
ABSTRACT
Recently, phage display technology has been announced as the recipient of Nobel Prize in Chemistry 2018. Phage display technique allows high affinity target-binding peptides to be selected from a complex mixture pool of billions of displayed peptides on phage in a combinatorial library and could be further enriched through the biopanning process; proving to be a powerful technique in the screening of peptide with high affinity and selectivity. In this review, we will first discuss the modifications in phage display techniques used to isolate various cancer-specific ligands by in situ, in vitro, in vivo, and ex vivo screening methods. We will then discuss prominent examples of solid tumor targeting-peptides; namely peptide targeting tumor vasculature, tumor microenvironment (TME) and over-expressed receptors on cancer cells identified through phage display screening. We will also discuss the current challenges and future outlook for targeting peptide-based therapeutics in the clinics.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Peptides are 2-dimensional, linear chains of amino acids, which are usually short (less than 50 AA) in length (Hayashi et al., 2012). They are either designed by rational computing methods or phage display screening to obtain peptides that binds with high specificity to the target of interest, with a possibility of modulating the target (Marqus et al., 2017). Compared to antibodies (~150 kDa), peptides are relatively small (~3–5 kDa) and therefore easy to synthesize and modified (Boohaker et al., 2012), have higher cell membrane penetration, and possess less immunogenicity. In cancer therapy, these peptides can be used as a targeting ligand assisting specific delivery of cytotoxic drug specifically into the tumor vasculature, tumor microenvironment or into the cancer cells. On the other hand, peptides could also be delivered intracellularly to target cancer specific upregulated transcription factors, oncogenes or enzymes (Jyothi, 2012; Marqus et al., 2017). The general comparison between antibody and peptide are summarized in Table 1.
Herein, we will review the utilization of phage display biopanning with modifications gearing towards in situ, in vitro, in vivo, ex vivo and in human application for high affinity peptide screening. We will also provide a comprehensive discussion on the latest discovery of tumor targeting-peptides; namely the peptides targeting (1) tumor vasculature, (2) tumor microenvironment (TME) and (3) over-expressed receptors on cancer cells.
Phage display technology and biopanning strategies
In 1985, George Smith first described phage display by demonstrating the ability of a filamentous phage to display peptide by fusing the library of peptide sequence into the virus’s capsid protein (Smith, 1985). Since the peptide was displayed on the viral surface, selection could be done to isolate those with the highest binding affinity towards a target. In the same year, Geroge Pieczenik filed a patent also describing the generation of phage display libraries in detail (US patent, 5866363). However, the application of this technology was pioneered by Greg Winter and his colleagues at the Scripps Research Institute for display of proteins (specifically antibodies) for therapeutic protein engineering. Due to their contribution in phage display technique development and the enormous implication of phage display technology, Smith and Winter were both awarded a quarter share of the 2018 Nobel Prize in chemistry, while the other half was awarded to Frances Arnold.
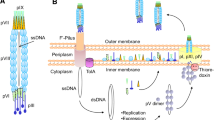
Phage-display is a powerful technology for screening and isolating target specific peptides. This method utilizes bacteriophage to display foreign peptides or antibodies on their surface through insertion of the gene encoding the corresponding polypeptides into the phage genome. For display of foreign polypeptides on the bacteriophage, the desired DNA sequence is inserted into the M13 phage pIII or pVIII gene (Fig. 1). The methodology using the major coat protein pVIII provides a multivalent display, however only short peptides (6–7 AA) could be displayed on pVIII gene. Therefore, most combinatorial libraries such as antibodies or proteins have been displayed using minor coat pIII. Since there could be only 3–5 copies of pIII protein per phage, this method limits the copy number but the length of foreign or synthetic polypeptides that can be expressed (Fig. 1).

A typical representation of M13 phage with about 1,000 nm in length and 5 nm wide. The major coat proteins are pIII (green), pVIII (purple) and pVII + pIX complex (yellow + red)
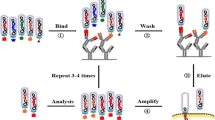
The phage selection method, referred to as biopanning, is an affinity selection process that isolates target-binding molecules. As explained in Fig. 2, generally phage display based biopanning consists of five screening steps for selection of peptides. The first step is “library construction & amplification” where polypeptide-displayed phage libraries were constructed via cloning of combinatorial DNA sequence (Fig. 2A). This library will be amplified prior to biopanning (Fig. 2B). The second step is the “target capturing step”, in which the phage library is incubated with target molecule for a specific time to allow binding (Fig. 2C). The third step is to “remove unbound & nonspecific phages” by using repetitive washing to remove any unbound and non-target specific phages (Fig. 2D). The fourth step is the “elution step”, in which target-bound phages are separated after a short incubation with low pH buffer or by competitive elution (Fig. 2E). Finally, in the fifth step “infection stage”, the eluted phages are infected in bacteria to amplify selected phages, making a new and more selective phage library that should be applied in a next round of biopanning (Fig. 2F and 2G).

The general scheme of phage display technique and biopanning selection of high affinity peptide. Peptide-based library is first obtained either commercially or specifically designed to cater for specific needs of each experiment
In general, three to five rounds of biopanning are necessary to isolate specific and high affinity peptide binders. Nonspecific phages are removed and phages with high affinity for the target are isolated by increasing the stringency in each round of biopanning by increasing the number of washing and decreasing the amount of target molecule. At the end of biopanning, phage ELISA and DNA sequencing are used for identification of individually specific phage with high affinity to target.
Ample research to isolate high affinity peptide by phage display screening
Although in situ phage display screening using immobilized antigen is capable of generating high affinity and specificity peptide (Kim et al., 2012b), to better mimic cellular and body condition, ample researches are being done on in vitro, in vivo (Liu et al., 2018), ex vivo (Sorensen and Kristensen, 2011) and even in cancer patient (Krag et al., 2006) screening for high affinity peptide in a heterogenous environment as this is a closer representation to their original condition.
Homogenous in situ screening
Homogenous in situ screening requires only the specific target to be coated on a 96-well (Fig. 3A). A single target exposure guarantees the isolation of target-specific peptide, without external interference from non-specific binding. This method is also the easiest, as all experiments could be carried out without living system (i.e., cell culture, animal model, patient samples). The disadvantages of in situ screening includes the risk of non-specific binding of the isolated peptide when exposed to in vitro or in vivo system. In addition, the target is artificially coated onto the plate, which could be misrepresent the actual secondary structure of the target in a living system, therefore increases the risk of isolating a peptide that only binds to the receptor in this particular setting (Kim et al., 2012b).

Various approaches in capturing high affinity peptide through phage display screening
In vitro cell screening
In vitro cell screening offers high-throughput approach for identifying multiple peptides that bind specifically to a single cell (i.e., cell lines or primary cells) and can be performed on adherent cells (live or fixed) (Fig. 3B). Advantages of using whole cell approach includes retaining their biological functions and activities, proper folding, 3-dimensional structure, receptor expression level and their association with neighboring proteins. Modified selection protocols could be used to isolate internalized peptides. Importantly, in vitro cell biopanning could identify novel cell surface receptors with unknown biological functions, which could be used to provide information on specific molecular changes (i.e., expression level of certain protein and their localization in normal vs. cancer cells) (Arap et al., 2002b; Zhao et al., 2007; Sun et al., 2012; Wu et al., 2016).
In vivo screening
By performing biopanning and selection in a living animal, organ-specific peptides could be isolated (Fig. 3C). Roushlati and co-workers first described in vivo phage display technology in 1996 (Pasqualini and Ruoslahti, 1996). For in vivo biopanning protocol, the biopanning selection is similar to that of the in vitro screening, the difference being the peptide phage library was introduced into the animal via systemic intravenous injection and allowed binding to occur within 1–2 h (as peptide-displayed phage is estimated to bound to target within 5–15 min (Laakkonen et al., 2002; Lee et al., 2007; Lo et al., 2008)), after which the animals will be perfused to remove unbound phages, sacrificed, and the desired organs will be collected and homogenized. Tissue-specific phage should increase after 3–5 rounds of biopanning (Rajotte et al., 1998; Lee et al., 2007; Chang et al., 2009). Through this approach, various types of tumor and malignant tissue vasculature have been identified (i.e., RGD-4C, NGR and GSL peptide (Koivunen et al., 1995; Pasqualini et al., 1997; Ruoslahti, 2000); detailed explanation below). One of the major pitfalls in using in vivo phage display technology is that the peptides may not be translated into human due to the differences of peptide binding between species (Wu et al., 2016).
Ex vivo screening
This method, first published in Nature in 2001, should only be applied to the selections of a specific rare cells in a heterogenous population (i.e., PBMCs in blood tumors) (Fig. 3D). Without sorting the cells, biopanning was performed on a glass slide containing the whole cell population. This method is advantageous for targeting a lower frequency of cells (<0.1% of the total population), as phages that binds non-selectively towards the other cells will be screened out. Once the phage was bound, UV irradiation was used so that the DNA of the phage particles on non-target cells is crosslinked by UV, while the phage on target cells were protected by a minute aluminum disc. Therefore, this method ensures that only non-crosslinked phage (target phage) were capable of replicating. The disadvantage of this method is that it is only optimized for antibody-based ligand selection, and thus not suitable for peptide selection. The yield of this method averages three antibodies per selection, which is very low compared to the other biopanning method (Sorensen and Kristensen, 2011).
In human screening
To diminish the compatibility of species difference between mice and human, phage display had been reported to be screened against human patients (Fig. 3E). The first in-human phage display screening was reported by Arap and colleagues in 2002. They reported a heptapeptide SMSIARL which could specifically home to prostate vasculature and exhibited 10–15 times more specificity to prostate compared to other organs (Arap et al., 2002b). Due to their success in proving safe usage of phage display in human, FDA approved similar techniques to be used by Krag and colleagues to screen tumor-specific peptide via phage display screening in terminal stage cancer patients (Krag et al., 2006).
Tumor targeting peptide
Tumor targeting peptide is a powerful tool that could be used in cancer diagnosis and treatment (Heppeler et al., 2000) as they have lower production cost and scale-up, easy to synthesize and yet they possess most if not all the merits of a targeting ligand: high affinity and specificity towards the target, with the advantage of high tumor penetration as compared to the large-sized antibody-based ligand (AlDeghaither et al., 2015). In the complexity of solid tumor, a peptide could be used to target the malfunctioned tumor vasculature, the dense extra-cellular matrix, tumor stromal cells, or overexpressed receptor on tumors. Herein, we will discuss some prominent examples of peptides identified through phage display biopanning techniques and their application in the biomedical field.
Peptide targeting tumor-microenvironment (TME)
Tumor microenvironment (TME) is a complex plethora of multiple components including tumor-associated vasculature, extra-cellular matrix, cancer associated fibroblast, tumor associated macrophages, immune cells (neutrophils, NK cells, T cells, B cells) and tumor cells (Binnewies et al., 2018) (Fig. 4). Often, these cells transformed into tumor-like phenotype as tumor progresses. For example, most tumor resident macrophages are M2-like (pro-tumoral) which means they are programmed to assist in tumor growth rather than having an M1-like (anti-tumoral) phenotype (Mantovani et al., 2017). These changes could be brought forth by constant communication with the other components in the TME through autocrine or paracrine manner. Therefore, by identifying peptide specific to these TME targets could generate drugs homing to TME that could efficiently normalize, modulate or disrupt the TME components. There are three points of intervention, namely (i) targeting tumor vasculature, (ii) targeting extra-cellular matrix, (iii) targeting tumor stromal cells (macrophages, cancer associated fibroblasts etc.).

Major components in the TME. (A) tumor vasculature components and extra-cellular matrix, (B) tumor stromal cells and (C) over-expressed receptors on tumor cells
Peptide targeting tumor vasculature
Angiogenesis is an event of the formation of new blood vessels and is vital in the event of tumor growth and progression. Due to the continuous formation of new blood vessels to feed the tumor, a hyper-vascular tumor could grow beyond the size of millimeter in diameter (Bergers et al., 1999). Therefore, stopping a tumor’s blood supply can dramatically reduce the tumor growth, and in some cases, even resulted in total tumor eradication (Ferrara and Alitalo, 1999; O’Reilly et al., 1999). The morphology of tumor vasculature is very different from normal tissue vasculature. Due to the on-going angiogenesis, tumor vasculatures consistently express angiogenic marker at high concentration (i.e., integrins, VEGFR) and are usually tortuous (Bergers et al., 1999), with pronounced hypoxic region. Tumor vasculatures are also “leaky” in nature and this might be related to pericyte deficiency (Ruoslahti, 2000), therefore Folkman hypothesized that angiogenesis inhibition could be used to treat solid tumors (Folkman, 1971).
