
Summary
Injury to the spinal cord is known to result in inflammation. To date, the preponderance of research has focused on the acute neuroinflammatory response, which begins immediately and is believed to terminate within hours to (at most) days after the injury. However, recent studies have demonstrated that postinjury inflammation is not restricted to the first few hours or days after injury, but can last for months to years after a spinal cord injury (SCI). These chronic studies have revealed that increased numbers of inflammatory cells, such as microglia and macrophages, and inflammatory factors, including cytokines, chemokines, and enzyme products are found at markedly delayed times after injury. Here we review experimental work on a selection of the novel inflammatory factors observed chronically after SCI, including the nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) oxidase enzyme and galectin-3. We will discuss the role of these proteins in inflammation with regard to both detrimental and beneficial effects of neuroinflammation after injury. Finally, the potential of these proteins to serve as therapeutic targets will be considered, and a novel therapeutic approach (i.e., the agonist for metabotropic glutamate receptor 5 [mGluR5], [RS]-2-Chloro-5-hydroxyphenylglycine [CHPG]) will be discussed. This review will demonstrate the expression and activity profiles, roles in potentiation of injury, and therapy studies of these inflammatory factors suggest that not only are these chronically expressed factors viable targets for SCI treatment, but that the therapeutic window is broader than has previously been thought.
Similar content being viewed by others

INTRODUCTION
Spinal cord injury (SCI) is initially caused by a mechanical insult promoting secondary biochemical and physiological changes. These changes contribute to permanent loss of sensory and motor function and lead to delayed cell death, glial scar formation, impaired regeneration, and neuroinflammation. However, SCI is not a static disorder, and evidence exists demonstrating progressive tissue loss with time, including chronic demyelination [1] and syringomyelia development [2]. The inflammation of the nervous system may serve as the mechanism by which SCI progresses from an acute to a chronic disorder. The immediate and ephemeral inflammation, occurring within the first hours to days after injury, is the acute stage. Chronic neuroinflammation, on the other hand, is a relatively new concept in the field, supported by findings of elevated inflammatory cell numbers in the injured spinal cord months to years after the initial insult [3, 4]. Overall, neuroinflammation involves the activation and recruitment of endogenous and systemic immune cells, including microglia, macrophages, and lymphocytes, and the expression of factors designed to respond to the injury and aid in repair (see Alexander and Popovich [5] for a full review). Microglia are the resident immune cells in the central nervous system and play an integral role in the neuroinflammatory response after SCI (see Loane and Byrnes [6] review).
To date, it is unclear if chronic neuroinflammation is deleterious or beneficial, and the current research is controversial. Studies have found that inhibition of inflammation or knockout of an inflammatory pathway can both improve [7] and deter [3] recovery. In fact, inflammatory cells themselves can produce both neurotoxic and neuroprotective substances [3]. A careful consideration of this dual role of inflammation will be given within this review.
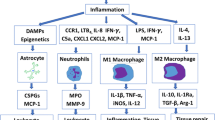
A number of inflammatory factors are involved in the response to injury of the spinal cord; most are known to be up-regulated acutely after injury and remain elevated or active for only a few hours [8]. However, a number of chronically up-regulated neuroinflammatory factors, including nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) oxidase and galectin-3, among others, have been identified in recent years. A notable finding for many of these factors is that they play a significant role in both potentiating the inflammatory response and contributing to resolution of inflammation. However, many of these factors have not been studied in detail with regard to SCI, nor have they been fully considered as therapeutic targets for SCI treatment. However, one therapy has been shown to target both NADPH oxidase and galectin-3, the metabotropic glutamate receptor 5 (mGluR5) agonist (RS)-2-Chloro-5-hydroxyphenylglycine (CHPG). Therefore, we now provide a detailed review of these chronic inflammatory factors, including a consideration of their role in inflammation and in SCIs, and the use of the novel anti-inflammatory mGluR5 agonist as a therapy to target these factors (FIG. 1).

Diagram of the the input of the novel chronic neuroinflammatory factors NADPH oxidase and galectin-3 on cell death, and the input of the novel anti-inflammatory treatment (RS)-2-Chloro-5-hydroxyphenylglycine (CHPG). Trauma results in microglial activation, including NADPH oxidase activity and galectin-3 expression. These proteins then act to both enhance inflammation and neurotoxicity, as well as promote their own expression and activity. The metabotropic glutamate receptor 5 (mGluR5) agonist CHPG has activities to block this neuroinflammation and may operate as a viable anti-inflammatory agent for chronic inflammation. (IL = interleukin; TGF = transforming growth factor; TNF = tumor necrosis factor.)
NADPH OXIDASE
Components of the NADPH oxidase enzyme and evidence of oxidative stress have been reported to be increased after SCI and remain elevated for chronic periods [9]. The NADPH oxidase enzyme is a protein complex that plays a key role in the production of reactive oxygen species (ROS) and is composed of 4 cytosolic subunits (gp40PHOX, p47PHOX, p67PHOX, and the GTP-binding protein p21-Rac1) and 2 membrane subunits (gp91PHOX and p22PHOX) (FIG. 2) [10]. There are several isoforms of NADPH oxidase, differing based on their enzymatic core. For NOX2, which is expressed in most phagocytic cells, such as microglia, the core is gp91PHOX [11]. Alternative forms include NOX1, NOX3, NOX4, NOX5, DUOX1, and DUOX2, and differ in their requirements for cytosolic and membrane subunits. In addition to NOX2, microglia have been shown to express functional NOX1 [12] and NOX4 [13]. Furthermore, NOX1, NOX2, and NOX4 are expressed in neurons, astrocytes, endothelial cells, pericytes and fibroblasts, among other cells [14, 15]. For the purpose of this review, we will focus on NOX2, but some of the research and therapy approaches may overlap with NOX1 and NOX4.

Diagram of the mechanism of NADPH oxidase-induced cell death. NADPH oxidase activation by aggregation of all components to the cell membrane results in production of superoxide (O2 -). Superoxide can then combine with nitric oxide (NO) to produce peroxynitrite (ONOO-), which interferes within mitochondrial activity and can induce cell death. Superoxide can also interact with the cell membrane to produce reactive aldehydes, which also induce cell death. ATP = adenosine triphosphate.
