides well-known M2 markers, including CD301, CD206, and RELMa, this study identified CD36 and lysosomal acid lipase as markers for the M2 metabolic phenotype.
Sex-Associated Modulation of Mononuclear Phagocytes
Clinical studies showed that males experience higher risk of stroke than females [162–164]. In rodent stroke, premenopausal females exhibit smaller infarct size than age-matched males [88, 165–168]. However, the risk of stroke is increased as females age [169–172]. More severe strokes and poorer recovery was observed in elderly women and aged rodents, regardless of the sex [162–164, 173–176]. A primary ovarian hormone, estrogen has been associated with the sex difference in ischemic stroke as treatment of estrogen in the male or ovariectomized animals reduced infarct and neuronal death following ischemia [177–180]. The neuroprotective effect of estrogen is related to anti-inflammation in young but proinflammation in the aged brain [181–184], reduced microglial proliferation, attenuated macrophage cytokine production, and recruitment of macrophages in ischemic injury [185]. Accordingly, reduced CD45hi/CD11b+ cells (activated microglia and infiltrated macrophages) in the brain and fewer spleen contractions have been related to the smaller injury in females [166]. Splenectomy was effective in reducing the injury size and CD45hi/CD11b+ cells in the ischemic brain only in male mice, supporting a role for peripheral mononuclear phagocytes to the sex differences in stroke [88]. While most stroke studies exclude female animals owing to the hormonal changes with the estrus cycle, evidence suggests sex as an important biological variable to be addressed in stroke research.
Modulatory Function of Mononuclear Phagocytes in Comorbidities
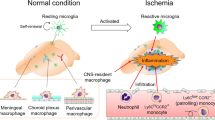
Stroke incidence is higher in populations with cardiovascular and cerebrovascular risk factors. Common comorbidities associated with stroke include aging, elevated blood pressure, and metabolic dysfunctions. These conditions are often associated with the exacerbation of ischemic outcomes and are predictive of worse recovery [186]. The inclusion of hyperlipidemia and diabetes in animal models of stroke has demonstrated that these conditions modulate postischemic inflammatory responses and lesion development [101, 152, 187, 188]. In addition, increased inflammatory factors such as cytokines, chemokines, and adhesion molecules in peripheral monocytes/macrophages in these conditions suggest that peripheral immune status modulates CNS injury. Systemic inflammation is also linked to CNS inflammatory status through priming and/or activation of mononuclear phagocytes in cerebrovascular diseases (Fig. 3). Evidence has shown that microglia exposed to systemic inflammatory stimuli have increased reactivity, leading to accelerated neuronal death and progression of CNS disease [189–191]. It has been suggested that primed microglia contribute to the increased response to a second inflammatory stimulus [192]. Thus, understanding the properties of mononuclear phagocytes in comorbid conditions prior to stroke will provide better insight about their modulatory function on stroke-induced brain injury.

Impact of comorbidities on immunity and central nervous system (CNS) injury. Comorbidity-induced systemic inflammatory response primes microglia in the CNS and increases the number of Ly-6Chi monocytes (monocytosis) in the periphery. Upon stroke, the primed microglia and increased proinflammatory monocytes deregulate the injury-induced inflammatory response. The inability to mount an optimal inflammatory response in the stroked hemisphere negatively influences stroke outcome
Aging
Aging is a nonmodifiable risk factor for stroke and it has an impact on immune cells, leading to immunosenescence of microglia and macrophages [193]. Studies using aged rodents demonstrated increased brain injury size and behavioral deficits, which were associated with modulation of the number or function of mononuclear phagocytes at the injury site [168, 194, 195]. Increased CD8+ T cells in the aged CNS were associated with compromised proinflammatory functions in microglia. The primed microglia increased inflammation and leukocyte recruitment in the brain following stroke [196]. Comparing mononuclear phagocytes between young and old mice, aged macrophages, and microglia exhibited reduced motility in response to laser-induced injury and extracellular ATP [197]. In demyelinating models, reduced migration of mononuclear cells [198], and impaired clearance of myelin debris in macrophages accompanied by an age-associated delay in remyelination have been observed [199–201]. Additionally, microglia isolated from aged mice displayed a reduced capability to engulf Aβ compared with young mice [202, 203]. Studies have shown that aging differentially affects the inflammatory response between microglia and peripheral macrophages. Aged microglia have an increased proinflammatory response and shift toward the M1 phenotype. Basal and stimulated secretion of TNF-α and IL-6 were increased in the ex vivo culture of senescent microglia [203], and hypertrophic morphological changes in the aged cells were observed [197, 204, 205]. Consistent with this evidence, an in vivo study showed that iNOS, TNF-α, and IL-6 were elevated, while arginase-1 was repressed in the midbrain of aged mice [206]. In contrast, peripheral macrophages exhibited less classical activation, leading to an attenuated proinflammatory response [207]. In a mouse model of age-related macular degeneration, ocular macrophages from old mice exhibited increased IL-10 but decreased IL-12 and TNF-α, suggesting that aged macrophages are polarized toward an M2-like phenotype [208]. Another study also reported increased numbers of M2 macrophages in the spleen, lymph nodes, and bone marrow of aged mice [209]. The observations of the deregulated pro- and anti-inflammatory balance in aged mononuclear phagocytes and their associated negative consequences suggest that strategies aimed at rejuvenating these immune cells would have benefits in reinstating injury-induced recovery in the aging brain [193].
Hypertension
Hypertension is the second most prevalent risk factor for stroke, affecting ~50 % of patients with stroke. Hypertension increases endothelial oxidative stress and inflammation and leads to worse stroke outcomes and higher mortality [210–220]. Studies investigating the underlying pathophysiology of hypertension indicates the involvement of neuroimmune interactions. Chronic systemic inflammation causes neurogenic hypertension mediated through neuroinflammation and oxidative stress in the rostral ventrolateral medulla [221]. Overexpression of junctional adhesion molecule-1 leads to leukocyte adherence to microvasculature, resulting in hypertension in previously normotensive rats [222]. Hypertension causes activation of astrocytes and microglia and elevates expression of adhesion molecules in endothelial cells [223–225]. Microglia activation has been observed in angiotensin-II-induced hypertensive rats and intracerebroventricular administration of minocycline abrogated the angiotensin-II-induced hypertension [226]. Hypertension also affects inflammatory responses in the periphery. A clinical study reported that the levels of IL-6 and TNF-α in plasma were correlated with blood pressure in human patients [227]. It also has been shown that hypertension spontaneously activates circulating monocytes and increases reactive oxygen species production in immune cells [228–230]. Using leukocyte-specific CCR2-deficient mice, Ishibashi et al. [231] revealed the importance of monocyte CCR2 in hypertension. In this study, the enhanced monocyte CCR2 expression observed in hypertensive mice was reduced by angiotensin-II inhibition, while angiotensin-II-induced vascular inflammation and remodeling were blunted in mice transplanted CCR2-deficient bone marrow.