Peptide targeting tumor endothelial cells (EC)
The peptides that home to tumor vasculature may also be useful in targeting therapies specifically to tumors. Tumors are critically dependent on blood supply; therefore, blocking or eliminating that supply can profoundly suppress tumor growth (Denekamp, 1993; Hanahan and Folkman, 1996; Bergers et al., 1999; Jain, 2001). Since blood vessels are easily accessible through IV administration, and they do not readily acquire mutations as cancer cells that leads to drug resistance (Kerbel, 1991; Boehm et al., 1997), targeting tumor ECs could be a promising approach for targeted drug delivery.
A classic example of vasculature targeting peptide is none other than the “RGD” peptide. Rouslahti and colleagues first isolated this peptide by phage display in vivo in the form of cyclic peptide CDCRGDCFC (RGD-4C). This peptide has been validated to selectively binds αvβ3 and αvβ5 integrins (Koivunen et al., 1995); and have shown to home to the vasculature of tumors (Pasqualini et al., 1997). Interestingly, RGD domain is also vital for the binding of vitronectin and fibronectin and to integrins, although it is now known that these molecules bind to different subset of integrin (Ruoslahti, 2003).
Arap et al. also developed a set of cyclic peptide CNGRC sharing “NGR” motifs (Arap et al., 1998). These peptides have been shown to bind to tumor vasculatures in breast carcinoma, melanoma and Kaposi’s sarcoma (Pasqualini et al., 1997; Arap et al., 1998; Pasqualini et al., 2000). Subsequently, many other publications followed, describing the isolation of tumor vasculature related targeting peptides (Table 2) (Landon and Deutscher, 2003; Zurita et al., 2003; Ruoslahti, 2004; Kelly et al., 2005; Su et al., 2005).
Peptide targeting MMPs
Matrix metalloproteinases (MMPs) family is among the molecules that are upregulated in tumor microenvironment, and has been known to be functionally important in angiogenesis (Koivunen et al., 1999). Not only that, MMPs are also involved in increasing cell motility and invasiveness (Birkedal-Hansen, 1995). Although MMPs are secreted proteins, they are able to mediate phage homing. This might be due to the binding of MMP-2 and MMP-9 to αvβ3 integrin (Brooks et al., 1996), thus forming a complex that is stable enough for the binding of phage. Apparently, the complex is stable enough for strong binding of the phage to the MMP. Interestingly, the selected phage bound to MMP-2 and MMP-9 also specifically homes to tumor vasculature (Koivunen et al., 1999), indicating that (i) that one, or both, of these MMPs is specifically expressed in tumor vasculature and (ii) they are available for phage binding from the circulation. Multiple peptides inhibiting MMP families have been isolated through phage display screening. Their sequence, activities and function are summarized in Table 2 (Ujula et al., 2010; Ndinguri et al., 2012).
Peptide targeting pericytes of angiogenic vessels
Pericytes secrete growth factors that stimulate EC proliferation. Pericytes also secrete proteases to modulate the surrounding ECM and guide EC migration (Gerhardt and Betsholtz, 2003; Armulik et al., 2005; Saunders et al., 2006; Stapor et al., 2014). Recently, more researches are pointing towards the importance of pericyte coverage in vessel remodeling, maturation, and stabilization (Ribeiro and Okamoto, 2015). Therefore, pericyte might be the overlooked player in angiogenesis and should be given more emphasis in anti-tumor targeted therapy.
Several rounds of biopanning led Burg et al. to identify two decapeptides (TAASGVRSMH and LTLRWVGLMS) specific to a transmembrane chondroitin sulfate proteoglycan NG2, which is expressed in pericytes of angiogenic vessels (Schlingemann et al., 1990; Burg et al., 1999). These peptides specifically homed to tumor vasculature in vivo but not to tumor vasculature in NG2 knockout mice, indicating the specificity and targeting capability of these peptides (Burg et al., 1999). Although the role of NG2 in angiogenesis is still unclear, NG2 is a cell surface receptor for type-VI collagen and also binds to PDGF-A, which could potentially stimulate this growth factor (Nishiyama et al., 1996). As a component in pericyte, NG2 is undetectable in endothelial cells (Burg et al., 1999), therefore blocking NG2 represents a specific pericyte targeting.
Peptides targeting extra-cellular matrix (ECM)
The role of ECM components is now recognized as an important determinant in the growth and progression of solid tumors (Wernert, 1997; Pupa et al., 2002). ECM is extensively remodeled in tumor progression through 2 main processes: (i) neosynthesis of ECM components (i.e., alternative splicing mechanism of fibronectin to include EDA and EDB domain in malignant tumor fibronectin) and (ii) degradation of ECM by hydrolytic enzymes (e.g., proteases) that are produced, activated or induced by neoplastic cells, therefore become more permissive environment for tumor growth (Kaspar et al., 2006).
Tumor-associated fibronectin Fibronectin serves as a coordinator between cancer cells and ECM, and is involved in cancer cell survival, proliferation, invasion and metastasis (Wierzbicka-Patynowski and Schwarzbauer, 2003). One of the most extensive changes in ECM remodeling is the addition of extra-domain A and B (EDA and EDB), which are alternatively spliced-in during the synthesis of tumor-associated fibronectin. These domains are undetectable in healthy adult but has been found in high concentrations in malignant tumors. Clinical evidences indicated that tumor-associated FN (also termed oncofetal FN), is overexpressed in many malignant cancers, including breast cancer (Ioachim et al., 2002; Bae et al., 2013), prostate cancer (Suer et al., 1996; Albrecht et al., 1999), bladder cancer (Arnold et al., 2016), oral squamous cell carcinoma (Lyons et al., 2001), head and neck squamous cell carcinoma (Mhawech et al., 2005), colorectal cancer (Inufusa et al., 1995) and lung cancer (Khan et al., 2005), and upregulated FN expression has been correlated with poor prognosis of the patients. Therefore, tumor-associated FN represents an ideal target for solid tumor targeting.
Through in situ phage display technology, Kim et al. developed an EDB binding scaffold-like peptide termed APTEDB (Kim et al., 2012b). APTEDB consists of a stabilizing scaffold and two target-binding regions, mimicking the morphology of a DNA leucine zipper. Taking advantage of the synergistic three-dimensional structure for optimal binding, APTEDB exhibited a high binding affinity (Kd ~65 nmol/L) to EDB and could be used as a targeting ligand to be conjugated to anti-cancer drugs for high tumor selectivity and reducing systemic toxicity (Kim et al., 2014; Kim et al., 2016), deliver biologics (i.e., oligonucleotides, siRNA and drugs) for solid tumor treatment (Saw et al., 2013; Saw et al., 2015; Saw et al., 2017) and to encapsulate superparamagnetic iron oxide particles for Magnetic Resonance Imaging of EDB over-expressing tumors (Park et al., 2012). In another study, Han et al. developed a cyclic nonapeptide (ZD2) with the sequence of CTVRTSADC that could be used for EDB specific targeting and imaging of prostate cancer. This linear peptide, which has Kd ~11 μmol/L binding affinity towards EDB, demonstrated excellent specific targeting to prostate cancer in vivo and could be utilized as an imaging agent for EDB-overexpressing prostate cancer (Han et al., 2015).
Tenascin C (TNC) TNC is a glycoprotein which forms a large structure body by assembling other ECM molecules and participates in cell adhesion, movement, permeation, survival, migration and differentiation (Chiquet-Ehrismann, 1990). As with tumor-associated FN, TNC is not usually expressed in normal cells except in immune tissues, such as bone marrow and thymus gland (Klein et al., 1993; Hemesath and Stefansson, 1994), but is specifically expressed in malignancy, inflammation and wound healing. It had been reported that the elevated expression of TNC depended on a malignancy in the tumor stroma of some malignancies, including oral cancer, sarcoma, breast cancer, and colon cancer, squamous cell carcinoma (Hindermann et al., 1999) chondrosarcoma (Ghert et al., 2001), breast cancer (Tsunoda et al., 2003) and colon cancers (Hanamura et al., 1997; Suzuki et al., 2017).
Kim et al. isolated a peptide that not only selectively bound to TNC in xenograft mouse tissue and patient tumors but also reduced TNC-induced cell rounding and migration. Due to the bulky size of TNC, they adopted two independent screening; the first using full-length TNC (expressed in eukaryotic cells) and the second using alternative spliced domain (expressed in bacteria). Out of a total of 35 clones, 19 had the same sequences (denoted peptide #1, FHKHKSPALSPV, 54.2% consensus) and another 13 clones were also identical (denoted peptide #2, FHKPFFPKGSAR, 37.1% consensus). The binding affinity of peptide #1 to TNC was 4.58 ± 1.4 µmol/L (Kim et al., 2012a).
Peptide targeting tumor associated macrophages (TAMs)
High density of TAMs has been correlated to poor prognosis in several types of cancers, including brain, breast, ovarian and pancreatic cancers, where the majority of these TAMs express M2-like phenotype (Kurahara et al., 2011; Medrek et al., 2012; Colvin, 2014; Zhou et al., 2015). Therefore, M2-like TAMs have been exploited as therapeutic targets, and positive outcomes were shown in selective depletion of this macrophage subpopulations (Georgoudaki et al., 2016). Small molecules such as folic acid (targeting folate receptor β) and mannose (targeting mannose receptor) have been conjugated to drugs or carriers for macrophage targeting and drug delivery (Hashida et al., 2001; Low et al., 2008; Yu et al., 2013). However, these receptors are not macrophage specific and they are also expressed in other cell types for example, mannose receptors are also expressed in dendritic cells (Sallusto et al., 1995)). Folic acid also binds different isoforms of folate receptors on tumor cells and normal epithelial cells (Ross et al., 1994), therefore diminishing the specificity effect of the ligand. In 2012, Segers et al. reported a novel peptide that binds selectively to scavenger receptor-A on macrophages in atherosclerotic plaques. Nevertheless, it was found that this receptor is also expressed on dendritic cells (Segers et al., 2012). Therefore, M2-like macrophage-specific peptide should be screened and developed for clinical application.
Cieslewicz et al. polarized murine bone marrow-derived macrophages with either IFN-γ and LPS or with IL-4 to generate both M1 and M2 cells for biopanning. After three rounds of phage panning, highly selective M2 macrophage-binding peptides were identified, and this peptide binds preferentially to M2 cells. Sequencing of the 10 clones obtained above revealed two unique sequences: YEQDPWGVKWWY (denoted M2pep Phage, consensus 80%), and HLSWLPDVVYAW (consensus 20%). M2pep Phage demonstrated higher affinity and selectivity towards M2; 10.8-fold higher binding to M2 macrophages over scramble-M2pep, as well as 5.7-fold higher binding to M2 over M1 macrophages. Furthermore, after intravenous administration, M2pep Phage was able to selectively binds M2-like TAMs in mouse colon carcinoma tumors (Cieslewicz et al., 2013).
Peptide targeting cancer associated fibroblasts (CAFs)
One of the dominant cell type in solid tumor is CAFs (Augsten, 2014). They are likely to be derived from the mesoderm and exhibited mesenchymal-like features (Kalluri and Weinberg, 2009). CAFs are often found in close vicinity or in direct contact with tumor cells (Kalluri and Weinberg, 2009). In normal condition, fibroblasts are likely to be quiescent or in resting state, yet became activated in response to growth factors, cytokines and mechanical stress (Kalluri and Weinberg, 2009; Rasanen and Vaheri, 2010; Shiga et al., 2015). Unlike tumor cells that presents diverse marker proteins on cell surface, CAFs selectively overexpressed certain proteins, such as fibroblast-activated protein-α (FAP-α) and α-smooth muscle actin (α-SMA) (Bhowmick et al., 2004; Kalluri and Zeisberg, 2006; Franco et al., 2010; Rasanen and Vaheri, 2010). Therefore, CAF targeting or responsive nanomaterial may be an efficient strategy to achieve improved antitumor efficacy.
Brinton et al. presented a new strategy for analysis by combining phage display and accompanying software: “PHage Analysis for Selective Targeted PEPtides” or PHASTpep, which they claimed to identify highly specific and selective peptides. Using this combination, they discovered and validated two peptide sequences (HTTIPKV and APPIMSV) targeted to pancreatic CAFs in mice. The Mander’s coefficient was high for both HTTIPKV (0.70) and APPIMSV (0.74) indicating phage clone binding to αSMA-positive CAFs in vivo (Brinton et al., 2016).