Activation of the NOX2 complex involves 2 steps: 1) up-regulation of expression of the individual protein components and 2) protein kinase C (PKC)- or mitogen activated protein kinase-mediated phosphorylation of the cytosolic components [16]. Phosphorylated subunits are then translocated to the membrane, where they assemble with the membrane subunits to form the active enzyme [17–20]. The active enzyme complex produces ROS, such as superoxide (O -2 ), which is converted into other chemically active superoxide-derived oxidants, such as hydrogen peroxide (H2O2), hydroxyl radicals, and peroxynitrite [21] (FIG. 2).
A time course evaluation reveals that at 6, 24, and 72 h postinjury, gp91PHOX and p67PHOX are significantly up-regulated into the spinal cord tissue [22, 23]. This increase is further elevated at 14 days postinjury, which is accompanied by an increase in the amount of nitrosylated tyrosine, a marker of oxidative stress [22]. Our group found that components of the NADPH oxidase enzyme system (including p22PHOX) are significantly up-regulated from 24 h to 7 days postinjury [24]. This up-regulation continues for at least 6 months after a spinal cord contusion injury in mice (FIG. 3). In human tissue, gp91PHOX-positive neutrophils have been identified in the lesion perimeter at 1 to 3 days postinjury, whereas microglia in the spinal cord are gp91PHOX-positive for months after injury [4]. From 5 days to several months postinjury, a portion of the phagocytic macrophages in the injured spinal cord were gp91PHOX-positive, although the majority of phagocytic cells were not. Interestingly, although neurons are known to also express some NADPH oxidase components, at 1 week after SCI it was found that most of the NOX2-positive cells in the injured spinal cord were Iba1+ microglia/macrophages rather than MAP2+ neurons [25].

Gene expression of the novel neuroinflammatory targets after spinal cord injury (SCI). Microarray data from http://pepr.cnmcresearch.org was mined to determine the expression of (a) galectin-3, (b) p22PHOX, and (c) gp91PHOX. Expression is significantly elevated, chronically after moderate SCI in mice for each marker in comparison with sham-injured tissue. (d) Sham-injured tissue and spinal cord-injured tissue stained with gp91 (red) and ToPro (blue) at 28 days after a moderate contusion injury in mice indicates elevation of this protein corresponding to the gene expression measurements. (Bar = 200 μm).
Various cytokines can induce the expression of NADPH oxidase components and activation of the enzyme. For example, TNFα and IFNγ can operate through PKC and the NFκB transcription factor to induce gp91PHOX expression [26, 27]. T-cell derived cytokines, such as interleukin-17A, can also induce NADPH oxidase activity [28]. Cytosolic phospholipase A2, which is induced in several injury models, may also induce NADPH oxidase activity by directly binding to the enzyme [29]. In turn, NADPH oxidase activity can induce NFκB activation [27], resulting in a self-propagating feedback loop.
In vitro experiments have shown that activated microglia can induce neuronal cell death, mediated through the NADPH oxidase enzyme. Knockout of gp91PHOX reduced neuronal apoptosis in response to injection of lipopolysaccharide (LPS, which is a bacterial cell wall component commonly used to initiate inflammatory cell activation) [30]. As neurons do not respond to LPS, it was proposed that this neuronal apoptosis resulted from indirect actions of microglia. A recent study by Hur et al. [31] further demonstrated that microglia exposed to ischemia induced neuronal cell apoptosis, which could be blocked by knocking out gp91PHOX. Furthermore, addition of TNFα and interleukin-1β to spinal cord explants resulted in NADPH oxidase activity and 3-nitrotyrosine (3-NT) accumulation in spinal cord motoneurons, but only when microglia were present [26].
T cells may also play a role in NADPH oxidase-mediated damage to the spinal cord. T cells produce cytokines that contribute to NADPH oxidase activation [28]. In addition, these cells have been shown to have increased proliferation in response to NADPH oxidase-derived superoxide [32]. As T cells have a delayed and potentially sustained presence in the injured spinal cord [33], this contribution to NADPH oxidase activity may play a significant role in potentiating the inflammatory response and in myelin debris clearance.
NADPH oxidase-dependent ROS production has a number of toxic effects on cells. The interaction of ROS and reactive nitrogen results in the formation of the highly reactive and toxic peroxynitrite (FIG. 2). Peroxynitrite can nitrosylate tyrosine residues [34]; the resultant 3-NT has been shown to be directly neurotoxic. Studies have shown that peroxynitrite markers, such as 3-NT, are up-regulated for at least 1 week after SCI [35]. 3-NT can inhibit mitochondrial activity, reduce adenosine triphosphate (ATP) production, and increase ROS production [36]. ROS can also interact with polyunsaturated fatty acids in cell membranes, producing reactive aldehydes that can bind to and interfere in normal protein function, causing additional toxicity [23].
Neurons are not the only cell type susceptible to inflammatory NADPH oxidase production in the spinal cord. Microglial NADPH oxidase activity and ROS generation has also been shown to induce oligodendrocyte cell death, which can have devastating consequences in SCI. Oligodendrocytes are responsible for myelination of axons in the spinal cord. Oligodendrocyte or oligodendrocyte precursor cell death results in a reduction of remyelination of newly sprouting or spared axons near the injury site, impairing recovery of function. Oligodendrocyte precursors have been shown to be susceptible to microglial-induce peroxynitrite production. LPS-stimulated microglia that are co-incubated with oligodendrocyte precursors can significantly reduce oligodendrocyte cell survival [37]. Endothelial cells, and thus blood-brain barrier (BBB) integrity, is also influenced by ROS production. An in vitro study with endothelial cells showed that increased exposure of endothelial cells to ROS resulted in an increased contractile function of the endothelial cells and increased adhesion molecules expression [28]. This effect can in turn induce an increase in cellular migration into the spinal cord, and can thus further the inflammatory response.
SCI can induce systemic inflammatory responses and changes in NADPH oxidase activity. The oxidative activity of cells in the peripheral blood is also altered by SCI. For example, Bao et al. [38] found that the NADPH oxidase activity of circulating monocytes, neutrophils, and lymphocytes was 4- to 6-fold higher in SCI-injured patients than in trauma control patients at 6 h to 2 weeks postinjury. ROS produced by NADPH oxidase may also play a role in postinjury neuropathic pain. A recent study by Kim et al. [25] demonstrated that postinjury allodynia is associated with an increase in microglial related NADPH oxidase component expression; pain could be alleviated by the addition of oxygen scavengers.