The relationship between hypertension and microglia phenotypes has recently been investigated. Both M1 (MHC II, CCR7, interferon-γ receptor, and iNOS) and M2 markers (CD36, mannose receptor, Tie2, and IL-4 receptor α) were upregulated in microglia from hypertensive mice, but not in monocytes, which may be explained by the neurogenic regulation of hypertension [232]. Other studies demonstrated that proinflammatory M1 macrophages are predominant in the aortas stimulated by angiotensin-II for 7 days [233, 234]; however, prolonged infusion of angiotensin-II for 14 to 28 days recruited Ly-6Chi monocytes that differentiated into M2 macrophages [235]. Microglia from hypertensive rats had reduced activation following stroke and lipopolysaccharide stimulation [236]. These results suggest that chronic systemic hypertension deregulates microglial responses and support the importance of injury-induced activation of microglia. In contrast, infiltrating peripheral leukocytes were correlated with the extent of injury in hypertension [237], demonstrating the detrimental function of periphery-derived mononuclear phagocytes. The involvement of mononuclear phagocytes in hypertension-induced exacerbation of stroke outcomes and mortality suggests an immunological explanation for this risk factor.
Hyperlipidemia
Cholesterols in the plasma and CNS do not exchange and their synthesis in the brain occurs in situ. However, in neurological and neurodegenerative conditions in which the integrity of the BBB is disrupted, chronic elevation of cholesterols and lipids in the periphery influences immune responses in the CNS. In hyperlipidemic mice, positron emission tomography imaging and immunostaining confirmed increased microglial phagocytosis, microgliosis, and higher expression of vascular adhesion molecules. In addition, larger numbers of activated myeloid phagocytes, T cells, and granulocytes in the choroid plexus were observed in hyperlipidemic mice [238]. Hyperlipidemia is associated with elevated IL-6, TNF-α, MCP-1, and CCL5 in the periphery [239]. Studies also suggest that hyperlipidemia shifts mononuclear phagocytes towards proinflammatory activated phenotypes. An extreme hyperlipidemic condition generated by feeding a high fat/cholesterol diet to ApoE knockout mice induced significant monocytosis in the spleen and blood, comprised mainly of the proinflammatory Ly-6Chi subset [240]. The hyperlipidemia-induced monocytosis occurs through increased production in the bone marrow, increased survival in the periphery, and reduced conversion from Ly-6Chi to the Ly-6Clo subset [240, 241]. Ly-6Chi monocytes actively accumulate into early atherosclerotic plaques, differentiate to M1 activated macrophages, and contribute to the progression of atherosclerosis [242].
The proinflammatory nature associated with peripheral hyperlipidemia affects acute stroke injury. Previous studies from our group reported that mice with elevated plasma cholesterols had larger infarcts and increased proinflammatory cytokine and chemokine expression in the ischemic hemisphere. The study also demonstrated that CD36 expressed in monocytes/macrophages is an important inflammatory mediator in hyperlipidemia-induced exacerbation of poststroke brain injury. The hyperlipidemic mice had higher CD36 expression in peripheral macrophages prior to stroke and had lipid-laden foam cells in the poststroke brain. Hyperlipidemic mice with targeted disruption of CD36 showed a reversal of the hyperlipidemia-associated phenotype in the brain (i.e., reduced infarct size, less foam cell formation, and less proinflammatory cytokine and chemokine expression) [152]. A subsequent study revealed the involvement of CD36 expression in peripheral mononuclear phagocytes and CNS for hyperlipidemia-induced exacerbation of infarct volume and brain edema. By exchanging bone marrow cells between CD36-expressing and CD36-deficient mice, the study demonstrated that CD36 regulates MCP-1/CCR2 expression in the host and macrophages, suggesting the involvement of CD36 in monocyte trafficking in hyperlipidemic conditions [101]. While the observed effect is likely mediated through the proinflammatory CCR2+ subset considering the major expansion of the CCR2+ (Ly-6Chi) subset in hyperlipidemic conditions, the relative contribution of the CCR2– (Ly-6Clow) subset and its function remain to be explored.
Diabetes
Obesity-induced insulin resistance is associated with developing diabetes, a prevalent risk factor for stroke. The diabetic condition is associated with chronic low-grade systemic inflammation. Despite the high prevalence of diabetes in patients with stroke [243], the contribution of this cluster of metabolic conditions on stroke pathology is poorly understood. Approximately 90 % of patients with diabetes are classified as having type 2 diabetes [244–246]. Studies demonstrate that adipose tissue-resident macrophages play an important role in the development of insulin resistance. Adipose tissue contains both M1 and M2 activated macrophages. Proinflammatory cytokines TNF-α, IL-6, and MCP-1 produced from M1 adipose tissue-resident macrophages contribute to insulin resistance [241]. Ly-6Chi monocytes are predominant in the blood of type 1 diabetic animals [247], and the monocytes isolated from patients with diabetes secrete higher levels of proinflammatory cytokines [248–250]. It is believed that the cytokines released from these cells influence peripheral inflammatory status.
Studies have also shown the influence of diabetic conditions on the CNS. Microglia activation with increased IFN-γ and IL-1β in the hippocampus and retina were observed in diabetic rats [251, 252]. Recently, we have reported the impact of diabetes on stroke outcomes. Diet-induced obese mice with insulin resistance and elevated fasting blood glucose levels had significant increases in infarct volume and edema [187]. The exacerbation was associated with increased CD36 in the brain. In vitro LPS stimulation of peritoneal macrophages from diabetic mice attenuated the inflammatory response [187]. The blunted inflammatory response in diabetic macrophages was also reported in db/db mice, with decreased microglial activation and proinflammatory cytokines in the stroked brain [188]. Microglial activation and the release of inflammatory factors are critical steps in eliciting a rapid inflammatory response in the context of injury. The impairment of mounting an effective acute inflammatory response in the diabetic condition was partly implicated for exacerbated injury in diabetic stroke. While there is not much evidence about how M1/M2 polarized macrophages affect ischemic stroke in diabetes, one study has shown the association of M2 activated macrophages with improved stroke repair in type 2 diabetic rats [253], suggesting the beneficial role of M2 macrophages in stroke repair in the diabetic condition. Thus, mounting an effective acute inflammatory response and subsequently resolving inflammation in a timely manner to limit expansion of the primary injury is essential in the context of chronic systemic metabolic dysfunction.