Urokinase plasminogen activator (uPA) receptor (uPAR) uPA is a serine protease largely produced in stromal fibroblast-like cells in melanoma, colon, breast, and prostate cancer. The uPA/uPAR interaction is important in early tumor development (i.e., cell adhesion and invasion). Goodson et al. isolated a uPAR specific peptide, AEPMPHSLNFSQYLWYT. This peptide was able to compete for binding of radiolabeled uPA fragment, therefore served as a potent antagonist for uPAR (Landon and Deutscher, 2003).
Plausible targets for the development of tumor-targeting peptide
CD10+GPR77+ CAFs
Recently we demonstrated that CD10+GPR77+ CAFs specifically define a subset of CAF that correlated with chemoresistance and poor survival in breast and lung cancer patients. Mechanistically, the activation of CD10+GPR77+ CAFs was driven by the consistent NF-κB activation, which is maintained via GPR77 (C5a receptor) complement signaling. Furthermore, CD10+GPR77+ CAFs could lead to successful engraftment of patient-derived xenografts (PDXs), while blocking these CAFs with a neutralizing anti-GPR77 antibody inhibited tumor formation while restoring chemosensitivity of the tumor. Therefore, targeting the CD10+GPR77+ CAF subset could present an effective therapeutic strategy against solid tumors (Su et al., 2018).
CD146
Also known as melanoma cell adhesion molecule (MCAM), CD146 is a member of cell adhesion molecules of the immunoglobulin (Ig) superfamily (Lehmann et al., 1989). CD146 has been known to be involved in angiogenesis, tumor metastasis, lymphocyte activation, morphogenesis during development and tissue regeneration (Ouhtit et al., 2009; Wang and Yan, 2013; Ye et al., 2013). As CD146 is mainly expressed on ECs, CD146 is required for endothelial cell proliferation, migration and tube formation (Kang et al., 2006; Zheng et al., 2009), playing critical roles in angiogenesis (Yan et al., 2003; Chan et al., 2005; Harhouri et al., 2010; Kebir et al., 2010; Tu et al., 2015). To date, antibody-drug conjugate (ADC) targeting CD146 have been developed, therefore suggesting CD146 targeting could mitigate tumor growth and metastasis (Rouleau et al., 2015).
PITPNM3
Phosphatidylinositol transfer protein, membrane-associated 3 (PITPNM3), also known as Nir1, is essential in CCL18-induced chemotaxis through calcium influx. The function of PITPNM3 could be completely diminished by GPCR pathway inhibitor pretreatment or via pertussis toxin (PTX). We first demonstrated that PITPNM3 is abundantly expressed in breast cancer cells (Chen et al., 2011). In another independent research, He et al. revealed that PITPNM3 was also upregulated in hepatocellular carcinoma (HCC) cells and tissues. While the silencing of PITPNM3 significantly attenuated the invasiveness and metastatic ability of HCC cells, the upregulation of PITPNM3 increased HCC cell mobility. Mechanism wise, the inhibition of PITPNM3 suppressed the activation of Pyk2, FAK, and Src, and also impaired integrin clustering; indicating that PITPNM3 is a key player in cancer migration and invasion, therefore is a promising target in cancer therapy (C. He et al., 2014).
Transmembrane 4L six family member 1 (TM4SF1)
TM4SF1 was first discovered as a tumor cell antigen and could be specifically recognized by mouse monoclonal antibody L6 (Hellstrom et al., 1986b; Marken et al., 1992). TM4SF1 is expressed abundantly on many cancer cells (Hellstrom et al., 1986a; Hellstrom et al., 1986b), on tumor blood vessel endothelial cells (Shih et al., 2009). TM4SF1 is also associated with pathologic angiogenesis, targeting TM4SF1 would provide a dual anticancer mechanism: simultaneously targeting tumor cells and the tumor vasculature (secondary mechanism) (Visintin et al., 2015).
Peptide targeting over-expressed receptors on tumor
In cancer treatment, overexpressed receptors are modulated by targeting agents such as antibodies, antibody fragments, peptides or small chemicals that could block their activities by directly binding these receptors, halting downstream mechanism therefore blocking cancer progression. Other approaches included exploiting receptor overexpression for the targeted delivery of anticancer drugs or biologically active molecules that are unable discriminate between cancer and normal cells. The ligand acts as their “eyes”, guiding them directly towards the overexpressed receptors on tumor cells, therefore specifically attacking malignant cells while sparing normal cells (Mendelsohn and Baselga, 2003).
In this section, we highlighted some prominent over-expressed receptors that have been widely used for cancer cell specific targeting. There is a myriad of targeting ligands that are currently known to be overexpressed in various cancer, differing in cancer types and subtypes, stages of cancer. It is quite a challenge to summarized all of these receptors in this review, therefore the selection was done on PubMed search with “receptor” and “targeting” filters. Figure 5 highlighted the Top-10 cancer-associated overexpressed receptors and their corresponding publications in PubMed until 2018. Comprehensive review of literature reveals that (with the exception of CD44 and Fas receptors), all other receptors had been used as targets for phage display biopanning and at least one peptide ligand has been developed for these receptors; which are highlighted in detail in the section below.

Top-10 cancer-associated overexpressed receptors and their corresponding publications in PubMed until 2018
ErbB family (EGFR & HER2)
In the ErbB family, there are four known members: ErbB1/EGFR/HER1 (only found in humans), ErbB2/HER2/Neu, ErbB3/HER3 and ErbB4/HER4 (Seshacharyulu et al., 2012). These receptors are transmembrane glycoproteins with molecular weights ranging from 170 to 185 kDa (Olayioye et al., 2000). EGFR are major contributors of a complex signaling cascade in cancer cells that modulates growth, signaling, differentiation, adhesion, migration and survival, therefore making EGFR an attractive candidate for anti-cancer targeting and therapy (Grandis and Sok, 2004). Specifically, EGFR has shown to play a key role in the development and growth of tumor cells, including cell proliferation and apoptosis (Wells, 1999).
In 2014, Wang, Zho and Joshi applied for an international patent (“Peptide reagents and methods for detection and targeting of dysplasia, early cancer and cancer”, Patent No. WO2016029125A1) for the screening and evaluation of EGFR-targeting peptide through in situ phage display screening, utilizing the PhD-7 heptapeptide random library and PhD-12 decapeptide random library provided by New England Biolab. The screening resulted in 17 EGFR-specific peptides: QRHKPRE, HAHRSWS, YLTMPTP, TYPISFM, KLPGWSG, IQSPHFF, YSIPKSS, SHRNRPRNTQPS, NRHKPREKTFTD, TAVPLKRSSVTI, GHTANRQPWPND, LSLTRTRHRNTR, RHRDTQNHRPTN, ARHRPKLPYTHT, KRPRTRNKDERR, SPMPQLSTLLTR and NHVHRMHATPAY; all showing selectivity and specificity towards EGFR. Nevertheless, the most prominent peptide sequences targeting EGFR so far are CMYIEALDKYAC (developed based on the structure of the natural EGF ligand) (Ai et al., 2011; Yang et al., 2016), YHWYGYTPQNVI (Kd~22 nmol/L, also known as “GE11”) (Li et al., 2005; Song et al., 2008; Ren et al., 2015; Fan et al., 2016) and LARLLT (also known as “D4”) (Song et al., 2009; Ongarora et al., 2012; Lin and Kao, 2014; Fontenot et al., 2016), was also found to have high specificity towards EGFR, though D4 peptide was developed using a structural model, not through phage display technology.
HER2 is encoded by the ErbB-2 proto-oncogene. The growth and differentiation of cells are regulated by the intracellular domain of HER2 (Yarden and Sliwkowski, 2001; Cho et al., 2003), while the extracellular domain of HER2 interacts with HER family members to form heterodimer complex that facilitates signal transduction (Burstein, 2005). HER2 is a major contributor to breast cancer and about 20%–30% of BC cases are HER2 positive (Lee-Hoeflich et al., 2008; Li et al., 2016). HER2 genes could be amplified to nearly about 2 million receptors on the surface of tumor cells (Kallioniemi et al., 1992; Gutierrez and Schiff, 2011). Therefore, HER2 emerged as a trustworthy drug target when addressing HER2+ cancers (Baselga and Swain, 2009; Rimawi et al., 2015), ovarian (Menderes et al., 2017; Zanini et al., 2017) and gastric cancers (Ruschoff et al., 2012; Abrahao-Machado and Scapulatempo-Neto, 2016). Karasseva et al. described the selection of HER2-binding peptides using phage display. The peptide, KCCYSL bound to purified HER2 with a Kd of 30 mmol/L, and selectively bound to breast and prostate cancer cell lines, but not to normal cells (Karasseva et al., 2002). Houimel et al. isolated three linear peptides specific to HER2 (MARSGL, MARAKE, MSRTMS), and from here derived a humanized pentameric ‘‘peptabody’’ (Pab) molecules (fusion of linear peptide to an antibody-like tail). All three Pab bound to ErbB-2 with Kd~6–16 nmol/L and inhibited HER2+ cancer cell growth and proliferation up to 40% (Houimel et al., 2001). Park et al. isolated bipodal peptide binder aptamer like peptide (aptide) specific to the extra-cellular domain of HER2 (APTHER2, Kd 89 mol/L). This APTHER2 was then conjugated onto superparamagnetic nanoparticles (SPION) for HER2-targeted specific magnetic resonance imaging (MRI) (Park et al., 2013).
VEGFR
Vascular endothelial cell growth factor (VEGF) is a protein tyrosine kinase, and a well-known mediator of angiogenesis which is predominately mostly mediated by VEGF receptor family (VEGRR1, 2, 3; neurophilin 1) (Ferrara et al., 2003; Hoeben et al., 2004). Ample evidences now show that VEGFR family could be exploited as a potent therapeutic target in cancers. Often, the overexpression of VEGF and VEGFR are associated with invasion and metastasis in many malignancies (Prewett et al., 1999), including colorectal (Amaya et al., 1997; Duff et al., 2006), breast (Price et al., 2001; Ryden et al., 2003; Wulfing et al., 2005; Ghosh et al., 2008) and non-small cell lung cancers (Koukourakis et al., 2000; Kajita et al., 2001).
Giordano et al. introduced “Biopanning and Rapid Analysis of Selective Interactive Ligands” (termed BRASIL) as a new approach in the screening, selection and sorting of high affinity peptides. The novelty of this method lies in the additional step of cell-surface-binding peptides sorting based on differential centrifugation. Cell suspension was first incubated with phage in an aqueous upper phase, which will then be centrifuged through a non-miscible organic lower phase. Giordano and colleagues claimed that this single-step organic phase separation is faster, with enhanced sensitivity and specificity comparing to current methods that primarily rely on multiple washing steps or limiting dilution. Using HUVEC cells as a selection, they isolated 21 phage clones bound to starved HUVECs and to VEGF-stimulated HUVECs. Fourteen clones (67%) had a >150% enhancement (range, 1.5–8.7-fold; median, 2.2-fold) binding upon VEGF stimulation. Sequence alignment analysis of 34 clones randomly chosen from the selected phage revealed that 24 clones (70%) of the phage recovered through BRASIL selection had peptide motifs that could be perfectly mapped to sequences present in VEGF family members. They selected two peptides (CPQPRPLC and CNIRRQGC) for in vitro binding assay on VEGF receptor- 1 (VEGFR-1). One of the selected peptides, CPQPRPLC phage bound to VEGFR at over 1,000-fold enrichment as compared to control (Giordano et al., 2001).
Folate receptor alpha (FRα)
Folate receptor alpha (FRα) is a 38-kDa glycoprotein, and is a receptor that binds to folates and mediates their intracellular transport(Henderson, 1990). FRα is significantly up-regulated in a many cancer such as ovarian cancer (OC), endometrial adenocarcinoma and non-small cell lung cancer (NSCLC) (Kane et al., 1988; Matsue et al., 1992; Kelemen, 2006). It is known that the expression of FRα is highly correlated with tumor grade, stage, malignancy and aggressiveness (Bueno et al., 2001; Hartmann et al., 2007), therefore suggesting that FRα is a promising target for tumor therapy and diagnosis.
Xing et al. reported a FRα specific 12-mer peptide C7 (MHTAPGWGYRLS, Kd~0.3 μmol/L) isolated through four rounds of biopanning by using a Ph.D.-12 phage library displaying random dodecapeptides. The tumor targeting ability of C7 was confirmed in in vivo phage homing experiment and fluorescence imaging. C7 was accumulated at the site of tumor tissue, indicating that the peptide has the ability to target tumor tissue without phage environment, indicating the probability of using this peptide for FRα targeted therapy (Xing et al., 2018).