There are several inhibitors of NADPH oxidase, although most tend to be nonspecific and have multiple off-target effects, rendering much of the chemical-inhibition based research controversial. The most commonly used inhibitors include diphenyleneiodonium (DPI) apocynin [11]. DPI directly inhibits all flavoenzymes, including nitric oxide synthase [37, 39]. DPI also inhibits all NOX isoforms and is believed to operate by modifying the heme component of NADPH oxidase, disrupting the ability of the enzyme to generate oxygen radicals [40, 41]. In vitro, reduction of NADPH oxidase activity can ameliorate microglial activation and reduce neuronal cell death. In microglia, DPI blocks NFκB activation, which reduces inducible nitric oxide synthase and cytokine production [42]. Inhibition of NADPH oxidase with DPI impairs peroxynitrite production and suppresses microglial-induced oligodendrocyte precursor cell death [37]. Furthermore, DPI treatment reduces endothelial cell adhesion molecule expression, leading to a reduction in permeability to circulating cells and improved BBB integrity [28]. One recent study has found that femtomolar concentrations of DPI inhibit NADPH oxidase in microglia, and reduce LPS-induced TNFα release and nitric oxide production [16]. Furthermore, this study found that LPS-induced microglial neurotoxicity could be blocked by DPI [16]. These effects were found to be preceded by a reduction in extracellular signal-related kinase (ERK) phosphorylation, suggesting that ultra-low DPI concentrations may act on the upstream events of NADPH oxidase activity (in addition to or instead of it) through direct activity at the NADPH oxidase enzyme.
Apocynin is a BBB permeable drug that reduces NADPH oxidase activity by inhibiting the membrane translocation of p47PHOX [43]. This drug may also have nonspecific ROS scavenging effects [11]. Application of apocynin inhibits microglial proliferation [24], cytokine production [12], ROS production [44], and microglial-mediate neurotoxicity [45]. Pre-treatment of microglia with apocynin prior to exposure to ischemic conditions or LPS in vitro blocks microglial-induced neurotoxicity [31, 46]. In vivo, using a brain ischemia model, apocynin at 2.5 mg/kg significantly reduced the infarct size and improved functional recovery [43]. It should be noted that apocynin at higher concentrations (3.75 and 5 mg/kg) could induce an increase in oxidative stress and impair recovery.
Despite the benefits observed with inhibition or knockout of NADPH oxidase, it is important to remember that NADPH oxidase can have beneficial effects in the injured spinal cord. For example, phagocytic activity is heavily influenced by ROS activity. In 2008, a study by Sun et al. [47] demonstrated that antioxidant treatment or apocynin administration, which reduced the ROS produced by microglia exposed to LPS, reduced microglial phagocytic activity. Microglial and macrophage activation are essential in myelin debris phagocytosis, which is important in Wallerian degeneration and the potential for regeneration [48]. Further, phagocytosis of myelin has been shown to induce an ROS burst in microglia that is related to a reduction in pro-inflammatory cytokine production [49]. In addition, Kunz et al. [50] demonstrated that NADPH oxidase activity played an essential role in cerebrovascular regulation, normalizing blood flow after ischemia via activity of peroxynitrite. Of course, it is also important to remember that NADPH oxidase-mediated ROS production is essential in eliminating microbial or other targets in elimination of debris.
These data suggest that NADPH oxidase generated ROS play a significant role in the alleviation of inflammatory response, as well as propagation of it, and future research needs to be done to be more fully understanding of the timing and related signals in this pathway.
GALECTIN-3
Galectin-3, also known as MAC-2, is a 30 kDa member of the lectins family that binds β-galactosides. All galectins contain conserved carbohydrate-recognition domains (CRDs) involved in carbohydrate binding. Based on structural features and the number of CRDs, galectins are classified as proto (one CRD), tandem-repeat-types (two homologous CRD), or chimera. Galectin-3 is the only galectin of the chimera type with a CRD that is connected to a nonlectin N-terminal [51, 52].
Galectin-3 can be localized within tissues in the extracellular, cytoplasmic, or nuclear compartments in monomeric or oligomeric forms [53]. Galectins lack of a signal sequence required for secretion through the classical secretory pathway [51, 54]; however, galectin-3 may be able to penetrate the lipid bilayer of liposomes in an energy-independent manner to be released into the extracellular space [55].
Galectin-3 appears to play a major role in control of inflammation. Galectin-3 gene and protein expression have been shown to increase in a mouse model for neonatal hypoxia-ischemia with galectin-3 found in activated microglia/macrophages, suggesting a role for galectin-3 in the inflammatory response [56]. Furthermore, measurements of galectin-3 in cerebrospinal fluid samples of amyotrophic lateral sclerosis patients have shown to be approximately twice as much as normal and disease controls [57]. Its inhibition has been shown to have anti-inflammatory properties in conditions, such as rheumatoid arthritis [58]. It has been hypothesized that galectin-3 is involved in the transition of a repetitive injury to chronic inflammation by facilitating the scarring processes and tissue fibrogenesis [53].
Galectin-3 expression is under the control of the AP1 transcription factor and NFκB, a proinflammatory transcription factor [59], and it is involved in monocyte chemotaxis [60], activation of NADPH oxidase [61, 62] , phagocytosis [63], and ROS release [64] (FIG. 1). NFκB has been shown to be activated after SCI as early as 30 minutes after injury [65], and its activation is an important step in the development of inflammation after SCI [66].
Gene expression of galectin-3 is delayed and prolonged after SCI, with an induction at 24 h after SCI, and a peak in expression between 14 and 28 days [24, 67]. We have found that galectin-3 mRNA expression after moderate SCI in mice remains at a level higher than sham through 6 months postinjury (FIG. 3). It has been hypothesized that galectin-3 is involved in the transition to chronic inflammation by facilitating the scarring processes and tissue fibrogenesis [53]. In a focal ischemia model, galectin-3 was up-regulated in OX-42+ microglia for up to 2 months after injury [68]. Galectin-3 was shown to be increased in the CA1 region of the hippocampus following transient ischemia [69]. In addition to macrophages and microglia, T cells have been reported to express galectin-3 [70].