Perspectives
With recent advances in our knowledge on peripheral immunity, studying the roles of mononuclear phagocytes in stroke has been an emerging area to understand interactions between the peripheral immune system and CNS disease. To define individual roles for resident microglia and infiltrating macrophages in stroke pathophysiology, several critical questions need to be answered. Because these 2 cell types express common antigens, devising a better tool to distinguish one from the other will greatly benefit the field. The presence of heterogeneity in subsets, one with a cytotoxic proinflammatory nature (M1) and the other with reparative, phagocytic, and anti-inflammatory properties (M2), has made it challenging to designate clearly the function of each cell type. Accumulating evidence that indicates potential conversion from one subtype to another in the stroked brain adds further complexity. There is a supporting view that the ischemic milieu drives microglia and monocyte/macrophage function and this occurs in a context-dependent manner as the ischemic environment changes over time. There is also a view that mounting a rapid inflammatory response in the acute phase is important for subsequent resolution of inflammation. The ability of M2 macrophages to resolve stroke-induced inflammation supports interventions that shift toward M2 polarization for repair/recovery. In-depth understanding on where each cell type and/or subset localizes in the brain, when they populate or accumulate in affected and surrounding tissues, and how each cell type/subset functions in response to injury (individually or in concert) are critical questions to be addressed in order to manipulate each subset to optimize the immune response in stroke. Importantly, characterization of the spatial, temporal, and functional understandings of mononuclear phagocytes in comorbid conditions would provide better translational strategies for potential interventions.
References
Lipton P. Ischemic cell death in brain neurons. Physiol Rev 1999;79:1431-1568.
Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol 2010;28:321-342.
Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol 1994;12:991-1045.
Matzinger P. The danger model: a renewed sense of self. Science 2002;296:301-305.
Marsh BJ, Williams-Karnesky RL, Stenzel-Poore MP. Toll-like receptor signaling in endogenous neuroprotection and stroke. Neuroscience 2009;158:1007-1020.
Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med 2011;17:796-808.
Jin R, Liu L, Zhang S, Nanda A, Li G. Role of inflammation and its mediators in acute ischemic stroke. J Cardiovasc Transl Res 2013;6:834-851.
Gelderblom M, Leypoldt F, Steinbach K, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009;40:1849-1857.
Fumagalli S, Perego C, Pischiutta F, Zanier ER, De Simoni MG. The ischemic environment drives microglia and macrophage function. Front Neurol 2015;6:81.
Chapman KZ, Dale VQ, Denes A, et al. A rapid and transient peripheral inflammatory response precedes brain inflammation after experimental stroke. J Cereb Blood Flow Metab 2009;29:1764-1768.
Emsley HC, Smith CJ, Gavin CM, et al. An early and sustained peripheral inflammatory response in acute ischaemic stroke: relationships with infection and atherosclerosis. J Neuroimmunol 2003;139:93-101.
Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab 2006;26:654-665.
Smith CJ, Emsley HC, Gavin CM, et al. Peak plasma interleukin-6 and other peripheral markers of inflammation in the first week of ischaemic stroke correlate with brain infarct volume, stroke severity and long-term outcome. BMC Neurol 2004;4:2.
Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci 2005;6:775-786.
Urra X, Cervera A, Villamor N, Planas AM, Chamorro A. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience 2009;158:1174-1183.
Chamorro A, Urra X, Planas AM. Infection after acute ischemic stroke: a manifestation of brain-induced immunodepression. Stroke 2007;38:1097-1103.
Shim R, Wong CH. Ischemia, Immunosuppression and infection-tackling the predicaments of post-stroke complications. Int J Mol Sci 2016;17.
Langhorne P, Stott DJ, Robertson L, et al. Medical complications after stroke: a multicenter study. Stroke 2000;31:1223-1229.
Vernino S, Brown RD, Jr., Sejvar JJ, Sicks JD, Petty GW, O'Fallon WM. Cause-specific mortality after first cerebral infarction: a population-based study. Stroke 2003;34:1828-1832.
Heuschmann PU, Kolominsky-Rabas PL, Misselwitz B, et al. Predictors of in-hospital mortality and attributable risks of death after ischemic stroke: the German Stroke Registers Study Group. Arch Intern Med 2004;164:1761-1768.
Ulm L, Harms H, Ohlraun S, Reimnitz P, Meisel A. Impact of infections on long-term outcome after severe middle cerebral artery infarction. J Neurol Sci 2012;319:15-17.
Wilson RD. Mortality and cost of pneumonia after stroke for different risk groups. J Stroke Cerebrovasc Dis 2012;21:61-67.
Ruhnau J, Schulze K, Gaida B, et al. Stroke alters respiratory burst in neutrophils and monocytes. Stroke 2014;45:794-800.
Seki Y, Sahara Y, Itoh E, Kawamura T. Suppressed neutrophil respiratory burst in patients with haemorrhagic stroke. J Clin Neurosci 2010;17:187-190.
Kim E, Yang J, Beltran CD, Cho S. Role of spleen-derived monocytes/macrophages in acute ischemic brain injury. J Cereb Blood Flow Metab 2014;34:1411-1419.
Offner H, Subramanian S, Parker SM, et al. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol 2006;176:6523-6531.
Easton AS. Neutrophils and stroke—can neutrophils mitigate disease in the central nervous system? Int Immunopharmacol 2013;17:1218-1225.
Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 2010;87:779-789.
Lossinsky AS, Buttle KF, Pluta R, Mossakowski MJ, Wisniewski HM. Immunoultrastructural expression of intercellular adhesion molecule-1 in endothelial cell vesiculotubular structures and vesiculovacuolar organelles in blood–brain barrier development and injury. Cell Tissue Res 1999;295:77-88.
Lossinsky AS, Shivers RR. Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. Review. Histol Histopathol 2004;19:535-564.
Stamatovic SM, Keep RF, Andjelkovic AV. Brain endothelial cell–cell junctions: how to "open" the blood brain barrier. Curr Neuropharmacol 2008;6:179-192.
Tambuyzer BR, Ponsaerts P, Nouwen EJ. Microglia: gatekeepers of central nervous system immunology. J Leukoc Biol 2009;85:352-370.
Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature 2010;468:253-262.
Sica A, Melillo G, Varesio L. Hypoxia: a double-edged sword of immunity. J Mol Med (Berl) 2011;89:657-665.
Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010;330:841-845.
Kierdorf K, Erny D, Goldmann T, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci 2013;16:273-280.
Ransohoff RM. Microglia and monocytes: 'tis plain the twain meet in the brain. Nat Neurosci 2011;14:1098-1100.
Mizutani M, Pino PA, Saederup N, Charo IF, Ransohoff RM, Cardona AE. The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. J Immunol 2012;188:29-36.
Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 2007;10:1538-1543.
Schulz C, Gomez Perdiguero E, Chorro L, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012;336:86-90.
Carson MJ, Bilousova TV, Puntambekar SS, Melchior B, Doose JM, Ethell IM. A rose by any other name? The potential consequences of microglial heterogeneity during CNS health and disease. Neurotherapeutics 2007;4:571-579.
Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990;39:151-170.
Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res 2005;81:302-313.
Salter MW, Beggs S. Sublime microglia: expanding roles for the guardians of the CNS. Cell 2014;158:15-24.
Ozen I, Deierborg T, Miharada K, et al. Brain pericytes acquire a microglial phenotype after stroke. Acta Neuropathol 2014;128:381-396.
Sakuma R, Kawahara M, Nakano-Doi A, et al. Brain pericytes serve as microglia-generating multipotent vascular stem cells following ischemic stroke. J Neuroinflammation 2016;13:57.
Zanier ER, Fumagalli S, Perego C, Pischiutta F, De Simoni MG. Shape descriptors of the "never resting" microglia in three different acute brain injury models in mice. Intensive Care Med Exp 2015;3:39.
Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 2005;8:752-758.
Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res 1992;587:250-256.
Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol 1992;149:2736-2741.
Colton CA, Gilbert DL. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett 1987;223:284-288.
Lee SC, Liu W, Roth P, Dickson DW, Berman JW, Brosnan CF. Macrophage colony-stimulating factor in human fetal astrocytes and microglia. Differential regulation by cytokines and lipopolysaccharide, and modulation of class II MHC on microglia. J Immunol 1993;150:594-604.
Liu B, Gao HM, Wang JY, Jeohn GH, Cooper CL, Hong JS. Role of nitric oxide in inflammation-mediated neurodegeneration. Ann N Y Acad Sci 2002;962:318-331.
Moss DW, Bates TE. Activation of murine microglial cell lines by lipopolysaccharide and interferon-gamma causes NO-mediated decreases in mitochondrial and cellular function. Eur J Neurosci 2001;13:529-538.
Sawada M, Kondo N, Suzumura A, Marunouchi T. Production of tumor necrosis factor-alpha by microglia and astrocytes in culture. Brain Res 1989;491:394-397.
Ritzel RM, Patel AR, Grenier JM, et al. Functional differences between microglia and monocytes after ischemic stroke. J Neuroinflammation 2015;12:106.
Kobayashi K, Imagama S, Ohgomori T, et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis 2013;4:e525.
Liu Z, Fan Y, Won SJ, et al. Chronic treatment with minocycline preserves adult new neurons and reduces functional impairment after focal cerebral ischemia. Stroke 2007;38:146-152.
Hoehn BD, Palmer TD, Steinberg GK. Neurogenesis in rats after focal cerebral ischemia is enhanced by indomethacin. Stroke 2005;36:2718-2724.
Kim BJ, Kim MJ, Park JM, et al. Reduced neurogenesis after suppressed inflammation by minocycline in transient cerebral ischemia in rat. J Neurol Sci 2009;279:70-75.
Montero M, Gonzalez B, Zimmer J. Immunotoxic depletion of microglia in mouse hippocampal slice cultures enhances ischemia-like neurodegeneration. Brain Res 2009;1291:140-152.
Vinet J, Weering HR, Heinrich A, et al. Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J Neuroinflammation 2012;9:27.
Faustino JV, Wang X, Johnson CE, et al. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J Neurosci 2011;31:12992-13001.
Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci 2007;27:2596-2605.
Szalay G, Martinecz B, Lenart N, et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun 2016;7:11499.
Grathwohl SA, Kalin RE, Bolmont T, et al. Formation and maintenance of Alzheimer's disease beta-amyloid plaques in the absence of microglia. Nat Neurosci 2009;12:1361-1363.
Gowing G, Philips T, Van Wijmeersch B, et al. Ablation of proliferating microglia does not affect motor neuron degeneration in amyotrophic lateral sclerosis caused by mutant superoxide dismutase. J Neurosci 2008;28:10234-10244.
Heppner FL, Greter M, Marino D, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med 2005;11:146-152.
Fogg DK, Sibon C, Miled C, et al. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 2006;311:83-87.
Leuschner F, Rauch PJ, Ueno T, et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med 2012;209:123-137.
Marchiori E, Escuissato DL, Irion KL, et al. Extramedullary hematopoiesis: findings on computed tomography scans of the chest in 6 patients. J Bras Pneumol 2008;34:812-816.
Robbins CS, Chudnovskiy A, Rauch PJ, et al. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation 2012;125:364-374.
van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. J Exp Med 1968;128:415-435.
Tsou CL, Peters W, Si Y, et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 2007;117:902-909.
Strauss-Ayali D, Conrad SM, Mosser DM. Monocyte subpopulations and their differentiation patterns during infection. J Leukoc Biol 2007;82:244-252.
Frankenberger M, Sternsdorf T, Pechumer H, Pforte A, Ziegler-Heitbrock HW. Differential cytokine expression in human blood monocyte subpopulations: a polymerase chain reaction analysis. Blood 1996;87:373-377.
Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003;19:71-82.
Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science 2010;327:656-661.
Yrlid U, Jenkins CD, MacPherson GG. Relationships between distinct blood monocyte subsets and migrating intestinal lymph dendritic cells in vivo under steady-state conditions. J Immunol 2006;176:4155-4162.
Miro-Mur F, Perez-de-Puig I, Ferrer-Ferrer M, et al. Immature monocytes recruited to the ischemic mouse brain differentiate into macrophages with features of alternative activation. Brain Behav Immun 2016;53:18-33.
Carlin LM, Stamatiades EG, Auffray C, et al. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell 2013;153:362-375.
Swirski FK, Nahrendorf M, Etzrodt M, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009;325:612-616.
Bao Y, Kim E, Bhosle S, Mehta H, Cho S. A role for spleen monocytes in post-ischemic brain inflammation and injury. J Neuroinflammation 2010;7:92.
Sahota P, Vahidy F, Nguyen C, et al. Changes in spleen size in patients with acute ischemic stroke: a pilot observational study. Int J Stroke 2013;8:60-67.
Vahidy FS, Parsha KN, Rahbar MH, et al. Acute splenic responses in patients with ischemic stroke and intracerebral hemorrhage. J Cereb Blood Flow Metab 2016;36:1012-1021.
Vendrame M, Gemma C, Pennypacker KR, et al. Cord blood rescues stroke-induced changes in splenocyte phenotype and function. Exp Neurol 2006;199:191-200.
Ajmo CT, Jr., Vernon DO, Collier L, et al. The spleen contributes to stroke-induced neurodegeneration. J Neurosci Res 2008;86:2227-2234.
Dotson AL, Wang J, Saugstad J, Murphy SJ, Offner H. Splenectomy reduces infarct volume and neuroinflammation in male but not female mice in experimental stroke. J Neuroimmunol 2015;278:289-298.
Ostrowski RP, Schulte RW, Nie Y, et al. Acute splenic irradiation reduces brain injury in the rat focal ischemic stroke model. Transl Stroke Res 2012;3:473-481.
