
Abstract
Alterations in the innate inflammatory response may underlie the pathophysiology of psychiatric diseases. Current antipsychotics modulate pro-/anti-inflammatory pathways, but their specific actions on these pathways remain only partly explored. This study was conducted to elucidate the regulatory role of paliperidone (1 mg/kg i.p.) on acute (6 h) and chronic (6 h/day for 21 consecutive days) restraint stress-induced alterations in 2 emerging endogenous anti-inflammatory/antioxidant mechanisms: nuclear factor erythroid-related factor 2 (NRF2)/antioxidant enzymes pathway, and the cytokine milieu regulating M1/M2 polarization in microglia, analyzed at the mRNA and protein levels in prefrontal cortex samples. In acute stress conditions, paliperidone enhanced NRF2 levels, possibly related to phosphoinositide 3-kinase upregulation and reduced kelch-Like ECH-associated protein 1 expression. In chronic conditions, paliperidone tended to normalize NRF2 levels through a phosphoinositide 3-kinase related-mechanism, with no effects on kelch-Like ECH-associated protein 1. Antioxidant response element-dependent antioxidant enzymes were upregulated by paliperidone in acute stress, while in chronic stress, paliperidone tended to prevent stress-induced downregulation of the endogenous antioxidant machinery. However, paliperidone increased transforming growth factor-β and interleukin-10 in favor of an M2 microglia profile in acute stress conditions, which was also corroborated by paliperidone-induced increased levels of the M2 cellular markers arginase I and folate receptor 2. This latter effect was also produced in chronic conditions. Immunofluorescence studies suggested an increase in the number of microglial cells expressing arginase I and folate receptor 2 in the stressed animals pretreated with paliperidone. In conclusion, the enhancement of endogenous antioxidant/anti-inflammatory pathways by current and new antipsychotics could represent an interesting therapeutic strategy for the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Psychological/physical stress exposure activates the innate immune system, producing an inflammatory response both in the peripheral and central nervous systems (CNS) [1]. The most studied pathway is the one driven by the activation of the Toll-like receptor 4 [2]. Consecutive steps in this transduction pathway are the activation of cytokines and the prototypic inflammatory nuclear transcription factor nuclear factor kappa B, which culminates in a potential deleterious overaccumulation of inflammatory and oxido-/nitrosative stress mediators in particular brain areas, such as the prefrontal cortex (PFC) [2]. In addition, chronic stress exposure affects synaptic plasticity, dendritic morphology, and neurogenesis in animals (reviewed in [3]), and induces both clinical and anatomical features of neurotoxic damage in humans [4].
The inflammatory response is a homeostatic mechanism essential for survival in stressful conditions, but it is also a costly process in terms of energy and resources and when uncontrolled/chronic could be pernicious for the normal structure and function of brain cellular populations [1]. There are several anti-inflammatory mechanisms (some of them acting synergistically in time and space) of control. One of these mechanisms involves the cyclopentenone prostaglandin 15-deoxy-prostaglandin J2 (15d-PGJ2) [5]. 15d-PGJ2 is the proposed endogenous ligand for the gamma isoform of peroxisome proliferator-activated nuclear receptors (PPARγ), a transcription factor whose main effect is to mitigate inflammation by repressing the expression of proinflammatory mediators [6]. PPARγ may be also activated by the antidiabetic thiazolidinedione drugs, which exert anti-inflammatory, antiexcitotoxic, and proenergetic effects in the brains of stressed rats [7, 8]. The antioxidant profile of 15d-PGJ2/PPARγ may be related to its ability to regulate the nuclear factor erythroid-related factor 2 (NRF2), in brain [9]. Inactive NRF2 is retained in the cytoplasm by association with the kelch-like ECH-associated protein 1 (KEAP1), but during inflammation or oxidative stress, the phosphorylation of some intracellular kinases, such as phosphoinositide 3-kinase (PI3K) and protein kinase B (AKT), elicits the translocation of NRF2 to the nucleus. Then, NRF2 binds to the antioxidant response elements (ARE) within the promoter of antioxidant enzymes, activating their transcription [10]. The most relevant of these enzymes are heme oxygenase-1 (HO1), nicotinamide adenine dinucleotide phosphate dehydrogenase quinone 1 (NQO1), superoxide dismutase (SOD), glutathione peroxidase (GPx) and catalase (CAT) [11].
The activation of PPARγ is also implicated in another important anti-inflammatory mechanism: the polarization profile of microglial cells in the evolution of inflammation-related CNS diseases. The use of PPARγ agonists induces the M2 phenotype in microglia in several animal models of neurodegeneration, such as Alzheimer’s and Parkinson’s [12, 13]. Microglial cells are a clear example of brain plasticity in response to the environment, in this case the cytokine milieu. Toll-like receptor-4 activation with lipopolysaccharide produces the synthesis and release of classical proinflammatory cytokines [tumor necrosis factor-α, interleukin (IL)-1β, IL-6, interferon-γ] and chemokines that polarize microglia into the proinflammatory M1 phenotype [14]. In contrast, microglia could change to the alternative anti-inflammatory M2 profile owing to the effects of the cytokines IL-4 and IL-13 [15]. M2 microglial cells expressed higher levels of arginase I (ArgI) and folate receptor 2 (FOLR2), and synthesize and release anti-inflammatory cytokines such as IL-10 and transforming growth factor (TGF)-β [16, 17]. The enhancement of the M2 profile affords neuroprotection in some neuropathological conditions and its role in neuropsychiatric diseases is currently emerging [18, 19].