Inhibition of galectin-3 has anti-inflammatory properties in a number of models and cell culture conditions, although SCI has not been tested yet. Several inhibitors of galectin-3 activity have been found, including lactose and modified citrus pectin. Modified citrus pectin, a galactose-rich polysaccharide that binds to galectin-3, has been found to reduce inflammation in rheumatoid arthritis [58]. Lactose incubation with neutrophils blocked galectin-3 mediated ROS production [64]. Furthermore, knockout of galectin-3 reduced tissue loss and measures of oxidative stress and inflammation in a neonatal hypoxia model [71], and reduced inflammatory cell presence in a thioglycollate broth stimulation [72].
This protein is involved in several other biological activities aside from inflammation, including regulation of cellular cycle, modulation of adhesion and tumor progression, and metastasis [73]; these activities can be modulated by collagenase and matrix metalloproteinases. Interestingly, metalloproteinases are also chronically up-regulated after SCI [74, 75]. However, these alternative activities and a recent study by Lalancette-Hebert et al. [76] demonstrate that galectin-3 may play a protective role after SCI. In this study, microglia expressing galectin-3 were selectively ablated, resulting in worsened recovery and larger infarct volume after an ischemic injury. In addition, galectin-3 is a mediator of the vascular endothelial growth factor-mediated angiogenic response and basic fibroblast growth factor-mediated angiogenic response. Angiogenesis assays revealed that galectin-3 inhibitors and knockout reduce vascular endothelial growth factor-mediated angiogenesis and basic fibroblast growth factor-mediated angiogenesis in vitro [77]. Therefore, treatment studies targeting galectin-3 require greater understanding of the role of this protein in both inflammation and angiogenesis.
NOVEL THERAPY: mGluR5 AGONIST CHPG
Within the central nervous system, metabotropic glutamate receptors (mGluRs) are well known to be expressed in many different cell types throughout the brain and spinal cord [78]. These receptors are G-protein coupled receptors that have been divided into 3 groups (groups I, II, and III) on the basis of signal transduction pathways and pharmacological profiles [79]. Briefly, group I mGluRs, including mGluR1 and 5, are typically postsynaptic and work through Gαq-proteins to release calcium and activate ERK1 and ERK2 downstream signaling pathways. Groups II (mGluR2 and 3) and III (mGluR4, 6, 7, 8), on the other hand, are typically presynaptic and are negatively coupled to adenylate cyclase to reduce calcium increases. Recent work has shown that mGluRs are expressed in several members of the immune system. Microglia, for example, express mGluR2, 4, 5, 6, and 8 [78, 80]. We have shown that mGluR5 is expressed in microglia, in vitro, in a SCI rodent model, and in the BV2 microglial cell line [81–83]. Lymphocytes also express groups I, II, and III mGluRs [84]; activation of mGluR group III with LAP4 induces an increase in ROS levels in these cells. In 2009, we presented a discussion of the role of mGluRs in neuroinflammation [79]; in this section we will expand on the research into mGluR involvement in neuroinflammation after SCI and the use of the mGluR5 agonist CHPG as a therapy.
Glutamate levels in the spinal cord peak at approximately 2 h after injury [85]. Although SCI does not induce a change in mGluR5, 3 or 4 gene expression in the lesion site from 30 minutes to 28 days postinjury, a slight reduction in mGluR5 expression at the lesion epicenter was found using Western blot at 24-h postinjury [86]. In addition, mGluR2 is increased at 7 days postinjury with mild injury [86]. Away from the lesion epicenter, studies have found changes in mGluR expression, with mGluR1 expression reduced rostral and caudal to the lesion at 1 day postinjury, and a reduction in mGluR2/3 from 7 to 60 days postinjury [86]. Gwak and Hulsebosch [87] found that mGluR1 and 5 proteins were increased in neurons and astrocytes for as much as 30 days after SCI, although they did not investigate possible microglial expression.
The expression and function of mGluR5 has been explored in microglia and in SCI, using both in vitro and in vivo models (Table 1). Blockade of group I mGluR with the antagonist AIDA in a neuronal/glial co-culture stretch-injury model resulted in reduced axonal signaling abnormalities and GFAP immunoreactivity [88]. Furthermore, N-methyl-D-aspartic acid-mediated currents were reduced in this culture model with mGluR5 agonist (RS)-2-Chloro-5-hydroxyphenylglycine (CHPG) administration, but only when glia were present [89]. While the exact contribution of different glial constituents was not clarified, it is possible that some of these neuroprotective effects were mediated by changes in microglial activity. Finally, in 2009, we showed that spinal cord and brain microglia exposed to the CHPG reduced their responses to LPS, including reductions in nitric oxide and ROS production [81–83] (FIG. 1). These effects could be reversed by knockout of the mGluR5 receptor or by addition of the mGluR5 antagonist 3-((2-Methyl-4-thiazolyl)ethynyl)pyridine (MTEP). Microglial-mediated neurotoxicity was also reduced by pre-treatment of microglia with CHPG in both primary microglia and the murine microglial cell line BV-2. A replication of these studies using the nonspecific mGluR group I agonist (S)-3,5-Dihydroxyphenylglycine (DHPG) demonstrated a reduction in LPS-induced microglial nitric oxide, TNFα, and glutamate release, as well as a reduction in the number of “activated microglia,” as measured by morphology [90].
Research into mGluR agonists and antagonists in vivo is limited. In a rat model of global cerebral ischemia, infusion of CHPG reduced infarct volume [91]. Alternatively, after a spinal cord contusion, the generic group I antagonists AIDA and LY367385 improved acute motor function performance and increased gray and white matter sparing, whereas the specific mGluR5 antagonist MPEP, had no effect on either function or white matter sparing [92]. Therefore, it is likely that the beneficial effects were mediated not by actions on mGluR5, but on mGluR1. This is supported by the finding that mGluR5 activation can improve recovery after SCI [81]. Infusion of the mGluR5 agonist CHPG for 7 days beginning 30 minutes after injury resulted in significant improvements in locomotor function performance, as well as reductions in lesion volume and increases in white matter sparing. These changes were potentially mediated by alleviation of inflammation, as measurements of proinflammatory cytokines and numbers of microglia were significantly reduced with CHPG treatment.