Lin JN, Lin CL, Lin MC, et al. Increased risk of hemorrhagic and ischemic strokes in patients with splenic injury and splenectomy: a nationwide cohort study. Medicine (Baltimore) 2015;94:e1458.
Chu HX, Broughton BR, Kim HA, Lee S, Drummond GR, Sobey CG. Evidence that Ly6C(hi) monocytes are protective in acute ischemic stroke by promoting M2 macrophage polarization. Stroke 2015;46:1929-1937.
Stevens SL, Bao J, Hollis J, Lessov NS, Clark WM, Stenzel-Poore MP. The use of flow cytometry to evaluate temporal changes in inflammatory cells following focal cerebral ischemia in mice. Brain Res 2002;932:110-119.
Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti MT. Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. FEBS J 2009;276:13-26.
Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 2003;183:25-33.
Bauer J, Huitinga I, Zhao W, Lassmann H, Hickey WF, Dijkstra CD. The role of macrophages, perivascular cells, and microglial cells in the pathogenesis of experimental autoimmune encephalomyelitis. Glia 1995;15:437-446.
Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 2007;38:1345-1353.
Schilling M, Strecker JK, Ringelstein EB, Schabitz WR, Kiefer R. The role of CC chemokine receptor 2 on microglia activation and blood-borne cell recruitment after transient focal cerebral ischemia in mice. Brain Res 2009;1289:79-84.
Schuette-Nuetgen K, Strecker JK, Minnerup J, Ringelstein EB, Schilling M. MCP-1/CCR-2-double-deficiency severely impairs the migration of hematogenous inflammatory cells following transient cerebral ischemia in mice. Exp Neurol 2012;233:849-858.
Chen Y, Hallenbeck JM, Ruetzler C, et al. Overexpression of monocyte chemoattractant protein 1 in the brain exacerbates ischemic brain injury and is associated with recruitment of inflammatory cells. J Cereb Blood Flow Metab 2003;23:748-755.
Hughes PM, Allegrini PR, Rudin M, Perry VH, Mir AK, Wiessner C. Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. J Cereb Blood Flow Metab 2002;22:308-317.
Kim E, Febbraio M, Bao Y, Tolhurst AT, Epstein JM, Cho S. CD36 in the periphery and brain synergizes in stroke injury in hyperlipidemia. Ann Neurol 2012;71:753-764.
Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol 2009;27:669-692.
David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci 2011;12:388-399.
Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 2013;13:709-721.
Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008;8:958-969.
Kadl A, Meher AK, Sharma PR, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res 2010;107:737-746.
Walker DG, Lue LF. Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther 2015;7:56.
Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 2007;204:3037-3047.
Fujiu K, Manabe I, Nagai R. Renal collecting duct epithelial cells regulate inflammation in tubulointerstitial damage in mice. J Clin Invest 2011;121:3425-3441.
Lin SL, Castano AP, Nowlin BT, Lupher ML, Jr., Duffield JS. Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J Immunol 2009;183:6733-6743.
Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol 2005;175:342-349.
Perego C, Fumagalli S, De Simoni MG. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J Neuroinflammation 2011;8:174.
Hu X, Li P, Guo Y, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012;43:3063-3070.
Gliem M, Mausberg AK, Lee JI, et al. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann Neurol 2012;71:743-752.
Michaud JP, Pimentel-Coelho PM, Tremblay Y, Rivest S. The impact of Ly6Clow monocytes after cerebral hypoxia-ischemia in adult mice. J Cereb Blood Flow Metab 2014;34:e1-e9.
Crane MJ, Daley JM, van Houtte O, Brancato SK, Henry WL, Jr., Albina JE. The monocyte to macrophage transition in the murine sterile wound. PLoS One 2014;9:e86660.
Wattananit S, Tornero D, Graubardt N, et al. Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J Neurosci 2016;36:4182-4195.
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006;124:783-801.
Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology 2010;51:621-632.
Bamboat ZM, Ocuin LM, Balachandran VP, Obaid H, Plitas G, DeMatteo RP. Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion. J Clin Invest 2010;120:559-569.
Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med 2006;203:2613-2625.
Ren Y, Silverstein RL, Allen J, Savill J. CD36 gene transfer confers capacity for phagocytosis of cells undergoing apoptosis. J Exp Med 1995;181:1857-1862.
Savill J. Recognition and phagocytosis of cells undergoing apoptosis. Br Med Bull 1997;53:491-508.
Stewart CR, Stuart LM, Wilkinson K, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol 2010;11:155-161.
Orsini F, De Blasio D, Zangari R, Zanier ER, De Simoni MG. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front Cell Neurosci 2014;8:380.
Benoit ME, Hernandez MX, Dinh ML, Benavente F, Vasquez O, Tenner AJ. C1q-induced LRP1B and GPR6 proteins expressed early in Alzheimer disease mouse models, are essential for the C1q-mediated protection against amyloid-beta neurotoxicity. J Biol Chem 2013;288:654-665.
Farber K, Cheung G, Mitchell D, et al. C1q, the recognition subcomponent of the classical pathway of complement, drives microglial activation. J Neurosci Res 2009;87:644-652.
Galvan MD, Foreman DB, Zeng E, Tan JC, Bohlson SS. Complement component C1q regulates macrophage expression of Mer tyrosine kinase to promote clearance of apoptotic cells. J Immunol 2012;188:3716-3723.
Hadas S, Reichert F, Rotshenker S. Dissimilar and similar functional properties of complement receptor-3 in microglia and macrophages in combating yeast pathogens by phagocytosis. Glia 2010;58:823-830.
Stevens B, Allen NJ, Vazquez LE, et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007;131:1164-1178.
Weinstein JR, Quan Y, Hanson JF, et al. IgM-dependent phagocytosis in microglia is mediated by complement receptor 3, not Fcalpha/mu receptor. J Immunol 2015;195:5309-5317.
Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 2007;447:869-874.
Serhan CN, Hong S, Gronert K, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med 2002;196:1025-1037.
Serhan CN, Yang R, Martinod K, et al. Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med 2009;206:15-23.
Campbell EL, Louis NA, Tomassetti SE, et al. Resolvin E1 promotes mucosal surface clearance of neutrophils: a new paradigm for inflammatory resolution. FASEB J 2007;21:3162-3170.
Canny G, Levy O, Furuta GT, et al. Lipid mediator-induced expression of bactericidal/ permeability-increasing protein (BPI) in human mucosal epithelia. Proc Natl Acad Sci U S A 2002;99:3902-3907.
Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol 2000;164:1663-1667.
Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol 2007;25:101-137.
Campbell L, Saville CR, Murray PJ, Cruickshank SM, Hardman MJ. Local arginase 1 activity is required for cutaneous wound healing. J Invest Dermatol 2013;133:2461-2470.