In the context of the inflammatory theory of psychiatric disease, activation of the innate immune system/inflammation could be an important process to take into consideration in the study of the etiology and physiopathology of stress-related psychiatric diseases, such as schizophrenia. Over the last few years there has been substantial interest in the role of inflammation in psychosis, based on epidemiological, peripheral, postmortem, genetic and therapeutic data [20, 21]. Recently, a meta-analysis of previous clinical trials of adjunctive nonsteroidal anti-inflammatory drugs and other immunomodulatory compounds in schizophrenia has reported modest beneficial effects [22]. Based on this evidence, future studies should have a change of focus. Current studies propose the control of inflammation via direct anti-inflammatory effects, but the pharmacological stimulation of antioxidant/anti-inflammatory pathways to potentiate the endogenous response against disease is emerging as a promising strategy.
Consequently, this study is aimed at the molecular level to investigate whether the therapeutic potential of the antipsychotic paliperidone could be related to its ability to regulate positively the endogenous antioxidant (NRF2/antioxidant enzymes) and anti-inflammatory (M1/M2 polarization in microglia) pathways that are compromised in the PFC of rats exposed to acute/chronic restraint stress exposure.
Methods
Animals
Forty-six young adult (12 weeks of age) male Wistar Hannover rats (HsdHan:Wist; Harlan Ibérica, Barcelona, Spain) weighing 225–250 g were used. All experimental protocols adhered to the guidelines of the Animal Welfare Committee of the Universidad Complutense, in accordance with European legislation (D2010/63/UE). The rats were housed in standard temperature and humidity conditions and in a 12-h light/dark cycle (lights on at 08:00), with free access to food and water. All animals were maintained under constant conditions for 7 days prior to the experiment.
Drug Administration and Experimental Design
The atypical antipsychotic paliperidone (3-[2-[4-(6-fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl]ethyl]-6,7,8,9-tetrahydro-9-hydroxy-2-methyl-4H-pyrido[1,2-a]pyrimidin-4-one) (MW 426.48) and other chemicals were purchased from Sigma-Aldrich (Madrid, Spain), or as indicated. Paliperidone was suspended in a saline solution with 0.1 % Tween 20 [Vehicle (Veh)] by sonication for 1 min.
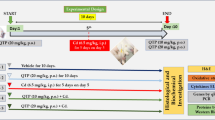
Two experimental designs were performed as follows. The first was an acute restraint stress model (6 h of restraint) using a plastic rodent restrainer (DecapiCone®, Braintree Scientific INC, Braintree, MA, USA) that allowed for a close fit to rats [23]. Control animals were not subjected to stress but were handled for a few seconds, and food and water were removed during the period of time that the stressed rats were kept in the restrainer. Vehicle or paliperidone was given by oral gavage (0.5 ml) immediately before placing the animal into the plastic restrainer. Groups were as follows: Veh + no stress (control); 1 mg/kg paliperidone (P) + no stress; Veh + stress; P + stress (groups 1–4 in Figure S1).
The second was a chronic restraint stress model (6 h/day for 21 days) [24]. The experimental groups were as follows: Veh + no stress (control); P + no stress; Veh + stress; P + stress (groups 5–8 in Figure S1).
All protocols started at 9:00 h, to avoid circadian changes in the stress response. All groups in the acute stress model contained 5 mice and 5–8 in chronic stress model. Brain samples were taken after the restraint session using sodium pentobarbital (320 mg/kg, i.p. Vetoquinol®, Madrid, Spain). Paliperidone was chosen in the present study considering previous reports have showed an immunomodulatory role of paliperidone in animal models based on acute/chronic stress exposure [25]. The dose of paliperidone (1 mg/kg) was chosen on the basis of previous in vivo determinations of signaling pathways related to oxidative stress, cytokines, and synaptic plasticity in the PFCof Sprague–Dawley rats [26].
To check whether the dose of paliperidone used in the 21-day chronic study is within the clinical range, plasma aliquots extracted at the end of the last session of stress were purified using mixed mode cation exchange solid-phase extraction columns, and paliperidone levels were measured using a sensitive high-performance liquid chromatography with electrospray tandem mass spectrometer as described previously [27]. The paliperidone concentration found was under the detection limit in control animals (<1.5 ng/mL). However, control paliperidone-treated animals showed a mean concentration of 17.45 ± 5.99 ng/mL; in stressed paliperidone-treated animals, it was 19.34 ± 3.83 ng/mL.
These values (41 and 45.3nM, respectively) are in the same range as the plasma levels obtained in a protocol of administration of risperidone (1 mg/kg) with an osmotic minipump for 7 days, achieving a Dopamine receptor 2 (D2) occupancy of 31 ± 9 % [28]. Thus, the dose achieved with our chronic administration protocol is below the clinically comparable range (60–80 % D2 occupancy).
Preparation of Biological Samples
Blood samples were obtained by cardiac puncture and centrifuged to extract plasma aliquots. After decapitation, the brain was removed from the skull. After careful removal of the meninges and blood vessels, PFC from both brain hemispheres were excised and stored at −80 °C until assayed.
Western Blot Analysis
To determine the expression levels of 4-hydroxynonenal (4-HNE), protein kinase B (AKT), phosphorylated AKT (pAKT), PI3K, NQO1, ArgI and FOLR2, brain PFC samples were homogenized by sonication in 400 μL phosphate-buffered saline (PBS; pH 7) mixed with a protease inhibitor cocktail (Complete®; Roche, Madrid, Spain), followed by centrifugation at 12,000 × g for 10 min at 4 °C. Protein levels of NRF2 were determined in nuclear extracts, and KEAP1 in cytosolic extract in PFC samples. Nuclear and cytosolic extracts were prepared according to published protocols [29]. Protein levels were measured using the Bradford method, based on the principle of protein-dye binding [30].
Blots were imaged using an Odyssey Fc System (Li-COR Biosciences, Lincoln, NE, USA) and were quantified by densitometry (ImageJ software; National Institutes of Health, Bethesda, MD, USA). In all the Western blot analyses, the housekeeping genes for β-actin, glyceraldehyde 3-phosphate dehydrogenase, or histone deacetylase 1 (for nuclear extracts) were used as a loading control (the blots are shown in the respective figures). Detailed information can be found in the Supplementary Material.