Importantly, mGluR5 agonist administration in vitro and in vivo resulted in reductions in galectin-3 and NADPH oxidase component expression [81, 82]. Furthermore, the effect of mGluR activity in both microglia and in SCI may be through an NADPH oxidase enzyme connection. Administration of the mGluR1 antagonist LY367385, which improves function after SCI [92], reduces expression of p67PHOX and gp91PHOX after transient cortical ischemia [93]. This was accompanied by a decrease in NADPH oxidase activity [93], which may be a result of less PKCδ activity; PKC can phosphorylate p47PHOX and induce its translocation to the membrane where it can interact with the other members of the NADPH oxidase enzyme and induce activity. Similarly, the mGluR5 agonist CHPG reduced p22PHOX and gp91PHOX expression, as well as NADPH oxidase activity in microglial cultures [83]. In addition, this study demonstrated that knockdown of NADPH oxidase components p22PHOX and gp91PHOX significantly reduced the effectiveness of CHPG in reducing LPS-induced microglial activation, suggesting that mGluR5 agonists affect microglial activity through the NADPH oxidase enzyme. In our study of SCI treatment with CHPG, both p22PHOX and gp91PHOX were significantly suppressed by treatment [81].
However, mGluR5 activation may play a role in neuropathic pain. Studies have shown that administration of CHPG to naïve animals results in cold hypersensitivity [94], and that administration of the mGluR5 antagonist MPEP can reduce allodynia and hyperalgesia [95, 96]. Although no evidence of pain was observed in the SCI study following CHPG administration [81], pain was not specifically tested for in the study. However, as pain may be mediated by microglial activation [97, 98], and mGluR5 agonist administration was found to reduce microglial activity, CHPG administration after SCI may actually reduce pain. Therefore, future studies of the effect of mGluR5 agonists on recovery after SCI must consider effects on pain and the contribution of microglial and neuronal responses to injury, and CHPG administration should be considered.
SUMMARY
There is now substantial evidence that inflammation continues for an extended period of time after SCI. This chronic inflammation includes the activation and infiltration of a number of inflammatory cells, including macrophages, T cells, B cells, and microglia. Microglia are the primary inflammatory response cell in the spinal cord; the factors previously identified have all been shown to be expressed on resting and activated microglia in the injured spinal cord [24, 81]. Furthermore, microglia have been shown to play both a neuroprotective and a neurotoxic role after injury (see review by Loane and Byrnes [6]). In fact, studies have shown that microglia, as with macrophages, have 2 separate phenotypes after injury: 1) M1, or pro-inflammatory and potentially neurotoxic, and 2) M2, or anti-inflammatory and protective [99]. Although this review does not focus on the expression of NADPH oxidase, galectin-3, or mGluR5 in M1 or M2 phenotypes, it is possible that these proteins play a role in microglial phenotypical characterization. However, we do discuss that the chronically expressed inflammatory factors may also play both a toxic and protective role, providing a mechanism by which microglia and other inflammatory cells have a dual purpose after injury. To date, M1:M2 identification in microglia has only been completed through 28 days postinjury, with the finding that M2 markers are not present at more delayed time points. Therefore, it is currently unknown which role is more prominent at more chronic time points after injury, or if any of these factors reviewed herein have a cellular dependence (i.e., if other cell types beyond microglia and macrophages play a significant role).
To date, the targets identified in this review have not been extensively studied as therapeutic targets for treating chronic SCI. Acute administration of the mGluR5 agonist has provided substantial improvement in recovery after SCI [81]; however chronic investigation of this therapy has not been investigated. Furthermore, a very recent study by Impellizzerri et al. [100] demonstrated an improvement in limb performance after acute administration of the NADPH oxidase inhibitor apocynin, but chronic administration has not been studied. Finally, no intervention strategy for galectin-3 in SCI has been completed to date. Nonetheless, the novel anti-inflammatory treatment of the mGluR5 agonist CHPG has been shown to affect both NADPH oxidase and galectin-3 expression after SCI and other disorders, and may have activities mediated through these mechanisms. However, chronic administration of CHPG has not been studied to date. Based on the chronic expression and activity profiles of the inflammatory-related proteins in this review, it is clear that more chronic administration time points should be considered. It is possible that treatment targeting neuroinflammation may be effective if delayed weeks and possibly months after the initial injury.
In conclusion, the expression and activity profiles, roles in potentiation of injury, and therapy studies of these inflammatory factors suggest that not only are these chronically expressed factors viable targets for SCI, but also there is a broader therapeutic window than has previously been thought possible. SCI is not a static disorder, and evidence exists demonstrating progressive tissue loss with a duration of time, including chronic demyelination [1] and syringomyelia development [2]. Inflammation can contribute to this chronic tissue loss and demyelination (see review by Loane and Byrnes [6]). However, it is important to remember that inflammation after SCI is complex and multifaceted. Therefore, future research must consider both the beneficial and detrimental effects of the target when using ablation or inhibitory treatments.
REFERENCES
Totoiu MO, Keirstead HS. Spinal cord injury is accompanied by chronic progressive demyelination. J Comp Neurol 2005;486:373–383.
Klekamp J. The pathophysiology of syringomyelia - historical overview and current concept. Acta Neurochir (Wien) 2002;144:649–664.
Beck KD, Nguyen HX, Galvan MD, Salazar DL, Woodruff TM, Anderson AJ. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain 2010;133(pt 2):433–447.
Fleming JC, Norenberg MD, Ramsay DA, et al. The cellular inflammatory response in human spinal cords after injury. Brain 2006;129(pt 12):3249–3269.
Alexander JK, Popovich PG. Neuroinflammation in spinal cord injury: therapeutic targets for neuroprotection and regeneration. Prog Brain Res 2009;175:125–137.
Loane DJ, Byrnes KR. Role of microglial activation in neurotrauma. Neurotherapeutics 2010;7(4):366–377.
Ma M, Wei P, Wei T, Ransohoff RM, Jakeman LB. Enhanced axonal growth into a spinal cord contusion injury site in a strain of mouse (129X1/SvJ) with a diminished inflammatory response. J Comp Neurol 2004;474:469–486.
Carlson SL, Parrish ME, Springer JE, Doty K, Dossett L. Acute inflammatory response in spinal cord following impact injury. Exp Neurol 1998;151:77–88.
Lee YS, Sindhu RK, Lin CY, Ehdaie A, Lin VW, Vaziri ND. Effects of nerve graft on nitric oxide synthase, NAD(P)H oxidase, and antioxidant enzymes in chronic spinal cord injury. Free Radic Biol Med 2004;36:330–339.