Fenn AM, Hall JC, Gensel JC, Popovich PG, Godbout JP. IL-4 signaling drives a unique arginase+/IL-1beta + microglia phenotype and recruits macrophages to the inflammatory CNS: consequences of age-related deficits in IL-4Ralpha after traumatic spinal cord injury. J Neurosci 2014;34:8904-8917.
Pesce JT, Ramalingam TR, Mentink-Kane MM, et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog 2009;5:e1000371.
Greenhalgh AD, Passos Dos Santos R, Zarruk JG, Salmon CK, Kroner A, David S. Arginase-1 is expressed exclusively by infiltrating myeloid cells in CNS injury and disease. Brain Behav Immun 2016;56:61-67.
Elliott MR, Chekeni FB, Trampont PC, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009;461:282-286.
Noda M, Doi Y, Liang J, et al. Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. J Biol Chem 2011;286:2308-2319.
Truman LA, Ford CA, Pasikowska M, et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 2008;112:5026-5036.
Neher JJ, Emmrich JV, Fricker M, Mander PK, Thery C, Brown GC. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci U S A 2013;110:E4098-4107.
Brown GC, Neher JJ. Eaten alive! Cell death by primary phagocytosis: 'phagoptosis'. Trends Biochem Sci 2012;37:325-332.
Fadok VA, Bratton DL, Henson PM. Phagocyte receptors for apoptotic cells: recognition, uptake, and consequences. J Clin Invest 2001;108:957-962.
Goh YC, Yap CT, Huang BH, et al. Heat-shock protein 60 translocates to the surface of apoptotic cells and differentiated megakaryocytes and stimulates phagocytosis. Cell Mol Life Sci 2011;68:1581-1592.
Sierra A, Abiega O, Shahraz A, Neumann H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci 2013;7:6.
Cho S, Kim E. CD36: a multi-modal target for acute stroke therapy. J Neurochem 2009;109(Suppl. 1):126-132.
Kim E, Tolhurst AT, Qin LY, Chen XY, Febbraio M, Cho S. CD36/fatty acid translocase, an inflammatory mediator, is involved in hyperlipidemia-induced exacerbation in ischemic brain injury. J Neurosci 2008;28:4661-4670.
Cho S, Park EM, Febbraio M, et al. The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J Neurosci 2005;25:2504-2512.
Kunz A, Abe T, Hochrainer K, et al. Nuclear factor-kappaB activation and postischemic inflammation are suppressed in CD36-null mice after middle cerebral artery occlusion. J Neurosci 2008;28:1649-1658.
Woo MS, Wang X, Faustino JV, et al. Genetic deletion of CD36 enhances injury after acute neonatal stroke. Ann Neurol 2012;72:961-970.
Zhao X, Sun G, Zhang J, et al. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol 2007;61:352-362.
Oh J, Riek AE, Weng S, et al. Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation. J Biol Chem 2012;287:11629-11641.
Xu W, Schlagwein N, Roos A, van den Berg TK, Daha MR, van Kooten C. Human peritoneal macrophages show functional characteristics of M-CSF-driven anti-inflammatory type 2 macrophages. Eur J Immunol 2007;37:1594-1599.
Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol 2011;6:275-297.
Rodriguez-Prados JC, Traves PG, Cuenca J, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol 2010;185:605-614.
Huang SC, Everts B, Ivanova Y, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 2014;15:846-855.
Appelros P, Stegmayr B, Terent A. Sex differences in stroke epidemiology: a systematic review. Stroke 2009;40:1082-1090.
Fukuda M, Kanda T, Kamide N, Akutsu T, Sakai F. Gender differences in long-term functional outcome after first-ever ischemic stroke. Intern Med 2009;48:967-973.
Niewada M, Kobayashi A, Sandercock PA, Kaminski B, Czlonkowska A, International Stroke Trial Collaborative G. Influence of gender on baseline features and clinical outcomes among 17,370 patients with confirmed ischaemic stroke in the international stroke trial. Neuroepidemiology 2005;24:123-128.
Alkayed NJ, Harukuni I, Kimes AS, London ED, Traystman RJ, Hurn PD. Gender-linked brain injury in experimental stroke. Stroke 1998;29:159-165.
Banerjee A, Wang J, Bodhankar S, Vandenbark AA, Murphy SJ, Offner H. Phenotypic changes in immune cell subsets reflect increased infarct volume in male vs. female mice. Transl Stroke Res 2013;4:554-563.
Brait VH, Jackman KA, Walduck AK, et al. Mechanisms contributing to cerebral infarct size after stroke: gender, reperfusion, T lymphocytes, and Nox2-derived superoxide. J Cereb Blood Flow Metab 2010;30:1306-1317.
Manwani B, Liu F, Scranton V, Hammond MD, Sansing LH, McCullough LD. Differential effects of aging and sex on stroke induced inflammation across the lifespan. Exp Neurol 2013;249:120-131.
Pathan M, Kittner SJ. Pregnancy and stroke. Curr Neurol Neurosci Rep 2003;3:27-31.
Putaala J, Curtze S, Hiltunen S, Tolppanen H, Kaste M, Tatlisumak T. Causes of death and predictors of 5-year mortality in young adults after first-ever ischemic stroke: the Helsinki Young Stroke Registry. Stroke 2009;40:2698-2703.
Rasura M, Spalloni A, Ferrari M, et al. A case series of young stroke in Rome. Eur J Neurol 2006;13:146-152.
Towfighi A, Saver JL, Engelhardt R, Ovbiagele B. A midlife stroke surge among women in the United States. Neurology 2007;69:1898-1904.
DiNapoli VA, Huber JD, Houser K, Li X, Rosen CL. Early disruptions of the blood-brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol Aging 2008;29:753-764.
Manwani B, Liu F, Xu Y, Persky R, Li J, McCullough LD. Functional recovery in aging mice after experimental stroke. Brain Behav Immun 2011;25:1689-1700.
Moore TL, Killiany RJ, Pessina MA, Moss MB, Finklestein SP, Rosene DL. Recovery from ischemia in the middle-aged brain: a nonhuman primate model. Neurobiol Aging 2012;33:619.
Popa-Wagner A, Carmichael ST, Kokaia Z, Kessler C, Walker LC. The response of the aged brain to stroke: too much, too soon? Curr Neurovasc Res 2007;4:216-227.
Dubal DB, Kashon ML, Pettigrew LC, et al. Estradiol protects against ischemic injury. J Cereb Blood Flow Metab 1998;18:1253-1258.
Jover T, Tanaka H, Calderone A, et al. Estrogen protects against global ischemia-induced neuronal death and prevents activation of apoptotic signaling cascades in the hippocampal CA1. J Neurosci 2002;22:2115-2124.