Real-time-Polymerase Chain Reaction Analysis
Total cytoplasmic RNA was prepared from samples of brain PFC using TRIZOL reagent (Invitrogen, Grand Island, NY, USA); aliquots were converted to complementary DNA using random hexamer primers. Quantitative changes in mRNA levels of HO1, NQO1, superoxide dismutase (SOD), GPx, CAT, IL-4, IL-10, TGF-β, glyceraldehyde 3-phosphate dehydrogenase and tubulin were estimated by real time-polymerase chain reaction (see Table S1 for details).
Antioxidant Enzyme Activity
To determine SOD and CAT activity, brain PFC samples were homogenized by sonication in 400 μL PBS (pH7) mixed with a protease inhibitor cocktail (Complete®; Roche), followed by centrifugation at 10,000 × g for 15 min at 4 °C. Supernatants were used for determinations, according to the manufacturer’s instruction. The sensitivity of the assay for SOD was 0.044 U/mL (K028-H1; Arbor Assays, Ann Arbor, MI, USA); for CAT it was 0.052 U/mL (K033-H1; Arbor Assays). The results were expressed as U/mg of protein.
Immunofluorescence
At the end of the stress session, mice were anesthetized and perfused via the ascending aorta with 4 % paraformaldehyde in 0.1 mol/L PBS, pH 7.4, and the brains were removed, postfixed overnight, and cryoprotected in 30 % sucrose for 24 h. Regularly spaced series of 30-μm-thick coronal sections were collected in cryoprotectant solution and stored at −40 °C until processing.
Sections were incubated with antisera for ArgI (ab91279, 1:300; Abcam, Cambridge, UK) and either 1) a mouse monoclonal anti-NeuN (ab104224, 1:1000; Abcam), used here as marker for neurons; 2) a goat polyclonal anti-ionized calcium binding adaptor molecule 1 (Iba1) (ab5076, 1:750; Abcam), used here as marker for parenchymal microglia. The respective primary antisera were incubated for 48 h at 4 °C. Subsequently, the sections were incubated for 1.5 h at room temperature with Alexa 555-conjugated donkey antirabbit IgG (1:1000; Life Technologies, Carlsbad, CA, USA), to localize ArgI, with Alexa 488-conjugated donkey antimouse IgG (1:1000; Life Technologies) for NeuN, and Alexa 488-conjugated donkey antigoat IgG (1:1000; Life Technologies) for Iba1. Control experiments included incubation of tissue sections from control and stressed animals with each antiserum singly and then with both secondary antisera to ensure that the latter did not cross-react with the inappropriate primary antiserum or with each other. Imaging was performed using an Olympus FV1200 confocal microscope (Olympus, Tokyo, Japan). For relative quantification of immunofluorescence, ten 40× fields were analyzed by experimental condition. For each field, the total number of Iba1-positive cells and ArgI-/Iba1-positive cells were counted using Image J software (National Institutes of Health). ArgI-positive microglia in each view was pooled and expressed in percentage.
Statistical Analyses
Data in the text and figures are expressed as mean ± SEM. For multiple comparisons, a 2-way ANOVA followed by the Tukey post-hoc test was used, considering as the first factor the presence or absence of stress and, as second, the presence or absence paliperidone treatment, and by 1-way ANOVA followed by Newman–Keuls post-hoc test when appropriate. A p-value < 0.05 was considered statistically significant. Data were analyzed using GraphPad Prism version 6 (GraphPad Software Inc., San Diego, CA, USA). All results of the 2-way ANOVA analyses (F values and dfs) are included in Tables S2 and S3.
Results
Effects of Paliperidone on the Brain NRF2 Regulatory Pathway After Acute and Chronic Restraint Stress Exposure
First, the index of oxidative cellular damage by lipid peroxidation-derived 4-HNE was studied. Post-test analysis showed that the 4-HNE content was significantly elevated under acute stress conditions (Fig. 1a). Two-way ANOVA analysis found a main effect for stress [F(1,12) = 16.11; p = 0.017].

Effects of paliperidone on brain nuclear factor erythroid-related factor 2 (NRF2) regulatory pathway after acute and chronic restraint stress exposure. Protein levels of (A, E) 4-hydroxynonenal (4-HNE), (B, F) NRF2, (C, G) phosphoinositide 3-kinase (PI3K), and (D, H) Kelch-like ECH-associated protein 1 (KEAP1) on the prefrontal cortex of rats in control and acute/chronic restraint stress conditions and pretreated with vehicle (Veh) or paliperidone (P). The densitometric data of the respective bands of interest are normalized by β-actin (lower band). *p < 0.05, **p < 0.01, ***p < 0.001 vs control (C) group; & p < 0.05 vs control + P group; # p < 0.05, ## p < 0.01, ### p < 0.001 vs acute/chronic stress + Veh. (Tukey’s post-hoc test or Newman–Keuls post-hoc test). Data represent the mean ± SEM
Next, the expression of the master regulator of the endogenous antioxidant response NRF2 was analyzed. A significant increase of NRF2 protein in the P + acute stress group compared with Veh + acute stress animals was found [F(3,12) = 4.022; p = 0.034 (Fig. 1b)]. Two-way ANOVA showed a main effect for paliperidone treatment and interaction in the analysis of NRF2 protein levels in nuclear extracts.
As NRF2 expression and activity is regulated by PI3K, AKT, and KEAP1, we next studied whether a paliperidone stimulatory effect on NRF2 could be related to effects on the content of its regulatory proteins. Regarding PI3K, a post-test showed a significant increase of PI3K levels in the P + acute stress group compared with P + control animals (Fig. 1c). A stress stimulatory effect on PI3K protein is only appreciable when the animals have been pretreated with paliperidone. Post-test confirmed this effect showing increasing levels in the Veh + acute stress animals compared with control group (Fig. S2a). The main effect for stress was found for both PI3K and pAKT/AKT ratio.