PD-L1
Immune checkpoint inhibition has demonstrated significant success in cancer treatment in recent years, as host immune response could recover from tumor evasion (Pardoll, 2012). By evoking the host’s innate immune response, patients can potentially negate the tumor’s ability to resist targeted therapy, eliminating the need for continuous lines of therapy (Tumeh et al., 2014). One of particular interest is the interaction between programmed cell death receptor 1 (PD-1) and its ligand, programmed cell death ligand 1 (PD-L1) (Zou et al., 2016). PD-L1 expression allow tumor cells to go unrecognized by immune T-cells as foreign. The overexpression on PD-L1 on tumor cells would interact with the PD-1 on the T-cell surface, inhibiting the T-cell to destroy the foreign (tumor) cell (J. Naidoo et al., 2014) Overexpression of PD-L1 has been reported in many different tumor types, such as melanoma (40%–100%), NSCLC (35%–95%), glioblastoma (100%), ovarian cancer (33%–80%), and colorectal adenocarcinoma (53%) (Chen et al., 2012).
Recently, Li et al. used a random bacterial surface display library to screen and identify the PD-L1 binding peptides, and further enriched the peptide binding with PD-L1 with magnetic-activated cell sorting (MACS) and fluorescence-activated cell sorting (FACS). From the initial 5 × 106 peptides library after one cycle of MACS, after eight cycles of FACS, the percentage of peptide in the sorting gate increased from 2.1% (40 nmol/L PD-L1) to 54.1% (10 nmol/L PD-L1). Sequencing of forty bacterial clones revealed nine different peptide sequences with the consensus sequence CWCWR, Kd~95 nmol/L. The soluble peptides of the CWCWR sequence were synthesized, and the binding specificity was tested in PD-L1 high-expressing MDA-MB-231 and low-expressing MDA-MB-435 breast cancer cell lines (Li et al., 2018).
c-MET
c-MET, also called tyrosine-protein kinase Met or hepatocyte growth factor receptor (HGFR), is a protein that is encoded by the MET gene. MET gene was discovered as a proto-oncogene more than two decades ago and it has been extensively studied (Cooper et al., 1984; Bottaro et al., 1991). Met could be activated via autocrine, paracrine, or genetic mutations that can lead to tumorigenesis, angiogenesis and metastasis (Rong et al., 1993; Rong et al., 1994; Takayama et al., 1997). Various studies have linked the overexpression of this C-Met-ligand-pair to most types of human solid tumors, including brain (Jung et al., 1994), breast (Altstock et al., 2000), ovary (Huntsman et al., 1999), thyroid (Di Renzo et al., 1992), pancreas (Ebert et al., 1994), stomach (Di Renzo et al., 1991), prostate (Humphrey et al., 1995) and nasopharyngeal carcinoma (Qian et al., 2002).
To isolate a specific c-Met-binding peptide, Zhao et al. screened for a Met-binding peptide (YLFSVHWPPLKA, Kd~64.2 nmol/L), designated Met-pep1. Met-pep1 binds to Met on the cell surface and thus competed with HGF for Met binding. Interestingly, Met-pep1 is internalized by the cells after binding, and inhibited human leiomyosarcoma SK-LMS-1 proliferation in vitro. In SK-LMS-1 mouse xenograft model, tumor-homing of Met-pep1 was evident as early as 1 h post-injection and remained visible in some animals as late as 24 h post injection (Zhao et al., 2007), indicating that Met-pep1 could be used as a diagnostic agent or a therapeutic carrier in c-MET overexpressing tumors.
CD133
CD133 is first identified as an antigenic marker for hematopoietic stem cells (Miraglia et al., 1997; Yin et al., 1997). CD133 is found to be expressed in several hematopoietic malignancies including acute myelogenous leukemia (Horn et al., 1999), chronic lymphocytic leukemia (Waller et al., 1999), and myelodysplastic syndromes (Green et al., 2000). Recently, CD133 has been reported to be overexpressed in several solid tumors including retinoblastoma (Hemmati et al., 2003), glioblastoma (Singh et al., 2003; Singh et al., 2004), prostate adenocarcinoma (Collins et al., 2005; Rizzo et al., 2005), kidney carcinoma (Florek et al., 2005), pancreatic cancer (Hermann et al., 2007) and colorectal cancers (O’Brien et al., 2007). Importantly, in glioblastoma and colorectal cancer, CD133-expressing cells are considered cancer stem cells (CSCs) as they mediate tumor initiation and metastasis (Singh et al., 2004; O’Brien et al., 2007; Ricci-Vitiani et al., 2007). These small population of CSCs are considered the tumor initiating cell population, and CSCs are often insensitive to chemotherapy and radiation treatment (Neuzil et al., 2007; Tang et al., 2007). Bao et al. showed that CD133+ glioma stem cells mediate radiation resistance in highly malignant gliomas (Bao et al., 2006). Therefore, targeting CD133+ would present an opportunity to eradicate tumor initiating cells, CSCs and tumor cells, also potentially drug-resistant cancer subpopulations (Smith et al., 2008).
Sun et al. identified a peptide binding specifically to mouse CD133, LS-7 (LQNAPRS, Kd~ ND). Co-localization of LS-7 was seen with CD133+ cells but not CD133- cells. LS-7 significantly inhibited cell migration of colon and breast cancer cells. In mice, in vivo treatment of LS-7 homed with high specificity towards CD133+ cells indicating CD133 could be a potential target for anti-motility and anti-metastasis strategy especially in cancer stem cell therapy (Sun et al., 2012).
Prostate-specific membrane antigen (PSMA)
PSMA is a 100 kDa type II transmembrane glycosylated protein and as the name implies, is overexpressed in nearly all prostate cancers cells, its expression is 100–1000 times higher in tumor tissues compared to normal tissues (Wright et al., 1995). The initial descriptions of an increase in PSMA expression in prostate cancer was associated with higher tumor grade with the presence of metastases (Bostwick et al., 1998; Sweat et al., 1998; Chang et al., 2001) suggesting that PSMA is a highly plausible target for PSMA-positive prostate cancer therapy and since has been adopted as a biomarker for diagnosis and imaging (Barve et al., 2014).
To screen for novel PSMA-specific peptide to be used as targeting ligands and targeted drug delivery to prostate cancer cells, Jin et al. identified PSMA-specific peptides through combinatorial phage display techniques. After five rounds of biopanning against recombinant human PSMA extracellular domain (ECD), GTI tripeptide was identified as the highest affinity peptides towards PSMA ECD, with Kd values of the GTI peptide to PSMA-positive LNCaP and C4-2 cells are 8.22 μmol/L and 8.91 μmol/L, respectively. Conjugation of GTI peptide with the proapoptotic peptide D(KLAKLAK)2 induced cell death in LNCaP cells. Also, GTI peptide shows the highest uptake in C4-2 xenografts, with minimal uptake in other organs (Jin et al., 2016).
Scarcity of intracellular targeting peptide: a case study for APTSTAT3
Most peptide therapeutics are peptides targeting intracellular checkpoints in tumor as these peptides could exert therapeutic effects per se via binding and inactive their targets. These peptides usually target transcription factor, enzymes or overexpressed oncogene that are not visible extracellularly. Oncogene-targeted therapeutic strategies have been shown to sensitize tumor cells to the effects of chemotherapy and radiotherapy, and act synergistically with the traditional chemo- and radiotherapeutics (Kumar et al., 1996; Milas et al., 2000; Yu and Hung, 2000; Argiris et al., 2004; Ropero et al., 2004). Nevertheless, compared to extracellular targeting peptide, publications related to intracellular targeting peptide in the suppression of oncogenes or transcription factor has not been on par, and this might be attributed to the inefficiency of the peptides to effectively cross the cellular membrane. However, if succeeded in overcoming this barrier, peptides could be much more effective than antibodies or their derivatives due to the absence of thiolated secondary structure, allowing peptides to retain their original secondary structure in exerting the targeting effect.
STAT3 has received much attention for the important role it plays in signaling pathways linked to cancers (Yu et al., 2009). In cancer cells notably, STAT3 tends to be constitutively activated and had been associated with tumorigenesis and malignancy. Constant STAT3 activation leads to the production of a number of cytokines that regulate proliferation, angiogenesis, survival, and metastasis (Yu et al., 2007). Therefore, many research groups have tried to develop STAT3 inhibitors that can block upstream or downstream elements in the STAT3 signaling pathway (Benekli et al., 2009; Yue and Turkson, 2009). We previously reported an identification of STAT3-binding peptide (APTSTAT3, Kd ~231 nmol/L). Conjugation of APTSTAT3 with a cell-penetrating peptide 9R (APTSTAT3-9R) was developed for enhanced cellular uptake. Not only APTSTAT3-9R blocked STAT3 phosphorylation, they also reduced the expression of STAT downstream molecules in various types of cancer cells (melanoma, breast, lung, liver and brain cancer) Furthermore, intra-tumoral injection of APTSTAT3-9R exerted potent antitumor activity in both xenograft and allograft tumor models. This study suggested a solid preclinical proof-of-concept for APTSTAT3 as a powerful agent for STAT3 inhibition for targeting broad array of cancers with constitutively activated STAT3 (Fig. 6).

Efficient intracellular delivery of peptides. (A–C) APTSTAT3 conjugated with cell penetrating peptide (CPP), 9R allowed high intracellular targeting of APTSTAT3. (D) Unlike antibodies, the absence of disulfide bond in the secondary structure of peptide ensures that APTSTAT3 remained biologically active in high glutathione (GSH) condition in the intracellular compartment. (E) treatment with APTSTAT3-9R prolonged mice survival
Challenges and future outlook
The utilization of peptide as a targeting could bring forth multiple advantages - such as highly specific, naturally degradable, easily synthesized, and simple tunability with a variety of linker chemistries, and potentially reduce side effects and toxicity (Wang et al., 2017). However, there are also various hurdles that needed to be overcome in order for these peptides to be developed in the clinics.
Increasing peptide avidity
The affinity of a peptide is used to describe the strength of a peptide-ligand interaction. Most peptides possess high affinity towards target (nanomolar to micromolar), which could be considered as high affinity. However, a short, singular linear ligand, peptides usually lack avidity, that is the ability to bind to the target via multiple interactions that can synergize their binding to enhance the affinity and also lead to enhancement of target residence time resulting in high local concentration of the targeted molecules (Vauquelin and Charlton, 2013). To overcome this barrier, most researchers decorated short linear peptides on the surface of nanocarriers to increase the probability of the peptide to interact with the specific ligands. Rouslahti et al. fused NGR peptide to TNFα, a highly toxic cytokine whose clinical application was limited due to its systemic toxicity. These targeted cytokines were effective even at 1,000-fold lower concentration that than usual dose, therefore diminishing the highly toxic side effects of TNFα. The success of this peptide-cytokine fusion could be attributed to the fact that the quaternary structure of TNFα is a trimer and the NGR peptide could be attached to each subunit, resulting in three NGR peptide: TNFα ratio; enhancing receptor binding of NGR peptide through an avidity effect (Ruoslahti, 2012). Similar strategy was adopted by Jeon et al., when they described an EDB-targeting aptide fused to mouse TNF-α (mTNFα-APTEDB) for systemic and targeted therapy of EDB-overexpressing fibrosarcoma (Jeon et al., 2017). mTNFα-APTEDB showed enhanced tumor inhibition properties than mTNFα alone or mTNFα linked to a nonrelevant aptide, without causing an appreciable toxicity as measured in body weight loss.
Reducing peptide aggregation and increasing peptide solubility
Peptide with 5 amino acids and less are usually water soluble and their solubility decreases with the length of the peptide. However, peptides screened through phage display biopanning ranged between 7–30 amino acids. In aqueous solution, these peptides could be conforming to a specific 3-dimentional structure that allowed specific binding with their receptors. In practice, solubilizing peptides could be challenging as improper solubilization could result in the loss of the peptide activity. For this reason, Xiao et al. conjugated betaine onto bacterial xanthine guanine phosphoribosyltransferase (CG-GPRT) protein and the HIV inhibitory peptide (CG-T20). Results indicated that betaine could successfully reduce the protein/peptide aggregation and increased the solubility of both the protein and the peptide (Xiao et al., 2008), therefore suggesting that betaine conjugation could be used for reducing peptide aggregation and increasing peptide solubility.