Rau SW, Dubal DB, Bottner M, Wise PM. Estradiol differentially regulates c-Fos after focal cerebral ischemia. J Neurosci 2003;23:10487-10494.
Simpkins JW, Rajakumar G, Zhang YQ, et al. Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J Neurosurg 1997;87:724-730.
Johnson AB, Sohrabji F. Estrogen's effects on central and circulating immune cells vary with reproductive age. Neurobiol Aging 2005;26:1365-1374.
Liu F, Benashski SE, Xu Y, Siegel M, McCullough LD. Effects of chronic and acute oestrogen replacement therapy in aged animals after experimental stroke. J Neuroendocrinol 2012;24:319-330.
Nordell VL, Scarborough MM, Buchanan AK, Sohrabji F. Differential effects of estrogen in the injured forebrain of young adult and reproductive senescent animals. Neurobiol Aging 2003;24:733-743.
Wise PM, Dubal DB, Wilson ME, Rau SW, Bottner M, Rosewell KL. Estradiol is a protective factor in the adult and aging brain: understanding of mechanisms derived from in vivo and in vitro studies. Brain Res Brain Res Rev 2001;37:313-319.
Vegeto E, Pollio G, Ciana P, Maggi A. Estrogen blocks inducible nitric oxide synthase accumulation in LPS-activated microglia cells. Exp Gerontol 2000;35:1309-1316.
Pinto A, Tuttolomondo A, Di Raimondo D, Fernandez P, Licata G. Cerebrovascular risk factors and clinical classification of strokes. Semin Vasc Med 2004;4:287-303.
Kim E, Tolhurst AT, Cho S. Deregulation of inflammatory response in the diabetic condition is associated with increased ischemic brain injury. J Neuroinflammation 2014;11:83.
Kumari R, Willing LB, Krady JK, Vannucci SJ, Simpson IA. Impaired wound healing after cerebral hypoxia-ischemia in the diabetic mouse. J Cereb Blood Flow Metab 2007;27:710-718.
Combrinck MI, Perry VH, Cunningham C. Peripheral infection evokes exaggerated sickness behaviour in pre-clinical murine prion disease. Neuroscience 2002;112:7-11.
Cunningham C, Campion S, Lunnon K, et al. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry 2009;65:304-312.
Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci 2005;25:9275-9284.
Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol 2014;10:217-224.
Rawji KS, Mishra MK, Michaels NJ, Rivest S, Stys PK, Yong VW. Immunosenescence of microglia and macrophages: impact on the ageing central nervous system. Brain 2016;139:653-661.
Doyle KP, Cekanaviciute E, Mamer LE, Buckwalter MS. TGFbeta signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J Neuroinflammation 2010;7:62.
Kharlamov A, Kharlamov E, Armstrong DM. Age-dependent increase in infarct volume following photochemically induced cerebral infarction: putative role of astroglia. J Gerontol A Biol Sci Med Sci 2000;55:B135-B141.
Ritzel RM, Crapser J, Patel AR, et al. Age-associated resident memory CD8 T cells in the central nervous system are primed to potentiate inflammation after ischemic brain injury. J Immunol 2016;196:3318-3330.
Damani MR, Zhao L, Fontainhas AM, Amaral J, Fariss RN, Wong WT. Age-related alterations in the dynamic behavior of microglia. Aging Cell 2011;10:263-276.
Zhao C, Li WW, Franklin RJ. Differences in the early inflammatory responses to toxin-induced demyelination are associated with the age-related decline in CNS remyelination. Neurobiol Aging 2006;27:1298-1307.
Kotter MR, Li WW, Zhao C, Franklin RJ. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci 2006;26:328-332.
Natrajan MS, de la Fuente AG, Crawford AH, et al. Retinoid X receptor activation reverses age-related deficiencies in myelin debris phagocytosis and remyelination. Brain 2015;138:3581-3597.
Ruckh JM, Zhao JW, Shadrach JL, et al. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell 2012;10:96-103.
Floden AM, Combs CK. Microglia demonstrate age-dependent interaction with amyloid-beta fibrils. J Alzheimers Dis 2011;25:279-293.
Njie EG, Boelen E, Stassen FR, Steinbusch HW, Borchelt DR, Streit WJ. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol Aging 2012;33:195 e191-e112.
Sheng JG, Mrak RE, Griffin WS. Enlarged and phagocytic, but not primed, interleukin-1 alpha-immunoreactive microglia increase with age in normal human brain. Acta Neuropathol 1998;95:229-234.
Tremblay ME, Zettel ML, Ison JR, Allen PD, Majewska AK. Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia 2012;60:541-558.
Tang Y, Li T, Li J, et al. Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson's disease. Cell Death Differ 2014;21:369-380.
Shaw SY, Tran K, Castoreno AB, et al. Selective modulation of autophagy, innate immunity, and adaptive immunity by small molecules. ACS Chem Biol 2013;8:2724-2733.
Kelly J, Ali Khan A, Yin J, Ferguson TA, Apte RS. Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J Clin Invest 2007;117:3421-3426.
Jackaman C, Radley-Crabb HG, Soffe Z, Shavlakadze T, Grounds MD, Nelson DJ. Targeting macrophages rescues age-related immune deficiencies in C57BL/6J geriatric mice. Aging Cell 2013;12:345-357.
Beason-Held LL, Moghekar A, Zonderman AB, Kraut MA, Resnick SM. Longitudinal changes in cerebral blood flow in the older hypertensive brain. Stroke 2007;38:1766-1773.
Ong SL, Zhang Y, Whitworth JA. Reactive oxygen species and glucocorticoid-induced hypertension. Clin Exp Pharmacol Physiol 2008;35:477-482.
Vaziri ND, Wang XQ, Oveisi F, Rad B. Induction of oxidative stress by glutathione depletion causes severe hypertension in normal rats. Hypertension 2000;36:142-146.
Chandramohan G, Bai Y, Norris K, Rodriguez-Iturbe B, Vaziri ND. Effects of dietary salt on intrarenal angiotensin system, NAD(P)H oxidase, COX-2, MCP-1 and PAI-1 expressions and NF-kappaB activity in salt-sensitive and -resistant rat kidneys. Am J Nephrol 2008;28:158-167.
Bravo Y, Quiroz Y, Ferrebuz A, Vaziri ND, Rodriguez-Iturbe B. Mycophenolate mofetil administration reduces renal inflammation, oxidative stress, and arterial pressure in rats with lead-induced hypertension. Am J Physiol Renal Physiol 2007;293:F616-F623.
Liao W, Wei Y, Yu C, et al. Prenatal exposure to zymosan results in hypertension in adult offspring rats. Clin Exp Pharmacol Physiol 2008;35:1413-1418.
Rodriguez-Iturbe B, Ferrebuz A, Vanegas V, Quiroz Y, Mezzano S, Vaziri ND. Early and sustained inhibition of nuclear factor-kappaB prevents hypertension in spontaneously hypertensive rats. J Pharmacol Exp Ther 2005;315:51-57.
