
Abstract
The ecological significance of fungi occurring asymptomatically inside living plant leaves is poorly understood. Given the broad saprotrophic potential of many endophytic fungi, we hypothesized that they persist in decaying litter for an extended period of time after leaf abscission. Fungal assemblages were assessed by high-throughput sequencing in autumn leaves of beech (Fagus sylvatica) and in the corresponding leaf litter in 388 samples from 22 beech forest plots in three widely distant regions of Germany. A considerable proportion of the leaf-endophytic fungi was also found in 1-year-old litter. Co-occurrence networks revealed that the fungi formed unstructured assemblages inside the living leaves, rather than well-structured communities. Previously endophytic fungi constituted an integral part of the fungal litter community and were by far the most active fungi in 1-year-old litter. We therefore consider these endophytic occurrences to represent transient stages. Composition of the aboveground microbiome appears therefore to be closely connected to the process of litter decomposition. Considering the respective linked fungal habitat will facilitate predicting nutrient and carbon cycling and storage in forest ecosystems as well as elucidating the ecology of leaf microbiomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
All macroorganisms are associated with microorganisms, the so-called microbiome. The microbiome has been shown to affect morphology, physiology, development, behaviour, health, immunology, reproduction and fitness of the host organisms (Rosenberg and Zilber-Rosenberg 2016). The holobiont, i.e. the macroorganism with its associated microbiome (Margulis and Fester 1991; Rohwer et al. 2002; Zilber-Rosenberg and Rosenberg 2008), was therefore suggested to represent the unit to be considered in ecological and evolutionary contexts (Hawksworth 1991). Plants are rooted in the environment and connected below ground by a network of mycorrhizal fungi (e.g., Simard et al. 1997; Beiler et al. 2010; Dickie et al. 2015). Individual plant holobionts are therefore not clearly delimitable (Peršoh 2015). The aboveground microbiome also includes a huge diversity of fungi, the majority of which is shared among host individuals and species (Saikkonen 2007; Rodriguez et al. 2009; Suryanarayanan et al. 2003; Suryanarayanan 2011, Sun et al. 2012), even though the aboveground plant organs are physically disconnected. Compositions of aboveground microbiomes nevertheless differ between hosts, due to individual microbes occurring at different abundances (Okane et al. 1998; Saikkonen 2007; Peršoh et al. 2010; Weig et al. 2013). Their composition, however, changes through time and space and is influenced by climatic and environmental conditions (Fisher et al. 1994; Collado et al. 1999; Gao et al. 2005; Jumpponen and Jones 2010; Cordier et al. 2012b; Peñuelas et al. 2012; Coince et al. 2013; Peršoh 2013, 2015). Due to their immense diversity, low host preference and temporal variability, microbiomes characteristic for a certain host are challenging to identify.
Plant leaves are colonized asymptomatically by two different types of endophytic fungi: While a huge diversity of fungi is found in all plants, grasses may be (in addition) systemically inhabited by Clavicipitacean endophytes which are vertically transmitted and produce secondary metabolites protecting the host from feeding damage (Porras-Alfaro and Bayman 2011; Purahong and Hyde 2011). By contrast, the non-Clavicipitacean leaf-endophytes were rarely found to have beneficial effects for the host plant (Hyde and Soytong 2008; Rodriguez et al. 2009; Khan et al. 2016). While several saprotrophic fungi were recently shown to be capable of biotrophic interactions with host plants (Smith et al. 2017), growing evidence suggests that some endophytic fungi become saprobes after leaf fall (Guo et al. 2001, 2003; Promputtha et al. 2002, 2005, 2007, 2010; Shirouzu et al. 2009; Suryanarayanan 2013; Saikkonen et al. 2015; Szink et al. 2016; Promputtha et al. 2017). Their endophytic occurrence might thus be just one stage in their life cycle. As pioneer colonizers, they may take advantage of easily accessible and readily utilizable chemical compounds in the freshly fallen leaves (Osono and Takeda 1999; Koide et al. 2005). They are thought to persist in litter for only a short period of time and quickly re-colonize living leaves to complete their life cycle (Osono 2006). However, many possess the enzymatic machinery to degrade complex structural plant compounds, such as cellulose and lignin (Kumaresan and Suryanarayanan 2002; Fukasawa et al. 2009; Promputtha et al. 2010; Yuan and Chen 2014). While part of these enzymes may facilitate invading plant hosts, their activity is also beneficial for saprotrophic decomposition of litter.