Cross AR, Segal AW. The NADPH oxidase of professional phagocytes —prototype of the NOX electron transport chain systems. Biochim Biophys Acta 2004;1657:1–22.
Jaquet V, Scapozza L, Clark RA, Krause KH, Lambeth JD. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid Redox Signal 2009;11:2535–2352.
Cheret C, Gervais A, Lelli A, et al. Neurotoxic activation of microglia is promoted by a nox1-dependent NADPH oxidase. J Neurosci 2008;28:12039–12051.
Li B, Bedard K, Sorce S, Hinz B, Dubois-Dauphin M, Krause KH. NOX4 expression in human microglia leads to constitutive generation of reactive oxygen species and to constitutive IL-6 expression. J Innate Immun 2009;1:570–581.
Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med 2009;47:1239–1253.
Sorce S, Krause KH. NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal 2009;11(10):2481–2504.
Qian L, Gao X, Pei Z, et al. NADPH oxidase inhibitor DPI is neuroprotective at femtomolar concentrations through inhibition of microglia over-activation. Parkinsonism Relat Disord 2007;13(suppl 3):S316-S320.
Choi SH, Lee da Y, Kim SU, Jin BK. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: role of microglial NADPH oxidase. J Neurosci 2005;25:4082–4090.
Bey EA, Xu B, Bhattacharjee A, et al. Protein kinase C delta is required for p47phox phosphorylation and translocation in activated human monocytes. J Immunol 2004;173:5730–5738.
Zhao X, Xu B, Bhattacharjee A, et al. Protein kinase Cdelta regulates p67phox phosphorylation in human monocytes. J Leukoc Biol Suppl 2005;77:414–420.
El Benna J, Han J, Park JW, Schmid E, Ulevitch RJ, Babior BM. Activation of p38 in stimulated human neutrophils: phosphorylation of the oxidase component p47phox by p38 and ERK but not by JNK. Arch Biochem Biophys 1996;334:395–400.
Patel M, Li QY, Chang LY, Crapo J, Liang LP. Activation of NADPH oxidase and extracellular superoxide production in seizure-induced hippocampal damage. J Neurochem 2005;92:123–131.
Vaziri ND, Lee YS, Lin CY, Lin VW, Sindhu RK. NAD(P)H oxidase, superoxide dismutase, catalase, glutathione peroxidase and nitric oxide synthase expression in subacute spinal cord injury. Brain Res 2004;995:76–83.
Bao F, Dekaban GA, Weaver LC. Anti-CD11d antibody treatment reduces free radical formation and cell death in the injured spinal cord of rats. J Neurochem 2005;94:1361–1373.
Byrnes KR, Garay J, Di Giovanni S, et al. Expression of two temporally distinct microglia-related gene clusters after spinal cord injury. Glia 2006;53:420–433.
Kim D, You B, Jo EK, Han SK, Simon MI, Lee SJ. NADPH oxidase 2-derived reactive oxygen species in spinal cord microglia contribute to peripheral nerve injury-induced neuropathic pain. Proc Natl Acad Sci U S A 2010;107:14851–14856.
Mir M, Asensio VJ, Tolosa L, et al. Tumor necrosis factor alpha and interferon gamma cooperatively induce oxidative stress and motoneuron death in rat spinal cord embryonic explants. Neuroscience 2009;162:959–971.
Brown GC, Neher JJ. Inflammatory Neurodegeneration and Mechanisms of Microglial Killing of Neurons. Mol Neurobiol 2010;41(2–3):242–247.
Huppert J, Closhen D, Croxford A, et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010;24:1023–1034.
Szaingurten-Solodkin I, Hadad N, Levy R. Regulatory role of cytosolic phospholipase A2alpha in NADPH oxidase activity and in inducible nitric oxide synthase induction by aggregated Abeta1-42 in microglia. Glia 2009;57:1727–1740.
Qin L, Liu Y, Wang T, et al. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem 2004;279:1415–1421.
Hur J, Lee P, Kim MJ, Kim Y, Cho YW. Ischemia-activated microglia induces neuronal injury via activation of gp91phox NADPH oxidase. Biochem Biophys Res Commun 2010;391:1526–1530.
van der Veen RC, Dietlin TA, Hofman FM, Pen L, Segal BH, Holland SM. Superoxide prevents nitric oxide-mediated suppression of helper T lymphocytes: decreased autoimmune encephalomyelitis in nicotinamide adenine dinucleotide phosphate oxidase knockout mice. J Immunol 2000;164:5177–5183.
Popovich PG, Wei P, Stokes BT. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol 1997;377:443–464.
Hall ED, Oostveen JA, Andrus PK, Anderson DK, Thomas CE. Immunocytochemical method for investigating in vivo neuronal oxygen radical-induced lipid peroxidation. J Neurosci Methods 1997;76:115–122.
Xiong Y, Rabchevsky AG, Hall ED. Role of peroxynitrite in secondary oxidative damage after spinal cord injury. J Neurochem 2007;100:639–649.
Ma TC, Mihm MJ, Bauer JA, Hoyt KR. Bioenergetic and oxidative effects of free 3-nitrotyrosine in culture: selective vulnerability of dopaminergic neurons and increased sensitivity of non-dopaminergic neurons to dopamine oxidation. J Neurochem 2007;103:131–144.
Li J, Baud O, Vartanian T, Volpe JJ, Rosenberg PA. Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proc Natl Acad Sci U S A 2005;102:9936–9941.
Bao F, Bailey CS, Gurr KR, et al. Increased oxidative activity in human blood neutrophils and monocytes after spinal cord injury. Exp Neurol 2009;215:308–316.
Stuehr DJ, Fasehun OA, Kwon NS, et al. Inhibition of macrophage and endothelial cell nitric oxide synthase by diphenyleneiodonium and its analogs. FASEB J 1991;5:98–103.
Doussiere J, Gaillard J, Vignais PV. The heme component of the neutrophil NADPH oxidase complex is a target for aryliodonium compounds. Biochemistry 1999;38:3694–3703.
O'Donnell BV, Tew DG, Jones OT, England PJ. Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil NADPH oxidase. Biochem J 1993;290(pt 1):41–9.
Min KJ, Pyo HK, Yang MS, Ji KA, Jou I, Joe EH. Gangliosides activate microglia via protein kinase C and NADPH oxidase. Glia 2004;48:197–206.