Acute stress induced an increase in KEAP1 in the vehicle-pretreated group compared with its control, and this increase was prevented by paliperidone treatment prior to stress [F(3,12) = 6.048; p = 0.0095 (Fig. 1d)]. Two-way ANOVA analysis of KEAP1 protein showed a main effect for stress and interaction.
In chronic stress conditions, stress exposure increased 4-HNE levels, an effect that was prevented by paliperidone treatment [F(3,22) = 4.771; p = 0.01 (Fig. 1e)]. Two-way ANOVA showed a main effect for treatment, stress, and interaction.
Stress induced a reduction of NRF2 content, this effect being partly prevented by preadministration of paliperidone after stress exposure. Two-way ANOVA showed a main effect for stress in NRF2 protein levels in nuclear extracts [F(1,14) = 13.49; p = 0.002 (Fig. 1 F)].
For PI3K protein levels, a highly significant stress-induced reduction was found, which was fully prevented by paliperidone treatment after stress exposure [F(3,22) = 18.33; p < 0.0001 (Fig. 1g)]. The main effects for stress, treatment, and interaction were found.
As in acute stress conditions, stress exposure increased the pAKT/AKT ratio in the Veh + acute stress animals compared with the control group (Fig. S2b). Two-way ANOVA showed a main effect for stress.
Finally, there were no changes in KEAP1 protein content in any groups of animals studied, although 2-way ANOVA showed a main effect for stress [F(1,22) = 5.54; p = 0.028 (Fig. 1h)].
Effects of Paliperidone on NRF2-dependent Antioxidant Enzymes After Acute and Chronic Restraint Stress Exposure
As NRF2 is an upstream regulator of the endogenous antioxidant response, we studied whether paliperidone was able to normalize/upregulate the expression/activity of the main antioxidant enzymes in control and acute/chronic stress conditions.
In acute stress conditions, an increase in NQO1 protein expression in paliperidone pretreated stressed animals compared with the paliperidone-treated control group and stressed animals was found [F(3,12) = 8.023; p = 0.0034 (Fig. 2a)]. Two-way ANOVA showed a main effect for stress, treatment, and interaction.

Effects of paliperidone on nuclear factor erythroid-related factor 2 (NRF2)-dependent antioxidant enzymes after acute and chronic restraint stress exposure. Protein levels of (A) the antioxidant enzyme nicotinamide adenine dinucleotide phosphate quinone 1 (NQO1), (B–D) mRNA levels of superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalase (CAT), (E) heme oxygenase-1 (HO1) mRNA levels, (F) SOD activity, (G) GPx mRNA levels and (H) CAT activity on the prefrontal cortex of rats in control and acute/chronic restraint stress conditions and pretreated with vehicle (Veh) or paliperidone (P). *p < 0.05 vs control (C) group; & p < 0.05 vs control + P group; # p < 0.05, ## p < 0.01 vs acute/chronic stress + veh. (Tukey’s post-hoc test or Newman–Keuls post-hoc test). Data represent the mean ± SEM
In the case of SOD and GPx mRNA levels, post-tests showed an increase in SOD and GPx in the group of stressed rats pretreated with paliperidone compared with the paliperidone control group and the vehicle-pretreated stressed animals (Fig. 2b, c). A main effect for treatment was present.
Regarding CAT mRNA levels, CAT content was higher in the paliperidone stressed animals than in the paliperidone control animals and vehicle pretreated stressed group [F(3,12) = 5.19; p = 0.0179 (Fig. 2d)]. There was a main effect of treatment and interaction.
There were no changes in SOD and CAT activity in any groups of animals studied (Fig. S2C, D), although 2-way ANOVA showed a main effect for stress in CAT activity [F(1,12) = 5.12; p = 0.043 (Fig. S2d)]. HO1 remained unaltered in all groups studied (data not shown).
In chronic stress conditions, a decrease in HO1 content induced by chronic stress was found (Fig. 2e). A main effect for stress exposure was found for HO1 mRNA levels.
No main factors or interaction were found for HO1 mRNA levels, but post-test showed a decrease in vehicle-pretreated animals compared with control that was prevented in the group of stressed animals treated with paliperidone (Fig. S2e).
SOD activity augmented in paliperidone-treated stressed animals compared with stressed animals [F(1,22) = 4.17; p = 0.02 (Fig. 2f)]. Two-way ANOVA analysis showed a main effect of treatment and interaction.
A similar scenario existed for GPx: post-tests showed an increase of GPx mRNA levels in the group of paliperidone-pretreated animals compared with the vehicle-treated group (Fig. 2g). There were no main factors or interaction found.
In the case of CAT activity, an increase in paliperidone-treated stressed animals compared with stressed animals was found [F(1,22) = 4.09; p = 0.02 (Fig. 2h)]. Two-way ANOVA analysis showed a main effect of treatment and interaction. Stress reduced CAT levels and this effect was normalized to control values by paliperidone [F(3,22) = 10.64; p = 0.0002 (Fig. S2f)]. Main effects for stress, treatment, and interaction were found.
Further analysis showed that NQO1 expression remained unaltered in all groups studied (data not shown).
Effects of Paliperidone on Microglia M2 Polarization After Acute and Chronic Restraint Stress Exposure
We first studied whether paliperidone pretreatment was able to modulate cytokine environment to favor the transition of microglia from the stress-induced M1 profile to the M2 profile. Thus, we analyzed the mRNA levels of the cytokines IL-4, TGF-β, and IL-10 by real-time polymerase chain reaction.
Acute stress increased IL-4 mRNA content that was fully prevented by paliperidone pretreatment [F(3,12) = 14.61; p = 0.0003 (Fig. 3a)]. There were main effects for stress, treatment, and interaction.