Overcoming poor cell permeability and increase cellular uptake
Since the discovery of natural CPPs (Tat and Penetratin), a number of synthetic peptides have since been added to this family; including short peptides comprising positive-charged amino acids such as arginine, lysine or histidine. To date, many reports on CPPs in their application as intracellular delivery vehicles, including small-molecule drugs (Lindgren et al., 2006), liposomes (Zhang et al., 2013), and biopharmaceuticals including oligonucleotides (Margus et al., 2012), peptides and proteins (Morris et al., 2001). When conjugated with TAT peptide, pro-apoptotic peptides (KLAKLAK)2 conjugate was taken up efficiently by mouse melanoma and human breast cancer cells in vitro. In the cells, the peptide conjugate further activated the endogenous caspase-3 which then cleaved the peptide resulting in release of the pro-apoptotic peptide (KLAKLAK)2. Not only this peptide induced apoptosis in these cells in vitro, they also inhibited the growth of mouse melanoma xenografts in mice (Kwon et al., 2008). This peptide conjugate also induced apoptosis in the various cancer cell lines such as melanoma, cervical carcinoma, non-small cell lung carcinoma, breast cancer (Yang et al., 2010).
Phage display biopanning technique has brought about an immense pool of high affinity and highly specific peptide ligand for solid tumor therapy. Many of these peptide-based targeting ligands have shown promising results in enhancing solid tumors therapy, including increasing tumor accumulation, highly specific tumor targeting and enhanced tumor inhibition effect when used in combination with anti-cancer drugs or biologics. However, for successful translation into the clinics, peptide-targeting ligand should be optimized for their affinity, avidity, water-solubility and target specificity. With the advancement of technology, one could now use a combined primary phage display screening and a secondary computational optimization method to develop an optimal peptide for targeting any receptor of interest in the field of solid tumor therapy.
References
Abrahao-Machado LF, Scapulatempo-Neto C (2016) HER2 testing in gastric cancer: an update. World J Gastroenterol 22:4619–4625
Ai S, Duan J, Liu X, Bock S, Tian Y, Huang Z (2011) Biological evaluation of a novel doxorubicin-peptide conjugate for targeted delivery to EGF receptor-overexpressing tumor cells. Mol Pharm 8:375–386
Albrecht M, Renneberg H, Wennemuth G, Moschler O, Janssen M, Aumuller G, Konrad L (1999) Fibronectin in human prostatic cells in vivo and in vitro: expression, distribution, and pathological significance. Histochem Cell Biol 112:51–61
AlDeghaither D, Smaglo BG, Weiner LM (2015) Beyond peptides and mAbs—current status and future perspectives for biotherapeutics with novel constructs. J Clin Pharmacol 55(Suppl 3):S4–20
Altstock RT, Stein GY, Resau JH, Tsarfaty I (2000) Algorithms for quantitation of protein expression variation in normal versus tumor tissue as a prognostic factor in cancer: Met oncogene expression, and breast cancer as a model. Cytometry 41:155–165
Amaya H, Tanigawa N, Lu C, Matsumura M, Shimomatsuya T, Horiuchi T, Muraoka R (1997) Association of vascular endothelial growth factor expression with tumor angiogenesis, survival and thymidine phosphorylase/platelet-derived endothelial cell growth factor expression in human colorectal cancer. Cancer Lett 119:227–235
Arap W, Pasqualini R, Ruoslahti E (1998) Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 279:377–380
Arap W, Haedicke W, Bernasconi M, Kain R, Rajotte D, Krajewski S, Ellerby HM, Bredesen DE, Pasqualini R, Ruoslahti E (2002a) Targeting the prostate for destruction through a vascular address. Proc Natl Acad Sci U S A 99:1527–1531
Arap W, Kolonin MG, Trepel M, Lahdenranta J, Cardo-Vila M, Giordano RJ, Mintz PJ, Ardelt PU, Yao VJ, Vidal CI et al (2002b) Steps toward mapping the human vasculature by phage display. Nat Med 8:121–127
Argiris A, Wang CX, Whalen SG, DiGiovanna MP (2004) Synergistic interactions between tamoxifen and trastuzumab (Herceptin). Clin Cancer Res 10:1409–1420
Armulik A, Abramsson A, Betsholtz C (2005) Endothelial/pericyte interactions. Circ Res 97:512–523
Arnold SA, Loomans HA, Ketova T, Andl CD, Clark PE, Zijlstra A (2016) Urinary oncofetal ED-A fibronectin correlates with poor prognosis in patients with bladder cancer. Clin Exp Metastasis 33:29–44
Augsten M (2014) Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front Oncol 4:62
Bae YK, Kim A, Kim MK, Choi JE, Kang SH, Lee SJ (2013) Fibronectin expression in carcinoma cells correlates with tumor aggressiveness and poor clinical outcome in patients with invasive breast cancer. Hum Pathol 44:2028–2037
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444:756–760
Barve A, Jin W, Cheng K (2014) Prostate cancer relevant antigens and enzymes for targeted drug delivery. J Control Release 187:118–132
Baselga J, Swain SM (2009) Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer 9:463–475
Benekli M, Baumann H, Wetzler M (2009) Targeting signal transducer and activator of transcription signaling pathway in leukemias. J Clin Oncol 27:4422–4432
Bergers G, Javaherian K, Lo KM, Folkman J, Hanahan D (1999) Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science 284:808–812
Bhowmick NA, Neilson EG, Moses HL (2004) Stromal fibroblasts in cancer initiation and progression. Nature 432:332–337
Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC et al (2018) Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 24:541–550
Birkedal-Hansen H (1995) Proteolytic remodeling of extracellular matrix. Curr Opin Cell Biol 7:728–735
Boehm T, Folkman J, Browder T, O’Reilly MS (1997) Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature 390:404–407
Boohaker RJ, Lee MW, Vishnubhotla P, Perez JM, Khaled AR (2012) The use of therapeutic peptides to target and to kill cancer cells. Curr Med Chem 19:3794–3804
Bostwick DG, Pacelli A, Blute M, Roche P, Murphy GP (1998) Prostate specific membrane antigen expression in prostatic intraepithelial neoplasia and adenocarcinoma: a study of 184 cases. Cancer 82:2256–2261
Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, Aaronson SA (1991) Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 251:802–804
Brinton LT, Bauknight DK, Dasa SS, Kelly KA (2016) PHASTpep: analysis software for discovery of cell-selective peptides via phage display and next-generation sequencing. PLoS ONE 11:e0155244
Brooks PC, Stromblad S, Sanders LC, von Schalscha TL, Aimes RT, Stetler-Stevenson WG, Quigley JP, Cheresh DA (1996) Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell 85:683–693
Bueno R, Appasani K, Mercer H, Lester S, Sugarbaker D (2001) The alpha folate receptor is highly activated in malignant pleural mesothelioma. J Thorac Cardiovasc Surg 121:225–233
Burg MA, Pasqualini R, Arap W, Ruoslahti E, Stallcup WB (1999) NG2 proteoglycan-binding peptides target tumor neovasculature. Cancer Res 59:2869–2874
Burstein HJ (2005) The distinctive nature of HER2-positive breast cancers. N Engl J Med 353:1652–1654
Chan B, Sinha S, Cho D, Ramchandran R, Sukhatme VP (2005) Critical roles of CD146 in zebrafish vascular development. Dev Dyn 232:232–244
Chang SS, Reuter VE, Heston WD, Gaudin PB (2001) Comparison of anti-prostate-specific membrane antigen antibodies and other immunomarkers in metastatic prostate carcinoma. Urology 57:1179–1183
Chang DK, Lin CT, Wu CH, Wu HC (2009) A novel peptide enhances therapeutic efficacy of liposomal anti-cancer drugs in mice models of human lung cancer. PLoS ONE 4:e4171
Chen J, Yao Y, Gong C, Yu F, Su S, Chen J, Liu B, Deng H, Wang F, Lin L et al (2011) CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell 19:541–555
Chen DS, Irving BA, Hodi FS (2012) Molecular pathways: next-generation immunotherapy-inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res 18:6580–6587
Chiquet-Ehrismann R (1990) What distinguishes tenascin from fibronectin? FASEB J 4:2598–2604
Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW Jr, Leahy DJ (2003) Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421:756–760
Cieslewicz M, Tang J, Yu JL, Cao H, Zavaljevski M, Motoyama K, Lieber A, Raines EW, Pun SH (2013) Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival. Proc Natl Acad Sci U S A 110:15919–15924
Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ (2005) Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 65:10946–10951
Colvin EK (2014) Tumor-associated macrophages contribute to tumor progression in ovarian cancer. Front Oncol 4:137
Cooper CS, Park M, Blair DG, Tainsky MA, Huebner K, Croce CM, Vande Woude GF (1984) Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 311:29–33
Denekamp J (1993) Review article: angiogenesis, neovascular proliferation and vascular pathophysiology as targets for cancer therapy. Br J Radiol 66:181–196
Di Renzo MF, Narsimhan RP, Olivero M, Bretti S, Giordano S, Medico E, Gaglia P, Zara P, Comoglio PM (1991) Expression of the Met/HGF receptor in normal and neoplastic human tissues. Oncogene 6:1997–2003
Di Renzo MF, Olivero M, Ferro S, Prat M, Bongarzone I, Pilotti S, Belfiore A, Costantino A, Vigneri R, Pierotti MA et al (1992) Overexpression of the c-MET/HGF receptor gene in human thyroid carcinomas. Oncogene 7:2549–2553
Duff SE, Jeziorska M, Rosa DD, Kumar S, Haboubi N, Sherlock D, O’Dwyer ST, Jayson GC (2006) Vascular endothelial growth factors and receptors in colorectal cancer: implications for anti-angiogenic therapy. Eur J Cancer 42:112–117
Ebert M, Yokoyama M, Friess H, Buchler MW, Korc M (1994) Coexpression of the c-met proto-oncogene and hepatocyte growth factor in human pancreatic cancer. Cancer Res 54:5775–5778
Essler M, Ruoslahti E (2002) Molecular specialization of breast vasculature: a breast-homing phage-displayed peptide binds to aminopeptidase P in breast vasculature. Proc Natl Acad Sci U S A 99:2252–2257
Fan M, Liang X, Yang D, Pan X, Li Z, Wang H, Shi B (2016) Epidermal growth factor receptor-targeted peptide conjugated phospholipid micelles for doxorubicin delivery. J Drug Target 24:111–119
Ferrara N, Alitalo K (1999) Clinical applications of angiogenic growth factors and their inhibitors. Nat Med 5:1359–1364
Ferrara N, Gerber HP, LeCouter J (2003) The biology of VEGF and its receptors. Nat Med 9:669–676
Florek M, Haase M, Marzesco AM, Freund D, Ehninger G, Huttner WB, Corbeil D (2005) Prominin-1/CD133, a neural and hematopoietic stem cell marker, is expressed in adult human differentiated cells and certain types of kidney cancer. Cell Tissue Res 319:15–26
Folkman J (1971) Tumor angiogenesis: therapeutic implications. N Engl J Med 285:1182–1186
Fontenot KR, Ongarora BG, LeBlanc LE, Zhou Z, Jois SD, Vicente MG (2016) Targeting of the epidermal growth factor receptor with mesoporphyrin IX–peptide conjugates. J Porphyr Phthalocyanines 20:352–366
Franco OE, Shaw AK, Strand DW, Hayward SW (2010) Cancer associated fibroblasts in cancer pathogenesis. Semin Cell Dev Biol 21:33–39
Georgoudaki AM, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Ostling J, Dahan R, Harris RA, Rantalainen M, Klevebring D et al (2016) Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep 15:2000–2011