Tang XN, Cairns B, Cairns N, Yenari MA. Apocynin improves outcome in experimental stroke with a narrow dose range. Neuroscience 2008;154:556–562.
Peng J, Stevenson FF, Oo ML, Andersen JK. Iron-enhanced paraquat-mediated dopaminergic cell death due to increased oxidative stress as a consequence of microglial activation. Free Radic Biol Med 2009;46:312–320.
Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson's disease. J Neurochem 2002;81:1285–1297.
Li B, Guo YS, Sun MM, et al. The NADPH oxidase is involved in lipopolysaccharide-mediated motor neuron injury. Brain Res 2008;1226:199–208.
Sun HN, Kim SU, Lee MS, et al. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-dependent activation of phosphoinositide 3-kinase and p38 mitogen-activated protein kinase signal pathways is required for lipopolysaccharide-induced microglial phagocytosis. Biol Pharm Bull 2008;31:1711–1715.
Vallieres N, Berard JL, David S, Lacroix S. Systemic injections of lipopolysaccharide accelerates myelin phagocytosis during Wallerian degeneration in the injured mouse spinal cord. Glia 2006;53:103–113.
Liu Y, Hao W, Letiembre M, et al. Suppression of microglial inflammatory activity by myelin phagocytosis: role of p47-PHOX-mediated generation of reactive oxygen species. J Neurosci 2006;26:12904–12913.
Kunz A, Park L, Abe T, et al. Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J Neurosci 2007;27:7083–7093.
Yang RY, Rabinovich GA, Liu FT. Galectins: structure, function and therapeutic potential. Expert Rev Mol Med 2008;10:e17.
Vasta GR. Roles of galectins in infection. Nat Rev Microbiol 2009;7:424–438.
Henderson NC, Sethi T. The regulation of inflammation by galectin-3. Immunol Rev 2009;230:160–71.
Elola MT, Wolfenstein-Todel C, Troncoso MF, Vasta GR, Rabinovich GA. Galectins: matricellular glycan-binding proteins linking cell adhesion, migration, and survival. Cell Mol Life Sci 2007;64:1679–1700.
Lukyanov P, Furtak V, Ochieng J. Galectin-3 interacts with membrane lipids and penetrates the lipid bilayer. Biochem Biophys Res Commun 2005;338:1031–1036.
Doverhag C, Hedtjarn M, Poirier F, et al. Galectin-3 contributes to neonatal hypoxic-ischemic brain injury. Neurobiol Dis 2010;38:36–46.
Zhou JY, Afjehi-Sadat L, Asress S, et al. Galectin-3 is a candidate biomarker for amyotrophic lateral sclerosis: discovery by a proteomics approach. J Proteome Res 2010;9:5133–5141.
Liu FT. Galectins: novel anti-inflammatory drug targets. Expert Opin Ther Targets 2002;6:461–468.
Dumic J, Lauc G, Flogel M. Expression of galectin-3 in cells exposed to stress-roles of jun and NF-kappaB. Cell Physiol Biochem 2000;10:149–158.
Sano H, Hsu DK, Yu L, et al. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J Immunol 2000;165:2156–2164.
Fernandez MR, Porte S, Crosas E, et al. Human and yeast zeta-crystallins bind AU-rich elements in RNA. Cell Mol Life Sci 2007;64:1419–1427.
Pesheva P, Kuklinski S, Schmitz B, Probstmeier R. Galectin-3 promotes neural cell adhesion and neurite growth. J Neurosci Res 1998;54:639–654.
Reichert F, Rotshenker S. Deficient activation of microglia during optic nerve degeneration. J Neuroimmunol 1996;70:153–161.
Karlsson A, Follin P, Leffler H, Dahlgren C. Galectin-3 activates the NADPH-oxidase in exudated but not peripheral blood neutrophils. Blood 1998;1;91:3430–3438.
Bethea JR, Castro M, Keane RW, Lee TT, Dietrich WD, Yezierski RP. Traumatic spinal cord injury induces nuclear factor-kappaB activation. J Neurosci 1998;18:3251–3260.
Sribnick EA, Samantaray S, Das A, et al. Postinjury estrogen treatment of chronic spinal cord injury improves locomotor function in rats. J Neurosci Res 2010;88:1738–1750.
Yan X, Liu J, Luo Z, et al. Proteomic profiling of proteins in rat spinal cord induced by contusion injury. Neurochem Int 2010;56:971–983.
Yan YP, Lang BT, Vemuganti R, Dempsey RJ. Galectin-3 mediates post-ischemic tissue remodeling. Brain Res 2009;1288:116–124.
Satoh K, Niwa M, Goda W, et al. Galectin-3 expression in delayed neuronal death of hippocampal CA1 following transient forebrain ischemia, and its inhibition by hypothermia. Brain Res 2011 (in press).
Chen HY, Fermin A, Vardhana S, et al. Galectin-3 negatively regulates TCR-mediated CD4+ T-cell activation at the immunological synapse. Proc Natl Acad Sci U S A 2009;106:14496–14501.
Doverhag C, Hedtjarn M, Poirier F, et al. Galectin-3 contributes to neonatal hypoxic-ischemic brain injury. Neurobiol Dis 2010;38(1):36–46.
Hsu DK, Yang RY, Pan Z, et al. Targeted disruption of the galectin-3 gene results in attenuated peritoneal inflammatory responses. Am J Pathol 2000;156:1073–1083.
Iacovazzi PA, Notarnicola M, Caruso MG, Guerra V, Frisullo S, Altomare DF. Serum levels of galectin-3 and its ligand 90 k/mac-2 bp in colorectal cancer patients. Immunopharmacol Immunotoxicol 2010;32:160–164.
Nagano S, Kim SH, Tokunaga S, Arai K, Fujiki M, Misumi K. Matrix metalloprotease-9 activity in the cerebrospinal fluid and spinal injury severity in dogs with intervertebral disc herniation. Res Vet Sci 2010 (in press).
Goussev S, Hsu JY, Lin Y, et al. Differential temporal expression of matrix metalloproteinases after spinal cord injury: relationship to revascularization and wound healing. J Neurosurg 2003;99(2 suppl):188–197.
Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci 2007;27:2596–2605.
Markowska AI, Liu FT, Panjwani N. Galectin-3 is an important mediator of VEGF- and bFGF-mediated angiogenic response. J Exp Med 2010;207:1981–1993.