Effects of paliperidone on microglia M2 polarization after acute and chronic restraint stress exposure. mRNA levels of the anti-inflammatory cytokines (A, F) interleukin (IL)-4, (B, G) transforming growth factor (TGF)-β, and (C, H) IL-10. Protein levels of M2 microglia cellular markers (D, I) arginase (Arg)I and (E, J) folate receptor 2 (FOLR2). ArgI/ionized calcium binding adaptor molecule 1 (Iba1)-positive cells quantification on the prefrontal cortex of rats in control and acute restraint stress conditions and pretreated with vehicle (Veh) or paliperidone (P). *p < 0.05, ***p <0.001 vs control (C) group; & p < 0.05, &&& p < 0.001 vs control + P group; # p < 0.05, ## p < 0.01, ### p < 0.001 vs acute/chronic stress + Veh. (Tukey’s post-hoc test or Newman–Keuls post-hoc test). Data represent the mean ± SEM
Regarding TGF-β, an increase in paliperidone pretreated stressed animals compared with the vehicle-treated stressed group was present [F(3,12) = 3.21; p = 0.06 (Fig. 3b)]. There was a main effect for treatment and interaction.
For IL-10, acute stress produced a marked decrease in IL-10 mRNA content that was normalized to control levels by pretreatment with paliperidone [F(3,12) = 15.14; p = 0.0002 (Fig. 3c)]. There were main effects for stress, treatment, and interaction.
Second, we studied by Western blot the direct effects of stress exposure on M2 phenotype markers ArgI and FOLR2 and the potential effects of paliperidone on microglia polarization profile. For the first M2 marker, ArgI, an increase in the group of stressed rats pretreated with paliperidone compared with vehicle-treated rats was found (Fig. 3d). There were main effects for treatment and interaction. Regarding FOLR2, as in the case of ArgI, an increase in the group of stressed rats pretreated with paliperidone compared with vehicle-treated rats and also to the paliperidone pretreated control group was present [F(3,12) = 5.01; p = 0.017 (Fig. 3e)]. Interaction was found.
Chronic stress exposure induced a decrease in IL-4 mRNA levels, which was fully prevented by paliperidone pretreatment in stress conditions [F(3,22) = 11.31; p < 0.0001 (Fig. 3f)]. Main effects for stress, treatment, and interaction were found.
There were no changes in TGF-β mRNA content in any groups studied (Fig. 3g).
For IL-10 mRNA levels, a stress-induced decrease in IL-10 mRNA levels was found (Fig. 3h). A main effect for chronic stress exposure was found.
Microglia M2 marker protein content also changed in the chronic stress protocol. Chronic stress reduced ArgI protein levels and paliperidone pretreatment fully prevented this downregulation [F(3,22) = 8.593; p = 0.0006 (Fig. 3i)]. Main effects for treatment and interaction were found. Regarding FOLR2, a significant increase in the stressed animals pretreated with paliperidone compared with the group of vehicle-treated animals was found [F(3,22) = 3.98; p = 0.028 (Fig. 3J)]. Interaction was found.
Finally, we checked by double immunofluorescence whether the observed increase in the expression of M2 phenotype markers occurred in microglial cells of the PFC. The qualitative approach trying to identify the cellular types where ArgI is expressed showed a predominant neuronal expression in the 4 groups studied (see Fig. S3). In addition, ArgI immunoreactivity was also found in microglia in control and stress conditions with a predominant cytoplasmic localization (Fig. 4). A more detailed analysis showed an increase in the number of microglial cells expressing ArgI in the acute stressed animals pretreated with paliperidone compared with the other groups [F(3,36) = 16.06; p < 0.0001; Figs 3k and 4l). Two-way ANOVA analysis showed main effects for stress, treatment, and interaction.

Immunofluorescence study of the effects of paliperidone on microglia M2 cellular markers after acute restraint stress exposure. Arginase (ArgI) immunoreactivity in 30-μm-thick sections through the brain prefrontal cortex of rats in (A–F) control and (G–L) acute restraint stress conditions, and pretreated with (A–C, G–I) vehicle or (D–F, J–L) paliperidone. Microglia cellular marker ionized calcium binding adaptor molecule 1 (appears in green and ArgI in red in all cases. In the particular cases, ArgI localizes to the respective cellular marker with areas of overlap appearing yellow/orange in the merged image (white arrows) Scale bar = 50 μm
Discussion
The enhancement of endogenous anti-inflammatory/antioxidant pathways could be considered a new adjuvant therapeutic strategy for the treatment of stress-related psychiatric disorders. In this study, we used an acute/chronic restraint stress model to elicit a strong proinflammatory/pro-oxidant status to check the ability of paliperidone to preserve/potentiate 2 major homeostatic mechanisms: NRF2-driven antioxidant pathway and the alternative polarization of microglia to the M2 anti-inflammatory profile in the PFC of rats (see Fig. S4). Further studies are needed to determinate whether this anti-inflammatory/antioxidant profile may be responsible for the protective effects of paliperidone pretreatment on abnormal rat behavior following exposure to the schizophrenia experimental model based on prenatal immune activation using polyriboinosinic-polyribocytidylic acid during pregnancy in rats [31]. It is important to note that other molecules and pathways could be related to the ability of paliperidone to regulate inflammatory homeostasis, as, for example, is the case of systemic levels of omega-3 polyunsaturated fatty acids, which appeared to be increased in rats chronically treated with risperidone or paliperidone [32].
In acute stress conditions, paliperidone increased NRF2 protein levels in a mechanism possibly related to a concomitant upregulation of PI3K, with a lack of effect on stress-induced increase of the other canonical NRF2 regulator, pAKT. It is also important to discuss the results found on KEAP1. Acute stress increases KEAP1 expression in cytoplasm, an effect that could be related to the nonsignificant reduction of NRF2 nuclear levels in stress conditions. Paliperidone pretreatment normalized KEAP1 to control levels and produced a significant increase in nuclear NRF2, which is directly related to the upregulation of antioxidant enzymes observed (see Fig. 2). As it has been described in previous pathological scenarios, stress protocols included [33], KEAP1 sequesters NRF2 in the cytoplasm, leading to ubiquitination and subsequent degradation by the 26S proteasome [34]. When cells are exposed to oxidative/nitrosative stress challenges, NRF2 escapes KEAP1-mediated repression and translocates to the nucleus activating ARE-dependent antioxidant enzymes [35].