Lawes CM, Vander Hoorn S, Rodgers A; International Society of Hypertension. Global burden of blood-pressure-related disease, 2001. Lancet 2008;371:1513-1518.
Lewington S, Clarke R, Qizilbash N, Peto R, Collins R; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002;360:1903-1913.
O'Donnell MJ, Xavier D, Liu L, et al. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): a case-control study. Lancet 2010;376:112-123.
Willmot M, Leonardi-Bee J, Bath PM. High blood pressure in acute stroke and subsequent outcome: a systematic review. Hypertension 2004;43:18-24.
Wu KL, Chan SH, Chan JY. Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. J Neuroinflammation 2012;9:212.
Waki H, Gouraud SS, Maeda M, Paton JF. Specific inflammatory condition in nucleus tractus solitarii of the SHR: novel insight for neurogenic hypertension? Auton Neurosci 2008;142:25-31.
Gouw AA, Seewann A, van der Flier WM, et al. Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatry 2011;82:126-135.
Kaiser D, Weise G, Moller K, et al. Spontaneous white matter damage, cognitive decline and neuroinflammation in middle-aged hypertensive rats: an animal model of early-stage cerebral small vessel disease. Acta Neuropathol Commun 2014;2:169.
Wardlaw JM, Smith C, Dichgans M. Mechanisms of sporadic cerebral small vessel disease: insights from neuroimaging. Lancet Neurol 2013;12:483-497.
Shi P, Diez-Freire C, Jun JY, et al. Brain microglial cytokines in neurogenic hypertension. Hypertension 2010;56:297-303.
Kim KI, Lee JH, Chang HJ, et al. Association between blood pressure variability and inflammatory marker in hypertensive patients. Circ J 2008;72:293-298.
Kim CH, Vaziri ND. Hypertension promotes integrin expression and reactive oxygen species generation by circulating leukocytes. Kidney Int 2005;67:1462-1470.
Yoon JW, Pahl MV, Vaziri ND. Spontaneous leukocyte activation and oxygen-free radical generation in end-stage renal disease. Kidney Int 2007;71:167-172.
Dorffel Y, Latsch C, Stuhlmuller B, et al. Preactivated peripheral blood monocytes in patients with essential hypertension. Hypertension 1999;34:113-117.
Ishibashi M, Hiasa K, Zhao Q, et al. Critical role of monocyte chemoattractant protein-1 receptor CCR2 on monocytes in hypertension-induced vascular inflammation and remodeling. Circ Res 2004;94:1203-1210.
Shen XZ, Li Y, Li L, et al. Microglia participate in neurogenic regulation of hypertension. Hypertension 2015;66:309-316.
Kossmann S, Hu H, Steven S, et al. Inflammatory monocytes determine endothelial nitric-oxide synthase uncoupling and nitro-oxidative stress induced by angiotensin II. J Biol Chem 2014;289:27540-27550.
Kossmann S, Schwenk M, Hausding M, et al. Angiotensin II-induced vascular dysfunction depends on interferon-gamma-driven immune cell recruitment and mutual activation of monocytes and NK-cells. Arterioscler Thromb Vasc Biol 2013;33:1313-1319.
Moore JP, Vinh A, Tuck KL, et al. M2 macrophage accumulation in the aortic wall during angiotensin II infusion in mice is associated with fibrosis, elastin loss, and elevated blood pressure. Am J Physiol Heart Circ Physiol 2015;309:H906-917.
De Geyter D, Stoop W, Zgavc T, et al. Spontaneously hypertensive rats display reduced microglial activation in response to ischemic stroke and lipopolysaccharide. J Neuroinflammation 2012;9:114.
Moller K, Posel C, Kranz A, et al. Arterial hypertension aggravates innate immune responses after experimental stroke. Front Cell Neurosci 2015;9:461.
Drake C, Boutin H, Jones MS, et al. Brain inflammation is induced by co-morbidities and risk factors for stroke. Brain Behav Immun 2011;25:1113-1122.
Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 2010;72:219-246.
Swirski FK, Libby P, Aikawa E, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest 2007;117:195-205.
Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007;117:175-184.
Khallou-Laschet J, Varthaman A, Fornasa G, et al. Macrophage plasticity in experimental atherosclerosis. PLoS One 2010;5:e8852.
Kernan WN, Inzucchi SE. Type 2 diabetes mellitus and insulin resistance: stroke prevention and management. Curr Treat Options Neurol 2004;6:443-450.
Qatanani M, Lazar MA. Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev 2007;21:1443-1455.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW, Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003;112:1796-1808.
Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003;112:1821-1830.
Hazra S, Jarajapu YP, Stepps V, et al. Long-term type 1 diabetes influences haematopoietic stem cells by reducing vascular repair potential and increasing inflammatory monocyte generation in a murine model. Diabetologia 2013;56:644-653.
Bradshaw EM, Raddassi K, Elyaman W, et al. Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing Th17 cells. J Immunol 2009;183:4432-4439.
Devaraj S, Glaser N, Griffen S, Wang-Polagruto J, Miguelino E, Jialal I. Increased monocytic activity and biomarkers of inflammation in patients with type 1 diabetes. Diabetes 2006;55:774-779.
Giulietti A, van Etten E, Overbergh L, Stoffels K, Bouillon R, Mathieu C. Monocytes from type 2 diabetic patients have a pro-inflammatory profile. 1,25-Dihydroxyvitamin D(3) works as anti-inflammatory. Diabetes Res Clin Pract 2007;77:47-57.
Hwang IK, Choi JH, Nam SM, et al. Activation of microglia and induction of pro-inflammatory cytokines in the hippocampus of type 2 diabetic rats. Neurol Res 2014;36:824-832.
Ibrahim AS, El-Remessy AB, Matragoon S, et al. Retinal microglial activation and inflammation induced by amadori-glycated albumin in a rat model of diabetes. Diabetes 2011;60:1122-1133.
Yan T, Venkat P, Chopp M, et al. Neurorestorative therapy of stroke in type 2 diabetes mellitus rats treated with human umbilical cord blood cells. Stroke 2015;46:2599-2606.
Acknowledgments
This work was supported by the National Institute of Health awards, NHLBI R01HL082511, NINDS R01NS07789 (SC), and R01NS095359-10 (SC).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
13311_2016_463_MOESM1_ESM.pdf
Required Author Forms Disclosure forms provided by the authors are available with the online version of this article. (PDF 1224 kb)
Rights and permissions
About this article

Cite this article
Kim, E., Cho, S. Microglia and Monocyte-Derived Macrophages in Stroke. Neurotherapeutics 13, 702–718 (2016). https://doi.org/10.1007/s13311-016-0463-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-016-0463-1