Gerhardt H, Betsholtz C (2003) Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res 314:15–23
Ghert MA, Jung ST, Qi W, Harrelson JM, Erickson HP, Block JA, Scully SP (2001) The clinical significance of tenascin-C splice variant expression in chondrosarcoma. Oncology 61:306–314
Ghosh S, Sullivan CA, Zerkowski MP, Molinaro AM, Rimm DL, Camp RL, Chung GG (2008) High levels of vascular endothelial growth factor and its receptors (VEGFR-1, VEGFR-2, neuropilin-1) are associated with worse outcome in breast cancer. Hum Pathol 39:1835–1843
Giordano RJ, Cardo-Vila M, Lahdenranta J, Pasqualini R, Arap W (2001) Biopanning and rapid analysis of selective interactive ligands. Nat Med 7:1249–1253
Goodson RJ, Doyle MV, Kaufman SE, Rosenberg S (1994) High-affinity urokinase receptor antagonists identified with bacteriophage peptide display. Proc Natl Acad Sci U S A 91:7129–7133
Grandis JR, Sok JC (2004) Signaling through the epidermal growth factor receptor during the development of malignancy. Pharmacol Ther 102:37–46
Green CL, Loken M, Buck D, Deeg HJ (2000) Discordant expression of AC133 and AC141 in patients with myelodysplastic syndrome (MDS) and acute myelogeneous leukemia (AML). Leukemia 14:770–772
Gutierrez C, Schiff R (2011) HER2: biology, detection, and clinical implications. Arch Pathol Lab Med 135:55–62
Han Z, Zhou Z, Shi X, Wang J, Wu X, Sun D, Chen Y, Zhu H, Magi-Galluzzi C, Lu ZR (2015) EDB fibronectin specific peptide for prostate cancer targeting. Bioconjug Chem 26:830–838
Hanahan D, Folkman J (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86:353–364
Hanamura N, Yoshida T, Matsumoto E, Kawarada Y, Sakakura T (1997) Expression of fibronectin and tenascin-C mRNA by myofibroblasts, vascular cells and epithelial cells in human colon adenomas and carcinomas. Int J Cancer 73:10–15
Harhouri K, Kebir A, Guillet B, Foucault-Bertaud A, Voytenko S, Piercecchi-Marti MD, Berenguer C, Lamy E, Vely F, Pisano P et al (2010) Soluble CD146 displays angiogenic properties and promotes neovascularization in experimental hind-limb ischemia. Blood 115:3843–3851
Hartmann LC, Keeney GL, Lingle WL, Christianson TJ, Varghese B, Hillman D, Oberg AL, Low PS (2007) Folate receptor overexpression is associated with poor outcome in breast cancer. Int J Cancer 121:938–942
Hashida M, Nishikawa M, Yamashita F, Takakura Y (2001) Cell-specific delivery of genes with glycosylated carriers. Adv Drug Deliv Rev 52:187–196
Hayashi MA, Ducancel F, Konno K (2012) Natural peptides with potential applications in drug development, diagnosis, and/or biotechnology. Int J Pept 2012:757838
He C, Su S, Chen F, Huang D, Zheng F, Huang W, Chen J, Cui X, Liu Q, Song E et al (2014) Overexpression of PITPNM3 promotes hepatocellular carcinoma cell metastasis. Chin Sci Bull 59:1326–1333
Hellstrom I, Beaumier PL, Hellstrom KE (1986a) Antitumor effects of L6, an IgG2a antibody that reacts with most human carcinomas. Proc Natl Acad Sci U S A 83:7059–7063
Hellstrom I, Horn D, Linsley P, Brown JP, Brankovan V, Hellstrom KE (1986b) Monoclonal mouse antibodies raised against human lung carcinoma. Cancer Res 46:3917–3923
Hemesath TJ, Stefansson K (1994) Expression of tenascin in thymus and thymic nonlymphoid cells. J Immunol 152:422–428
Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI (2003) Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A 100:15178–15183
Henderson GB (1990) Folate-binding proteins. Annu Rev Nutr 10:319–335
Heppeler A, Froidevaux S, Eberle AN, Maecke HR (2000) Receptor targeting for tumor localisation and therapy with radiopeptides. Curr Med Chem 7:971–994
Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C (2007) Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 1:313–323
Hindermann W, Berndt A, Borsi L, Luo X, Hyckel P, Katenkamp D, Kosmehl H (1999) Synthesis and protein distribution of the unspliced large tenascin-C isoform in oral squamous cell carcinoma. J Pathol 189:475–480
Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA (2004) Vascular endothelial growth factor and angiogenesis. Pharmacol Rev 56:549–580
Horn PA, Tesch H, Staib P, Kube D, Diehl V, Voliotis D (1999) Expression of AC133, a novel hematopoietic precursor antigen, on acute myeloid leukemia cells. Blood 93:1435–1437
Houimel M, Schneider P, Terskikh A, Mach JP (2001) Selection of peptides and synthesis of pentameric peptabody molecules reacting specifically with ErbB-2 receptor. Int J Cancer 92:748–755
Humphrey PA, Zhu X, Zarnegar R, Swanson PE, Ratliff TL, Vollmer RT, Day ML (1995) Hepatocyte growth factor and its receptor (c-MET) in prostatic carcinoma. Am J Pathol 147:386–396
Huntsman D, Resau JH, Klineberg E, Auersperg N (1999) Comparison of c-met expression in ovarian epithelial tumors and normal epithelia of the female reproductive tract by quantitative laser scan microscopy. Am J Pathol 155:343–348
Inufusa H, Nakamura M, Adachi T, Nakatani Y, Shindo K, Yasutomi M, Matsuura H (1995) Localization of oncofetal and normal fibronectin in colorectal cancer. Correlation with histologic grade, liver metastasis, and prognosis. Cancer 75:2802–2808
Ioachim E, Charchanti A, Briasoulis E, Karavasilis V, Tsanou H, Arvanitis DL, Agnantis NJ, Pavlidis N (2002) Immunohistochemical expression of extracellular matrix components tenascin, fibronectin, collagen type IV and laminin in breast cancer: their prognostic value and role in tumour invasion and progression. Eur J Cancer 38:2362–2370
Jain RK (2001) Delivery of molecular and cellular medicine to solid tumors. Adv Drug Deliv Rev 46:149–168
Jeon H, Kim D, Choi M, Kang S, Kim JY, Kim S, Jon S (2017) Targeted Cancer Therapy Using Fusion Protein of TNFalpha and Tumor-Associated Fibronectin-Specific Aptide. Mol Pharm 14:3772–3779
Jin W, Qin B, Chen Z, Liu H, Barve A, Cheng K (2016) Discovery of PSMA-specific peptide ligands for targeted drug delivery. Int J Pharm 513:138–147
Jung W, Castren E, Odenthal M, Vande Woude GF, Ishii T, Dienes HP, Lindholm D, Schirmacher P (1994) Expression and functional interaction of hepatocyte growth factor-scatter factor and its receptor c-met in mammalian brain. J Cell Biol 126:485–494
Jyothi T (2012) Cancer treatment using peptides: current therapies and future prospects. J Amino Acids 2012:13
Kajita T, Ohta Y, Kimura K, Tamura M, Tanaka Y, Tsunezuka Y, Oda M, Sasaki T, Watanabe G (2001) The expression of vascular endothelial growth factor C and its receptors in non-small cell lung cancer. Br J Cancer 85:255–260
Kallioniemi OP, Kallioniemi A, Kurisu W, Thor A, Chen LC, Smith HS, Waldman FM, Pinkel D, Gray JW (1992) ERBB2 amplification in breast cancer analyzed by fluorescence in situ hybridization. Proc Natl Acad Sci U S A 89:5321–5325
Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119:1420–1428
Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6:392–401
Kane MA, Elwood PC, Portillo RM, Antony AC, Najfeld V, Finley A, Waxman S, Kolhouse JF (1988) Influence on immunoreactive folate-binding proteins of extracellular folate concentration in cultured human cells. J Clin Invest 81:1398–1406
Kang Y, Wang F, Feng J, Yang D, Yang X, Yan X (2006) Knockdown of CD146 reduces the migration and proliferation of human endothelial cells. Cell Res 16:313–318
Karasseva NG, Glinsky VV, Chen NX, Komatireddy R, Quinn TP (2002) Identification and characterization of peptides that bind human ErbB-2 selected from a bacteriophage display library. J Protein Chem 21:287–296
Kaspar M, Zardi L, Neri D (2006) Fibronectin as target for tumor therapy. Int J Cancer 118:1331–1339
Kebir A, Harhouri K, Guillet B, Liu JW, Foucault-Bertaud A, Lamy E, Kaspi E, Elganfoud N, Vely F, Sabatier F et al (2010) CD146 short isoform increases the proangiogenic potential of endothelial progenitor cells in vitro and in vivo. Circ Res 107:66–75
Kelemen LE (2006) The role of folate receptor alpha in cancer development, progression and treatment: cause, consequence or innocent bystander? Int J Cancer 119:243–250
Kelly KA, Allport JR, Tsourkas A, Shinde-Patil VR, Josephson L, Weissleder R (2005) Detection of vascular adhesion molecule-1 expression using a novel multimodal nanoparticle. Circ Res 96:327–336
Kerbel RS (1991) Inhibition of tumor angiogenesis as a strategy to circumvent acquired resistance to anti-cancer therapeutic agents. BioEssays 13:31–36
Khan ZA, Caurtero J, Barbin YP, Chan BM, Uniyal S, Chakrabarti S (2005) ED-B fibronectin in non-small cell lung carcinoma. Exp Lung Res 31:701–711
Kim MY, Kim OR, Choi YS, Lee H, Park K, Lee CT, Kang KW, Jeong S (2012a) Selection and characterization of tenascin C targeting peptide. Mol Cells 33:71–77
Kim S, Kim D, Jung HH, Lee IH, Kim JI, Suh JY, Jon S (2012b) Bio-inspired design and potential biomedical applications of a novel class of high-affinity peptides. Angew Chem Int Ed Engl 51:1890–1894
Kim H, Lee Y, Lee IH, Kim S, Kim D, Saw PE, Lee J, Choi M, Kim YC, Jon S (2014) Synthesis and therapeutic evaluation of an aptide-docetaxel conjugate targeting tumor-associated fibronectin. J Control Release 178:118–124
Kim H, Lee Y, Kang S, Choi M, Lee S, Kim S, Gujrati V, Kim J, Jon S (2016) Self-assembled nanoparticles comprising aptide-SN38 conjugates for use in targeted cancer therapy. Nanotechnology 27:48LT01
Klein G, Beck S, Muller CA (1993) Tenascin is a cytoadhesive extracellular matrix component of the human hematopoietic microenvironment. J Cell Biol 123:1027–1035
Koivunen E, Wang B, Ruoslahti E (1995) Phage libraries displaying cyclic peptides with different ring sizes: ligand specificities of the RGD-directed integrins. Biotechnology (N Y) 13:265–270
Koivunen E, Arap W, Valtanen H, Rainisalo A, Medina OP, Heikkila P, Kantor C, Gahmberg CG, Salo T, Konttinen YT et al (1999) Tumor targeting with a selective gelatinase inhibitor. Nat Biotechnol 17:768–774
Koukourakis MI, Giatromanolaki A, Thorpe PE, Brekken RA, Sivridis E, Kakolyris S, Georgoulias V, Gatter KC, Harris AL (2000) Vascular endothelial growth factor/KDR activated microvessel density versus CD31 standard microvessel density in non-small cell lung cancer. Cancer Res 60:3088–3095
Krag DN, Shukla GS, Shen GP, Pero S, Ashikaga T, Fuller S, Weaver DL, Burdette-Radoux S, Thomas C (2006) Selection of tumor-binding ligands in cancer patients with phage display libraries. Cancer Res 66:7724–7733
Kumar R, Mandal M, Lipton A, Harvey H, Thompson CB (1996) Overexpression of HER2 modulates bcl-2, bcl-XL, and tamoxifen-induced apoptosis in human MCF-7 breast cancer cells. Clin Cancer Res 2:1215–1219
Kurahara H, Shinchi H, Mataki Y, Maemura K, Noma H, Kubo F, Sakoda M, Ueno S, Natsugoe S, Takao S (2011) Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J Surg Res 167:e211–e219
Kwon MK, Nam JO, Park RW, Lee BH, Park JY, Byun YR, Kim SY, Kwon IC, Kim IS (2008) Antitumor effect of a transducible fusogenic peptide releasing multiple proapoptotic peptides by caspase-3. Mol Cancer Ther 7:1514–1522
Laakkonen P, Porkka K, Hoffman JA, Ruoslahti E (2002) A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat Med 8:751–755
Landon LA, Deutscher SL (2003) Combinatorial discovery of tumor targeting peptides using phage display. J Cell Biochem 90:509–517
Lee TY, Lin CT, Kuo SY, Chang DK, Wu HC (2007) Peptide-mediated targeting to tumor blood vessels of lung cancer for drug delivery. Cancer Res 67:10958–10965
Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, Sliwkowski MX, Stern HM (2008) A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res 68:5878–5887
Lehmann JM, Riethmuller G, Johnson JP (1989) MUC18, a marker of tumor progression in human melanoma, shows sequence similarity to the neural cell adhesion molecules of the immunoglobulin superfamily. Proc Natl Acad Sci U S A 86:9891–9895
Li Z, Zhao R, Wu X, Sun Y, Yao M, Li J, Xu Y, Gu J (2005) Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J 19:1978–1985
Li J, Wang H, Li J, Bao J, Wu C (2016) Discovery of a potential HER2 inhibitor from natural products for the treatment of HER2-Positive Breast cancer. Int J Mol Sci 17:E1055
Li C, Zhang N, Zhou J, Ding C, Jin Y, Cui X, Pu K, Zhu Y (2018) Peptide Blocking of PD-1/PD-L1 Interaction for Cancer Immunotherapy. Cancer Immunol Res 6:178–188
Lin WJ, Kao LT (2014) Cytotoxic enhancement of hexapeptide-conjugated micelles in EGFR high-expressed cancer cells. Expert Opin Drug Deliv 11:1537–1550
Lindgren M, Rosenthal-Aizman K, Saar K, Eiriksdottir E, Jiang Y, Sassian M, Ostlund P, Hallbrink M, Langel U (2006) Overcoming methotrexate resistance in breast cancer tumour cells by the use of a new cell-penetrating peptide. Biochem Pharmacol 71:416–425
Liu X, Peng J, He J, Li Q, Zhou J, Liang X, Tang S (2018) Selection and identification of novel peptides specifically targeting human cervical cancer. Amino Acids 50:577–592
Lo A, Lin CT, Wu HC (2008) Hepatocellular carcinoma cell-specific peptide ligand for targeted drug delivery. Mol Cancer Ther 7:579–589
Low PS, Henne WA, Doorneweerd DD (2008) Discovery and development of folic-acid-based receptor targeting for imaging and therapy of cancer and inflammatory diseases. Acc Chem Res 41:120–129
Lyons AJ, Bateman AC, Spedding A, Primrose JN, Mandel U (2001) Oncofetal fibronectin and oral squamous cell carcinoma. Br J Oral Maxillofac Surg 39:471–477
Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P (2017) Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 14:399–416
Margus H, Padari K, Pooga M (2012) Cell-penetrating peptides as versatile vehicles for oligonucleotide delivery. Mol Ther 20:525–533
Marken JS, Schieven GL, Hellstrom I, Hellstrom KE, Aruffo A (1992) Cloning and expression of the tumor-associated antigen L6. Proc Natl Acad Sci U S A 89:3503–3507
Marqus S, Pirogova E, Piva TJ (2017) Evaluation of the use of therapeutic peptides for cancer treatment. J Biomed Sci 24:21
Matsue H, Rothberg KG, Takashima A, Kamen BA, Anderson RG, Lacey SW (1992) Folate receptor allows cells to grow in low concentrations of 5-methyltetrahydrofolate. Proc Natl Acad Sci U S A 89:6006–6009
Medrek C, Ponten F, Jirstrom K, Leandersson K (2012) The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer 12:306
Mendelsohn J, Baselga J (2003) Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J Clin Oncol 21:2787–2799
Menderes G, Bonazzoli E, Bellone S, Black JD, Lopez S, Pettinella F, Masserdotti A, Zammataro L, Litkouhi B, Ratner E et al (2017) Efficacy of neratinib in the treatment of HER2/neu-amplified epithelial ovarian carcinoma in vitro and in vivo. Med Oncol 34:91
Mhawech P, Dulguerov P, Assaly M, Ares C, Allal AS (2005) EB-D fibronectin expression in squamous cell carcinoma of the head and neck. Oral Oncol 41:82–88
Milas L, Mason K, Hunter N, Petersen S, Yamakawa M, Ang K, Mendelsohn J, Fan Z (2000) In vivo enhancement of tumor radioresponse by C225 antiepidermal growth factor receptor antibody. Clin Cancer Res 6:701–708
Miraglia S, Godfrey W, Yin AH, Atkins K, Warnke R, Holden JT, Bray RA, Waller EK, Buck DW (1997) A novel five-transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood 90:5013–5021
Morris MC, Depollier J, Mery J, Heitz F, Divita G (2001) A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat Biotechnol 19:1173–1176
Naidoo J, Page DB, Wolchok JD (2014) Immune Checkpoint Blockade. Hematol Oncol Clin North Am 28:585–600
Ndinguri MW, Bhowmick M, Tokmina-Roszyk D, Robichaud TK, Fields GB (2012) Peptide-based selective inhibitors of matrix metalloproteinase-mediated activities. Molecules 17:14230–14248
Neuzil J, Stantic M, Zobalova R, Chladova J, Wang X, Prochazka L, Dong L, Andera L, Ralph SJ (2007) Tumour-initiating cells vs. cancer ‘stem’ cells and CD133: what’s in the name? Biochem Biophys Res Commun 355:855–859
Nishiyama A, Lin XH, Giese N, Heldin CH, Stallcup WB (1996) Interaction between NG2 proteoglycan and PDGF alpha-receptor on O2A progenitor cells is required for optimal response to PDGF. J Neurosci Res 43:315–330
O’Brien CA, Pollett A, Gallinger S, Dick JE (2007) A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445:106–110
Olayioye MA, Neve RM, Lane HA, Hynes NE (2000) The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J 19:3159–3167
Ongarora BG, Fontenot KR, Hu X, Sehgal I, Satyanarayana-Jois SD, Vicente MG (2012) Phthalocyanine-peptide conjugates for epidermal growth factor receptor targeting. J Med Chem 55:3725–3738
O’Reilly MS, Pirie-Shepherd S, Lane WS, Folkman J (1999) Antiangiogenic activity of the cleaved conformation of the serpin antithrombin. Science 285:1926–1928
Ouhtit A, Gaur RL, Abd Elmageed ZY, Fernando A, Thouta R, Trappey AK, Abdraboh ME, El-Sayyad HI, Rao P, Raj MG (2009) Towards understanding the mode of action of the multifaceted cell adhesion receptor CD146. Biochim Biophys Acta 1795:130–136
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12:252–264
Park J, Kim S, Saw PE, Lee IH, Yu MK, Kim M, Lee K, Kim YC, Jeong YY, Jon S (2012) Fibronectin extra domain B-specific aptide conjugated nanoparticles for targeted cancer imaging. J Control Release 163:111–118
Park J, Park S, Kim S, Lee I-H, Saw PE, Lee K, Kim Y-C, Kim Y-J, Farokhzad OC, Jeong YY et al (2013) HER2-specific aptide conjugated magneto-nanoclusters for potential breast cancer imaging and therapy. J Mater Chem B 1:4576–4583
Pasqualini R, Ruoslahti E (1996) Organ targeting in vivo using phage display peptide libraries. Nature 380:364–366
Pasqualini R, Koivunen E, Ruoslahti E (1997) Alpha v integrins as receptors for tumor targeting by circulating ligands. Nat Biotechnol 15:542–546
Pasqualini R, Koivunen E, Kain R, Lahdenranta J, Sakamoto M, Stryhn A, Ashmun RA, Shapiro LH, Arap W, Ruoslahti E (2000) Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res 60:722–727
Prewett M, Huber J, Li Y, Santiago A, O’Connor W, King K, Overholser J, Hooper A, Pytowski B, Witte L et al (1999) Antivascular endothelial growth factor receptor (fetal liver kinase 1) monoclonal antibody inhibits tumor angiogenesis and growth of several mouse and human tumors. Cancer Res 59:5209–5218
Price DJ, Miralem T, Jiang S, Steinberg R, Avraham H (2001) Role of vascular endothelial growth factor in the stimulation of cellular invasion and signaling of breast cancer cells. Cell Growth Differ 12:129–135
Pupa SM, Menard S, Forti S, Tagliabue E (2002) New insights into the role of extracellular matrix during tumor onset and progression. J Cell Physiol 192:259–267
Qian CN, Guo X, Cao B, Kort EJ, Lee CC, Chen J, Wang LM, Mai WY, Min HQ, Hong MH et al (2002) Met protein expression level correlates with survival in patients with late-stage nasopharyngeal carcinoma. Cancer Res 62:589–596
Rajotte D, Arap W, Hagedorn M, Koivunen E, Pasqualini R, Ruoslahti E (1998) Molecular heterogeneity of the vascular endothelium revealed by in vivo phage display. J Clin Invest 102:430–437
Rasanen K, Vaheri A (2010) Activation of fibroblasts in cancer stroma. Exp Cell Res 316:2713–2722
Ren H, Gao C, Zhou L, Liu M, Xie C, Lu W (2015) EGFR-targeted poly(ethylene glycol)-distearoylphosphatidylethanolamine micelle loaded with paclitaxel for laryngeal cancer: preparation, characterization and in vitro evaluation. Drug Deliv 22:785–794
Ribeiro AL, Okamoto OK (2015) Combined effects of pericytes in the tumor microenvironment. Stem Cells Int 2015:868475
Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R (2007) Identification and expansion of human colon-cancer-initiating cells. Nature 445:111–115
Rimawi MF, Schiff R, Osborne CK (2015) Targeting HER2 for the treatment of breast cancer. Annu Rev Med 66:111–128
Rizzo S, Attard G, Hudson DL (2005) Prostate epithelial stem cells. Cell Prolif 38:363–374
Rong S, Jeffers M, Resau JH, Tsarfaty I, Oskarsson M, Vande Woude GF (1993) Met expression and sarcoma tumorigenicity. Cancer Res 53:5355–5360
Rong S, Segal S, Anver M, Resau JH, Vande Woude GF (1994) Invasiveness and metastasis of NIH 3T3 cells induced by Met-hepatocyte growth factor/scatter factor autocrine stimulation. Proc Natl Acad Sci U S A 91:4731–4735
Ropero S, Menendez JA, Vazquez-Martin A, Montero S, Cortes-Funes H, Colomer R (2004) Trastuzumab plus tamoxifen: anti-proliferative and molecular interactions in breast carcinoma. Breast Cancer Res Treat 86:125–137
Ross JF, Chaudhuri PK, Ratnam M (1994) Differential regulation of folate receptor isoforms in normal and malignant tissues in vivo and in established cell lines. Physiologic and clinical implications. Cancer 73:2432–2443
Rouleau C, Gianolio DA, Smale R, Roth SD, Krumbholz R, Harper J, Munroe KJ, Green TL, Horten BC, Schmid SM et al (2015) Anti-endosialin antibody–drug conjugate: potential in sarcoma and other malignancies. Mol Cancer Ther 14:2081–2089
Ruoslahti E (2000) Targeting tumor vasculature with homing peptides from phage display. Semin Cancer Biol 10:435–442
Ruoslahti E (2003) The RGD story: a personal account. Matrix Biol 22:459–465
Ruoslahti E (2004) Vascular zip codes in angiogenesis and metastasis. Biochem Soc Trans 32:397–402
Ruoslahti E (2012) Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv Mater 24:3747–3756
Ruschoff J, Hanna W, Bilous M, Hofmann M, Osamura RY, Penault-Llorca F, van de Vijver M, Viale G (2012) HER2 testing in gastric cancer: a practical approach. Mod Pathol 25:637–650
Ryden L, Linderholm B, Nielsen NH, Emdin S, Jonsson PE, Landberg G (2003) Tumor specific VEGF-A and VEGFR2/KDR protein are co-expressed in breast cancer. Breast Cancer Res Treat 82:147–154
Sallusto F, Cella M, Danieli C, Lanzavecchia A (1995) Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J Exp Med 182:389–400
Saunders WB, Bohnsack BL, Faske JB, Anthis NJ, Bayless KJ, Hirschi KK, Davis GE (2006) Coregulation of vascular tube stabilization by endothelial cell TIMP-2 and pericyte TIMP-3. J Cell Biol 175:179–191
Saw PE, Kim S, Lee I-H, Park J, Yu M, Lee J, Kim J-I, Jon S (2013) Aptide-conjugated liposome targeting tumor-associated fibronectin for glioma therapy. J Mater Chem B 1:4723–4726
Saw PE, Park J, Lee E, Ahn S, Lee J, Kim H, Kim J, Choi M, Farokhzad OC, Jon S (2015) Effect of PEG pairing on the efficiency of cancer-targeting liposomes. Theranostics 5:746–754
Saw PE, Park J, Jon S, Farokhzad OC (2017) A drug-delivery strategy for overcoming drug resistance in breast cancer through targeting of oncofetal fibronectin. Nanomedicine 13:713–722
Schlingemann RO, Rietveld FJ, de Waal RM, Ferrone S, Ruiter DJ (1990) Expression of the high molecular weight melanoma-associated antigen by pericytes during angiogenesis in tumors and in healing wounds. Am J Pathol 136:1393–1405
Segers FM, Yu H, Molenaar TJ, Prince P, Tanaka T, van Berkel TJ, Biessen EA (2012) Design and validation of a specific scavenger receptor class AI binding peptide for targeting the inflammatory atherosclerotic plaque. Arterioscler Thromb Vasc Biol 32:971–978
Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK, Batra SK (2012) Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets 16:15–31
Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, Takeyama H (2015) Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers (Basel) 7:2443–2458
Shih SC, Zukauskas A, Li D, Liu G, Ang LH, Nagy JA, Brown LF, Dvorak HF (2009) The L6 protein TM4SF1 is critical for endothelial cell function and tumor angiogenesis. Cancer Res 69:3272–3277
Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB (2003) Identification of a cancer stem cell in human brain tumors. Cancer Res 63:5821–5828
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB (2004) Identification of human brain tumour initiating cells. Nature 432:396–401
Smith GP (1985) Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228:1315–1317
Smith LM, Nesterova A, Ryan MC, Duniho S, Jonas M, Anderson M, Zabinski RF, Sutherland MK, Gerber HP, Van Orden KL et al (2008) CD133/prominin-1 is a potential therapeutic target for antibody-drug conjugates in hepatocellular and gastric cancers. Br J Cancer 99:100
Song S, Liu D, Peng J, Sun Y, Li Z, Gu JR, Xu Y (2008) Peptide ligand-mediated liposome distribution and targeting to EGFR expressing tumor in vivo. Int J Pharm 363:155–161
Song S, Liu D, Peng J, Deng H, Guo Y, Xu LX, Miller AD, Xu Y (2009) Novel peptide ligand directs liposomes toward EGF-R high-expressing cancer cells in vitro and in vivo. FASEB J 23:1396–1404
Sorensen MD, Kristensen P (2011) Selection of antibodies against a single rare cell present in a heterogeneous population using phage display. Nat Protoc 6:509–522
Stapor PC, Sweat RS, Dashti DC, Betancourt AM, Murfee WL (2014) Pericyte dynamics during angiogenesis: new insights from new identities. J Vasc Res 51:163–174
Su JL, Lai KP, Chen CA, Yang CY, Chen PS, Chang CC, Chou CH, Hu CL, Kuo ML, Hsieh CY et al (2005) A novel peptide specifically binding to interleukin-6 receptor (gp80) inhibits angiogenesis and tumor growth. Cancer Res 65:4827–4835
Su S, Chen J, Yao H, Liu J, Yu S, Lao L, Wang M, Luo M, Xing Y, Chen F et al (2018) CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 172:841–856.e816
Suer S, Sonmez H, Karaaslan I, Baloglu H, Kokoglu E (1996) Tissue sialic acid and fibronectin levels in human prostatic cancer. Cancer Lett 99:135–137
Sun J, Zhang C, Liu G, Liu H, Zhou C, Lu Y, Zhou C, Yuan L, Li X (2012) A novel mouse CD133 binding-peptide screened by phage display inhibits cancer cell motility in vitro. Clin Exp Metastasis 29:185–196
Suzuki H, Sasada M, Kamiya S, Ito Y, Watanabe H, Okada Y, Ishibashi K, Iyoda T, Yanaka A, Fukai F (2017) The promoting effect of the extracellular matrix peptide TNIIIA2 derived from tenascin-C in colon cancer cell infiltration. Int J Mol Sci 18:E181
Sweat SD, Pacelli A, Murphy GP, Bostwick DG (1998) Prostate-specific membrane antigen expression is greatest in prostate adenocarcinoma and lymph node metastases. Urology 52:637–640
Takayama H, LaRochelle WJ, Sharp R, Otsuka T, Kriebel P, Anver M, Aaronson SA, Merlino G (1997) Diverse tumorigenesis associated with aberrant development in mice overexpressing hepatocyte growth factor/scatter factor. Proc Natl Acad Sci U S A 94:701–706
Tang C, Ang BT, Pervaiz S (2007) Cancer stem cell: target for anti-cancer therapy. FASEB J 21:3777–3785
Tsunoda T, Inada H, Kalembeyi I, Imanaka-Yoshida K, Sakakibara M, Okada R, Katsuta K, Sakakura T, Majima Y, Yoshida T (2003) Involvement of large tenascin-C splice variants in breast cancer progression. Am J Pathol 162:1857–1867
Tu T, Zhang C, Yan H, Luo Y, Kong R, Wen P, Ye Z, Chen J, Feng J, Liu F et al (2015) CD146 acts as a novel receptor for netrin-1 in promoting angiogenesis and vascular development. Cell Res 25:275–287
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V et al (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515:568–571
Ujula T, Huttunen M, Luoto P, Peräkylä H, Simpura I, Wilson I, Bergman M, Roivainen A (2010) Matrix metalloproteinase 9 targeting peptides: syntheses, 68Ga-labeling, and preliminary evaluation in a rat melanoma xenograft model. Bioconjug Chem 21:1612–1621
Vauquelin G, Charlton SJ (2013) Exploring avidity: understanding the potential gains in functional affinity and target residence time of bivalent and heterobivalent ligands. Br J Pharmacol 168:1771–1785
Visintin A, Knowlton K, Tyminski E, Lin CI, Zheng X, Marquette K, Jain S, Tchistiakova L, Li D, O’Donnell CJ et al (2015) Novel Anti-TM4SF1 Antibody-Drug Conjugates with Activity against Tumor Cells and Tumor Vasculature. Mol Cancer Ther 14:1868–1876
Waller CF, Martens UM, Lange W (1999) Philadelphia chromosome-positive cells are equally distributed in AC133+ and AC133- fractions of CD34+ peripheral blood progenitor cells from patients with CML. Leukemia 13:1466–1467
Wang Z, Yan X (2013) CD146, a multi-functional molecule beyond adhesion. Cancer Lett 330:150–162
Wang J, Masehi-Lano JJ, Chung EJ (2017) Peptide and antibody ligands for renal targeting: nanomedicine strategies for kidney disease. Biomater Sci 5:1450–1459
Wells A (1999) EGF receptor. Int J Biochem Cell Biol 31:637–643
Wernert N (1997) The multiple roles of tumour stroma. Virchows Arch 430:433–443
Wierzbicka-Patynowski I, Schwarzbauer JE (2003) The ins and outs of fibronectin matrix assembly. J Cell Sci 116:3269–3276
Wright GL Jr, Haley C, Beckett ML, Schellhammer PF (1995) Expression of prostate-specific membrane antigen in normal, benign, and malignant prostate tissues. Urol Oncol 1:18–28
Wu CH, Liu IJ, Lu RM, Wu HC (2016) Advancement and applications of peptide phage display technology in biomedical science. J Biomed Sci 23:8
Wulfing P, Kersting C, Buerger H, Mattsson B, Mesters R, Gustmann C, Hinrichs B, Tio J, Bocker W, Kiesel L (2005) Expression patterns of angiogenic and lymphangiogenic factors in ductal breast carcinoma in situ. Br J Cancer 92:1720–1728
Xiao J, Burn A, Tolbert TJ (2008) Increasing solubility of proteins and peptides by site-specific modification with betaine. Bioconjug Chem 19:1113–1118
Xing L, Xu Y, Sun K, Wang H, Zhang F, Zhou Z, Zhang J, Zhang F, Caliskan B, Qiu Z et al (2018) Identification of a peptide for folate receptor alpha by phage display and its tumor targeting activity in ovary cancer xenograft. Sci Rep 8:8426
Yan X, Lin Y, Yang D, Shen Y, Yuan M, Zhang Z, Li P, Xia H, Li L, Luo D et al (2003) A novel anti-CD146 monoclonal antibody, AA98, inhibits angiogenesis and tumor growth. Blood 102:184–191
Yang H, Liu S, Cai H, Wan L, Li S, Li Y, Cheng J, Lu X (2010) Chondroitin sulfate as a molecular portal that preferentially mediates the apoptotic killing of tumor cells by penetratin-directed mitochondria-disrupting peptides. J Biol Chem 285:25666–25676
Yang F, Ai W, Jiang F, Liu X, Huang Z, Ai S (2016) Preclinical Evaluation of an Epidermal Growth Factor Receptor-Targeted Doxorubicin-Peptide Conjugate: toxicity, Biodistribution, and Efficacy in Mice. J Pharm Sci 105:639–649
Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2:127–137
Ye Z, Zhang C, Tu T, Sun M, Liu D, Lu D, Feng J, Yang D, Liu F, Yan X (2013) Wnt5a uses CD146 as a receptor to regulate cell motility and convergent extension. Nat Commun 4:2803
Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, Olweus J, Kearney J, Buck DW (1997) AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 90:5002–5012
Yu D, Hung MC (2000) Role of erbB2 in breast cancer chemosensitivity. BioEssays 22:673–680
Yu H, Kortylewski M, Pardoll D (2007) Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 7:41–51
Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9:798–809
Yu SS, Lau CM, Barham WJ, Onishko HM, Nelson CE, Li H, Smith CA, Yull FE, Duvall CL, Giorgio TD (2013) Macrophage-specific RNA interference targeting via “click”, mannosylated polymeric micelles. Mol Pharm 10:975–987
Yue P, Turkson J (2009) Targeting STAT3 in cancer: how successful are we? Expert Opin Investig Drugs 18:45–56
Zanini E, Louis LS, Antony J, Karali E, Okon IS, McKie AB, Vaughan S, El-Bahrawy M, Stebbing J, Recchi C et al (2017) The Tumor-Suppressor Protein OPCML Potentiates Anti-EGFR- and Anti-HER2-Targeted Therapy in HER2-Positive Ovarian and Breast Cancer. Mol Cancer Ther 16:2246–2256
Zhang Q, Tang J, Fu L, Ran R, Liu Y, Yuan M, He Q (2013) A pH-responsive alpha-helical cell penetrating peptide-mediated liposomal delivery system. Biomaterials 34:7980–7993
Zhao P, Grabinski T, Gao C, Skinner RS, Giambernardi T, Su Y, Hudson E, Resau J, Gross M, Vande Woude GF et al (2007) Identification of a met-binding peptide from a phage display library. Clin Cancer Res 13:6049–6055
Zheng C, Qiu Y, Zeng Q, Zhang Y, Lu D, Yang D, Feng J, Yan X (2009) Endothelial CD146 is required for in vitro tumor-induced angiogenesis: the role of a disulfide bond in signaling and dimerization. Int J Biochem Cell Biol 41:2163–2172
Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X et al (2015) Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol 17:170–182
Zou W, Wolchok JD, Chen L (2016) PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med 8:328rv4
Zurita AJ, Arap W, Pasqualini R (2003) Mapping tumor vascular diversity by screening phage display libraries. J Control Release 91:183–186
ACKNOWLEDGEMENTS
This work was supported by grants from the National Key Research and Development Program of China (2016YFC1302300), the Natural Science Foundation of China (Grant Nos. 81720108029, 81621004, 81490750, 81874226 and 81803020), Guangdong Science and Technology Department (2016B030229004), Guangzhou Science Technology and Innovation Commission (201803040015). The research is partly supported by Fountain-Valley Life Sciences Fund of University of Chinese Academy of Sciences Education Foundation.
ABBREVIATIONS
α-SMA, α-smooth muscle actin; ADC, antibody-drug conjugate; CAFs, cancer associated fibroblasts; CSCs, cancer stem cells; EC, endothelial cells; ECD, extracellular domain; FACS, fluorescence-activated cell sorting; FAP-α, fibroblast-activated protein-α; FRα, folate receptor alpha; HCC, hepatocellular carcinoma; HGFR, hepatocyte growth factor receptor; MACS, magnetic-activated cell sorting; MCAM, melanoma cell adhesion molecule; MMPs, matrix metalloproteinases; MRI, magnetic resonance imaging; NSCLC, non-small cell lung cancer; OC, ovarian cancer; PDXs, patient-derived xenografts; PITPNM3, phosphatidylinositol transfer protein, membrane-associated 3; PSMA, prostate-specific membrane antigen; PTX, pertussis toxin; TAMs, tumor associated macrophages; TME, tumor microenvironment; TNC, tenascin C; uPA, urokinase plasminogen activator; uPAR, urokinase plasminogen activator receptor; VEGF, vascular endothelial cell growth factor.
COMPLIANCE WITH ETHICS GUIDELINES
Phei Er Saw and Er-Wei Song declare that they have no conflict of interest.This article does not contain any studies with human or animal subjects performed by the any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Saw, P.E., Song, EW. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell 10, 787–807 (2019). https://doi.org/10.1007/s13238-019-0639-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-019-0639-7