After chronic stress exposure, paliperidone partly blocked the stress-induced downregulation of NRF2, there was no significant effect on KEAP1 and paliperidone maintained the expression of PI3K and pAKT at control levels, suggesting the intent to enhance NRF2 expression and activity.
There are other inflammation-related molecules capable of regulating NRF2 expression. Nuclear factor kappa B is a negative regulator of NRF2 by binding to response elements in the NRF2 gene promoter [36]. In addition, the p65 subunit blocks NRF2 binding to CREB protein and recruits histone deacetylase 3, a co-repressor of ARE [37]. Another example is the anti-inflammatory prostaglandin 15d-PGJ2, which positively regulates the NRF2 pathway producing a conformational change in KEAP1 protein that allows the translocation of NRF2 to nucleus [38]. Furthermore, NRF2 knockdown in astrocytes abrogated the neuroprotective effect of 15d-PGJ2, as well as 15d-PGJ2-facilitated ARE-dependent antioxidant enzyme gene induction [9]. Both pathways are regulated by paliperidone in animal models of acute/chronic stress and could be implicated, at least in part, in the effects reported here [25, 29].
The antioxidant potential of paliperidone shown here could be therapeutically relevant, considering that many studies have reported altered antioxidant enzymes levels and signs of oxidative stress both in brain and peripheral nervous system and CNS of patients diagnosed with schizophrenia (reviewed in [21]). In agreement with our results, a recent meta-analysis focusing on peripheral markers showed that total antioxidant status in the blood was found to be decreased after a first psychotic episode and increased in longitudinal studies in patients receiving antipsychotic treatment [39].
To our knowledge, our results are the first to suggest a direct role of paliperidone in the regulation of microglia polarization (measured by the detection of specific Arg1 and FOLR2 cellular markers) towards a M2 profile in stress-induced proinflammatory/oxidant conditions. However, more detailed quantitative immunohistochemical studies are needed to determine in detail the magnitude of the effects reported here both in acute/chronic stress-induced inflammatory conditions.
Our results also show a potential role for paliperidone as a regulator of the pro-M2 cytokine environment, although these results need to be corroborated at protein level. Paliperidone elicited normalization to control levels of IL-4 mRNA content both in acute/chronic stress conditions (despite of the opposite effects of the different stresses) and increased TGFβ in acute stress conditions. In this vein, antipsychotics activated the TGF-β pathway effector SMAD3 in brain samples [40].
IL-10 is a cytokine that reflects M2 polarization of microglia and its synthesis and release inhibits the production of proinflammatory cytokines by M1 microglia [41]. Increased IL-10 levels have been found in the cerebrospinal fluid of patients with schizophrenia [42], but the stimulatory effect of paliperidone on IL-10 mRNA levels in PFC samples shown herein needs to be corroborated in the cerebrospinal fluid or postmortem brain tissue of patients treated with risperidone/paliperidone or other antipsychotics.
The results reported herein study are remarkable, considering that some authors have hypothesized that microglia are the main sources of inflammatory mediators and oxidative/nitrosative stress in schizophrenia [43], although some controversy still exists. Activated M1 microglia produce prostaglandins, chemokines, cytokines, complement proteins, proteinases, reactive oxygen species, and reactive nitrogen species, the sustained production of which can have a deleterious effect on susceptible cell populations by enhancing oxidative stress and activating cell death pathways through stimulation of kinases and caspase cascades [43]. Postmortem evidence is growing: brain microglial activation has been suggested in postmortem and positron emission tomography studies using (R)-[11C]PK11195, a ligand that recognizes the translocator protein [44–47]. Other postmortem studies have found increased numbers and structural degenerative impairments of human leukocyte antigen–antigen D related+ microglia in schizophrenia [48–50]. There is also some evidence from schizophrenia animal models showing progressive microglial activation, increased inducible nitric oxide synthase expression, and oxidative/nitrosative stress markers in the offspring of rats exposed to the viral mimetic polyriboinosinic-polyribocytidylic acid during pregnancy, which are prevented by the administration of clozapine [51]. In addition, some antipsychotics inhibit the release of nitric oxide and cytokines from activated microglial cells, possibly through the suppression of [Ca2+]i elevation in microglial cells [43, 52]. Thus, there is a growing perception of the role of microglia in the pathophysiology of schizophrenia and other stress-related psychiatric diseases, as well as the regulatory role of antipsychotics on microglia activation and possibly polarization to other anti-inflammatory profiles in different states of the natural course of the disease [41].
Considering the natural evolution of the disease and the existence of different subgroups of patients with diverse grades of inflammation and diverse grades of response to current treatments, the use of an alternative and complementary experimental design where antipsychotics were given to animals after stress exposure, with pre-existing or differing levels of inflammation, could increase the clinical relevance of our results. In addition, correlation of the molecular effects elicited by paliperidone with behavioral deficits affected by the restraint stress needs to be studied in future complementary studies. In this vein, chronic treatment with risperidone (0.1 mg/kg) improved stress-induced behavioral perturbations such as memory impairment and anxiety-like behavior in elevated plus maze and Y-maze tests in an animal model of post-traumatic stress disorder, based on the exposure to a stress–re-stress protocol (restraint stress + forced swimming) [53].
The paliperidone levels achieved in our chronic stress protocol suggest that the antioxidant/anti-inflammatory effects reported here could be due to an intermittent or subclinical dose of paliperidone and not directly related to antagonism of D2 receptors. In this vein, other authors have found neuroprotective effects of a subclinical dose of risperidone in a rodent model of prenatal infection [54]. This could have relevance for the off-label use of paliperidone in specific populations at doses that are below those required for full blockade of positive symptoms.