Ferraguti F, Shigemoto R. Metabotropic glutamate receptors. Cell Tissue Res 2006;326:483–504.
Byrnes KR, Loane DJ, Faden AI. Metabotropic glutamate receptors as targets for multipotential treatment of neurological disorders. Neurotherapeutics 2009;6:94–107.
Biber K, Laurie DJ, Berthele A, et al. Expression and signaling of group I metabotropic glutamate receptors in astrocytes and microglia. J Neurochem 1999;72:1671–1680.
Byrnes KR, Stoica B, Riccio A, Pajoohesh-Ganji A, Loane DJ, Faden AI. Activation of metabotropic glutamate receptor 5 improves recovery after spinal cord injury in rodents. Ann Neurol 2009;66:63–74.
Byrnes KR, Stoica B, Loane DJ, Riccio A, Davis MI, Faden AI. Metabotropic glutamate receptor 5 activation inhibits microglial associated inflammation and neurotoxicity. Glia 2009;57:550–560.
Loane DJ, Stoica BA, Pajoohesh-Ganji A, Byrnes KR, Faden AI. Activation of metabotropic glutamate receptor 5 modulates microglial reactivity and neurotoxicity by inhibiting NADPH oxidase. J Biol Chem 2009;284:15629–15639.
Boldyrev AA, Carpenter DO, Johnson P. Emerging evidence for a similar role of glutamate receptors in the nervous and immune systems. J Neurochem 2005;95:913–918.
Mills CD, Xu GY, McAdoo DJ, Hulsebosch CE. Involvement of metabotropic glutamate receptors in excitatory amino acid and GABA release following spinal cord injury in rat. J Neurochem 2001;79:835–848.
Mills CD, Fullwood SD, Hulsebosch CE. Changes in metabotropic glutamate receptor expression following spinal cord injury. Exp Neurol 2001;170:244–257.
Gwak YS, Hulsebosch CE. Upregulation of Group I metabotropic glutamate receptors in neurons and astrocytes in the dorsal horn following spinal cord injury. Exp Neurol 2005;195:236–243.
Floyd CL, Rzigalinski BA, Sitterding HA, Willoughby KA, Ellis EF. Antagonism of group I metabotropic glutamate receptors and PLC attenuates increases in inositol trisphosphate and reduces reactive gliosis in strain-injured astrocytes. J Neurotrauma 2004;21:205–216.
Lea PM, Custer SJ, Vicini S, Faden AI. Neuronal and glial mGluR5 modulation prevents stretch-induced enhancement of NMDA receptor current. Pharmacol Biochem Behav 2002;73:287–298.
Farso MC, O'Shea RD, Beart PM. Evidence group I mGluR drugs modulate the activation profile of lipopolysaccharide-exposed microglia in culture. Neurochem Res 2009;34:1721–1728.
Bao WL, Williams AJ, Faden AI, Tortella FC. Selective mGluR5 receptor antagonist or agonist provides neuroprotection in a rat model of focal cerebral ischemia. Brain Res 2001;922:173–179.
Mills CD, Johnson KM, Hulsebosch CE. Group I metabotropic glutamate receptors in spinal cord injury: roles in neuroprotection and the development of chronic central pain. J Neurotrauma 2002;19:23–42.
Murotomi K, Takagi N, Mizutani R, et al. mGluR1 antagonist decreased NADPH oxidase activity and superoxide production after transient focal cerebral ischemia. J Neurochem 2010;114(6):1711–1719.
Urban MO, Hama AT, Bradbury M, Anderson J, Varney MA, Bristow L. Role of metabotropic glutamate receptor subtype 5 (mGluR5) in the maintenance of cold hypersensitivity following a peripheral mononeuropathy in the rat. Neuropharmacology 2003;44:983–993.
Osikowicz M, Mika J, Makuch W, Przewlocka B. Glutamate receptor ligands attenuate allodynia and hyperalgesia and potentiate morphine effects in a mouse model of neuropathic pain. Pain 2008;139:117–126.
Zhu CZ, Wilson SG, Mikusa JP, et al. Assessing the role of metabotropic glutamate receptor 5 in multiple nociceptive modalities. Eur J Pharmacol 2004;506:107–118.
Carlton SM, Du J, Tan HY, et al. Peripheral and central sensitization in remote spinal cord regions contribute to central neuropathic pain after spinal cord injury. Pain 2009;147:265–276.
Gwak YS, Hulsebosch CE. Remote astrocytic and microglial activation modulates neuronal hyperexcitability and below-level neuropathic pain after spinal injury in rat. Neuroscience 2009;161:895–903.
Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 2009;29:13435–13444.
Impellizzeri D, Mazzon E, Esposito E, Paterniti I, Bramanti P, Cuzzocrea S. Effect of Apocynin, an inhibitor of NADPH oxidase, in the inflammatory process induced by an experimental model of spinal cord injury. Free Radic Res 2010;45(2):221–236.
Balazs R, Miller S, Romano C, de Vries A, Chun Y, Cotman CW. Metabotropic glutamate receptor mGluR5 in astrocytes: pharmacological properties and agonist regulation. J Neurochem 1997;69:151–163.
Faden AI, O'Leary DM, Fan L, Bao W, Mullins PG, Movsesyan VA. Selective blockade of the mGluR1 receptor reduces traumatic neuronal injury in vitro and improvesoOutcome after brain trauma. Exp Neurol 2001;167:435–444.
Banasik T, Jasinski A, Pilc A, Majcher K, Brzegowy P. Application of magnetic resonance diffusion anisotropy imaging for the assessment neuroprotecting effects of MPEP, a selective mGluR5 antagonist, on the rat spinal cord injury in vivo. Pharmacol Rep 2005;57:861–866.
Acknowledgments
The authors would like to thank Eric Hoffman and Josephine Chen for data obtained from the Public Expression Profiling Resource (PEPR) portal (http://pepr.cnmcresearch.org). This work was supported by a grant from the National Capital Area Rehabilitation and Research Network (pilot grant to K.R.B.). Full conflict of interest disclosure is available in the electronic supplemental materials for this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 510 kb)
Rights and permissions
About this article
Cite this article
Pajoohesh-Ganji, A., Byrnes, K.R. Novel Neuroinflammatory Targets in the Chronically Injured Spinal Cord. Neurotherapeutics 8, 195–205 (2011). https://doi.org/10.1007/s13311-011-0036-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-011-0036-2