In conclusion, the therapeutic benefits of paliperidone could also be related to its ability to regulate positively endogenous antioxidant and anti-inflammatory pathways; thus, its proper administration in time and dose, and possible the use of adjuvant compounds that enhance these and other homeostatic pathways, could result in a better management of psychotic and other stress-related psychiatric pathologies.
References
Garcia-Bueno B, Caso JR, Leza JC. Stress as a neuroinflammatory condition in brain: damaging and protective mechanisms. Neurosci Biobehav Rev 2008;32:1136–1151.
Garate I, Garcia-Bueno B, Madrigal JL, et al. Stress-induced neuroinflammation: role of the Toll-like receptor-4 pathway. Biol Psychiatry 2013;73:32–43.
Radley J, Morilak D, Viau V, Campeau S. Chronic stress and brain plasticity: mechanisms underlying adaptive and maladaptive changes and implications for stress-related CNS disorders. Neurosci Biobehav Rev 2015;58:79–91.
Bremner JD, Randall P, Scott TM, et al. MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatry 1995;152:973–981.
Scher JU, Pillinger MH. 15d-PGJ2: the anti-inflammatory prostaglandin? Clin Immunol 2005;114:100–109.
Feinstein DL. Therapeutic potential of peroxisome proliferator-activated receptor agonists for neurological disease. Diabetes Technol Ther 2003;5:67–73.
Garcia-Bueno B, Caso JR, Perez-Nievas BG, Lorenzo P, Leza JC. Effects of peroxisome proliferator-activated receptor gamma agonists on brain glucose and glutamate transporters after stress in rats. Neuropsychopharmacology 2007;32:1251–1260.
Garcia-Bueno B, Madrigal JL, Lizasoain I, Moro MA, Lorenzo P, Leza JC. Peroxisome proliferator-activated receptor gamma activation decreases neuroinflammation in brain after stress in rats. Biol Psychiatry 2005;57:885–894.
Haskew-Layton RE, Payappilly JB, Xu H, Bennett SA, Ratan RR. 15-Deoxy-Delta12,14-prostaglandin J2 (15d-PGJ2) protects neurons from oxidative death via an Nrf2 astrocyte-specific mechanism independent of PPARgamma. J Neurochem 2013;124:536–547.
Zenkov NK, Menshchikova EB, Tkachev VO. Keap1/Nrf2/ARE redox-sensitive signaling system as a pharmacological target. Biochemistry (Mosc) 2013;78:19–36.
van Muiswinkel FL, Kuiperij HB. The Nrf2-ARE Signalling pathway: promising drug target to combat oxidative stress in neurodegenerative disorders. Curr Drug Targets CNS Neurol Disord 2005;4:267–281.
Mandrekar-Colucci S, Karlo JC, Landreth GE. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer's disease. J Neurosci 2012;32:10117–10128.
Pisanu A, Lecca D, Mulas G, et al. Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-gamma agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson's disease. Neurobiol Dis 2014;71:280–291.
Li Z, Ma L, Kulesskaya N, Voikar V, Tian L. Microglia are polarized to M1 type in high-anxiety inbred mice in response to lipopolysaccharide challenge. Brain Behav Immun 2014;38:237–248.
Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol 2013;39:3–18.
Ekdahl CT. Microglial activation—tuning and pruning adult neurogenesis. Front Pharmacol 2012;3:41.
Hu X, Leak RK, Shi Y, et al. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol 2015;11:56–64.
Reus GZ, Fries GR, Stertz L, et al. The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 2015;300:141–154.
Franco R, Fernandez-Suarez D. Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol 2015;131:65–86.
Kirkpatrick B, Miller BJ. Inflammation and schizophrenia. Schizophr Bull 2013;39:1174–1179.
Leza JC, Bueno B, Bioque M, et al. Inflammation in schizophrenia: A question of balance. Neurosci Biobehav Rev 2015;55:612–626.
Sommer IE, de WL, Begemann M, Kahn RS. Nonsteroidal anti-inflammatory drugs in schizophrenia: ready for practice or a good start? A meta-analysis. J Clin Psychiatry 2012;73:414–419.
Leza JC, Salas E, Sawicki G, Russell JC, Radomski MW. The effects of stress on homeostasis in JCR-LA-cp rats: the role of nitric oxide. J Pharmacol Exp Ther 1998;286:1397–1403.
Madrigal JL, Olivenza R, Moro MA, et al. Glutathione depletion, lipid peroxidation and mitochondrial dysfunction are induced by chronic stress in rat brain. Neuropsychopharmacology 2001;24:420–429.
Macdowell KS, Caso JR, Martin-Hernandez D, Madrigal JI, Leza JC, Garcia-Bueno B. Paliperidone prevents brain toll-like receptor 4 pathway activation and neuroinflammation in rat models of acute and chronic restraint stress. Int J Neuropsychopharmacol 2014;18:pii:oyu070.
Corena-McLeod MP, Oliveros A, Charlesworth C, et al. Paliperidone as a mood stabilizer: a pre-frontal cortex synaptoneurosomal proteomics comparison with lithium and valproic acid after chronic treatment reveals similarities in protein expression. Brain Res 2008;1233:8–19.
Remmerie BM, Sips LL, de Vries R, et al. Validated method for the determination of risperidone and 9-hydroxyrisperidone in human plasma by liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2003;783:461–472.
Kapur S, VanderSpek SC, Brownlee BA, Nobrega JN. Antipsychotic dosing in preclinical models is often unrepresentative of the clinical condition: a suggested solution based on in vivo occupancy. J Pharmacol Exp Ther 2003;305:625–631.
Macdowell KS, Garcia-Bueno B, Madrigal JL, et al. Risperidone normalizes increased inflammatory parameters and restores anti-inflammatory pathways in a model of neuroinflammation. Int J Neuropsychopharmacol 2013;16:121–135.
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–254.
Richtand NM, Ahlbrand R, Horn P, Stanford K, Bronson SL, McNamara RK. Effects of risperidone and paliperidone pre-treatment on locomotor response following prenatal immune activation. J Psychiatr Res 2011;45:1194–1201.
McNamara RK, Jandacek R, Rider T, Tso P, Cole-Strauss A, Lipton JW. Differential effects of antipsychotic medications on polyunsaturated fatty acid biosynthesis in rats: Relationship with liver delta6-desaturase expression. Schizophr Res 2011;129:57–65.
Djordjevic J, Djordjevic A, Adzic M, Mitic M, Lukic I, Radojcic MB. Alterations in the Nrf2-Keap1 signaling pathway and its downstream target genes in rat brain under stress. Brain Res 2015;1602:20–31.
Dhakshinamoorthy S, Jaiswal AK. Functional characterization and role of INrf2 in antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene 2001;20:3906–3917.
Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 1997;236:313–322.
Nair S, Doh ST, Chan JY, Kong AN, Cai L. Regulatory potential for concerted modulation of Nrf2- and Nfkb1-mediated gene expression in inflammation and carcinogenesis. Br J Cancer 2008;99:2070–2082.
Liu GH, Qu J, Shen X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim Biophys Acta 2008;1783:713–727.
Oh JY, Giles N, Landar A, Darley-Usmar V. Accumulation of 15-deoxy-delta(12,14)-prostaglandin J2 adduct formation with Keap1 over time: effects on potency for intracellular antioxidant defence induction. Biochem J 2008;411:297–306.
Flatow J, Buckley P, Miller BJ. Meta-analysis of oxidative stress in schizophrenia. Biol Psychiatry 2013;74(6):400–409.
Cohen T, Sundaresh S, Levine F. Antipsychotics activate the TGFbeta pathway effector SMAD3. Mol Psychiatry 2013;18:347–357.
Nakagawa Y, Chiba K. Role of microglial m1/m2 polarization in relapse and remission of psychiatric disorders and diseases. Pharmaceuticals (Basel) 2014;7:1028–1048.
Maxeiner HG, Marion SE, Kurfiss ST, Brettschneider J, Tumani H, Bechter K. Cerebrospinal fluid and serum cytokine profiling to detect immune control of infectious and inflammatory neurological and psychiatric diseases. Cytokine 2014;69:62–67.
Monji A, Kato TA, Mizoguchi Y, et al. Neuroinflammation in schizophrenia especially focused on the role of microglia. Prog Neuropsychopharmacol Biol Psychiatry 2013;42:115–121.
Busse S, Busse M, Schiltz K, et al. Different distribution patterns of lymphocytes and microglia in the hippocampus of patients with residual versus paranoid schizophrenia: further evidence for disease course-related immune alterations? Brain Behav Immun 2012;26:1273–1279.
Doorduin J, de Vries EF, Willemsen AT, de Groot JC, Dierckx RA, Klein HC. Neuroinflammation in schizophrenia-related psychosis: a PET study. J Nucl Med 2009;50:1801–1807.
van Berckel BN, Bossong MG, Boellaard R, et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry 2008;64:820–822.
Steiner J, Bielau H, Brisch R, et al. Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res 2008;42:151–157.
Steiner J, Mawrin C, Ziegeler A, et al. Distribution of HLA-DR-positive microglia in schizophrenia reflects impaired cerebral lateralization. Acta Neuropathol 2006;112:305–316.
Wierzba-Bobrowicz T, Lewandowska E, Lechowicz W, Stepien T, Pasennik E. Quantitative analysis of activated microglia, ramified and damage of processes in the frontal and temporal lobes of chronic schizophrenics. Folia Neuropathol 2005;43:81–89.
Radewicz K, Garey LJ, Gentleman SM, Reynolds R. Increase in HLA-DR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenics. J Neuropathol Exp Neurol 2000;59:137–150.
Ribeiro BM, do Carmo MR, Freire RS, et al. Evidences for a progressive microglial activation and increase in iNOS expression in rats submitted to a neurodevelopmental model of schizophrenia: reversal by clozapine. Schizophr Res 2013;151:12–19.
Mizoguchi Y, Kato TA, Horikawa H, Monji A. Microglial intracellular Ca(2+) signaling as a target of antipsychotic actions for the treatment of schizophrenia. Front Cell Neurosci 2014;8:370.
Krishnamurthy S, Garabadu D, Joy KP. Risperidone ameliorates post-traumatic stress disorder-like symptoms in modified stress re-stress model. Neuropharmacology 2013;75:62–77.
Piontkewitz Y, Arad M, Weiner I. Risperidone administered during asymptomatic period of adolescence prevents the emergence of brain structural pathology and behavioral abnormalities in an animal model of schizophrenia. Schizophr Bull 2011;37:1257–1269.
Acknowledgments
This work was supported by the Spanish Ministry of Science and Innovation (SAF07-63138), the Instituto de Salud Carlos III (FIS 10/00123 & 13/1102), the Centro de Investigación en Red de Salud Mental, CIBERSAM, Foundation Santander-UCM (GR 58/08), and the Fundación Mutua Madrileña 2011. B.G.B. is a Ramón y Cajal post-doctoral fellow (MEC). We thank the Facility of Innovative Technologies in Clinical Sciences Fc, Medicine, Complutense Univeristy (F. Bandrés) for the help in determining paliperidone levels by liquid chromatography tandem mass spectrometry.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Rights and permissions
About this article

Cite this article
MacDowell, K.S., Caso, J.R., Martín-Hernández, D. et al. The Atypical Antipsychotic Paliperidone Regulates Endogenous Antioxidant/Anti-Inflammatory Pathways in Rat Models of Acute and Chronic Restraint Stress. Neurotherapeutics 13, 833–843 (2016). https://doi.org/10.1007/s13311-016-0438-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-016-0438-2