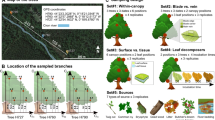
We therefore hypothesized that a considerable proportion of endophytic fungi persists in fallen leaves for a prolonged period of time (i.e. for at least 1 year). As an integral part of the early litter decomposing community, these fungi might represent a part of the holobiont microbiome which may fulfil functional roles when detached from the macroorganism. We analysed the fungal assemblages in beech-dominated forest plots across three distinct regions in Germany. The fungal microbiome in autumn leaves of beech (Fagus sylvatica) was compared to the corresponding community in 1-year-old leaf litter in the following year (Fig. 1). We analysed integration of co-occurring fungi in the microbiomes of living and decaying leaves by calculating their embeddedness in the respective co-occurrence networks. Contribution of former endophytic fungi to fungal activity in litter was assessed by targeting the short-lived precursor rRNA in addition to the rRNA gene.

Study design. Foliar and endophytic fungal assemblages were assessed in autumn 2014 (and 2015; not depicted) in five leaves of each of five beech trees in 22 forest plots in three regions of Germany ①. The corresponding fungal community in decaying (1-year-old) litter was assessed in 2015 ②. While leaf litter has often been thought to be mostly colonized by soil fungi (brown symbols) according to the current ideas, we hypothesized that some former endophytic fungi (green symbols) persist in decaying leaves for at least 1 year. Plant-indigenous endophytes (yellow symbols) were not differentiated from other endophytes for the litter analyses. Exclusively epiphyllous fungi (blue symbols) were hypothesized to played a minor role in the litter community, in agreement with current ideas. Colours code for fungal guilds and symbols for species
Materials and methods
Study area and sample collection
To reliably assess the interconnectivity between endophytic and litter degrading fungal assemblages, we chose an adequate spatial scale for tracing ecosystem processes while focusing on a single tree species, i.e. European Beech (Fagus sylvatica). The studied sites were beech-dominated forest plots selected in the frame of the German Biodiversity Exploratory project for large-scale and long-term functional biodiversity research (Fischer et al. 2010). In each of three widely separated (> 300 km) regions (Hainich-Dün, Schorfheide-Chorin and Swabian Alb), three unmanaged and three age-class forest plots were selected (Table S1). In addition, three plots in selection-cut forests were selected in Hainich-Dün and a fourth age-class forest plot in the Swabian Alb. Each plot is 100 m × 100 m in size and separated from the nearest neighbouring plot by 15 km (± 9 km).
Falling beech leaves were collected in autumn 2014 in nets by shaking trees and/or twigs at five positions (corners and centre) per plot (Table S2). In autumn 2015, still attached and greenish leaves were picked from the trees. From each of the 5 replicate collections, five leaves were selected, stacked, and 15 disks (Ø 6 mm) were obtained using a cork borer. These were placed in 15 mL reaction tubes with 10 mL of silica gel beads for rapid drying at ambient temperature. Another five leaves were stored at 4 °C and surface sterilized within 12 h. The surface sterilization was achieved by successively washing the leaves in 700 mL of water for 1 min, 500 mL of 70% ethanol (2 min), 500 mL of 1.4% NaOCl (5 min), 500 mL of 70% ethanol (1 min), and 3 times in 500 mL of double distilled water. Disks from the surface sterilized leaves were dried in silica gel as detailed above.
One-year-old beech leaf litter was collected in autumn 2015 at the corresponding five positions per plot (Table S2). Disks were obtained as detailed above, but immediately transferred to 2 mL screw cap tubes and placed in liquid nitrogen in the field and stored at −80 °C in the laboratory until further processing.
Nucleic acid extraction and RNA processing
DNA from two dried disks was extracted using the gDNA Plant Kit (Invitrogen™, ChargeSwitch®) as recommended by the manufacturer, but with all volumes scaled down to 10%. Cell disruption was achieved in a FastPrep®-24 Instrument (MP Biomedicals) at 6 m s−1 for 60 s after adding a bead mixture (0.03 g of Ø 0.1–0.25 mm, 0.06 g of Ø 0.25–0.5 mm and 5–6 glass beads Ø 1.25–1.55 mm) to the samples.
To cope with interfering humic substances, nucleic acids from litter were extracted (under RNase-free conditions) by adapting and combining two protocols (Peršoh et al. 2008; Paulin et al. 2013). A bead mixture (0.15 g of Ø 0.1–0.25 mm, 0.12 g of Ø 0.25–0.5 mm and 0.12 g of Ø 1.25–1.55 mm glass beads and 3 Ø 3 mm steel beads) and 1535 μL of an extraction buffer supplemented with Salmon Sperm DNA [700 μL Tris–buffer (0.1 M, pH 5.5); 10 μL Salmon Sperm DNA sodium salt (Roth) (10 g L−1); 325 μL 4× extraction buffer (0.4 M LiCl, 200 mM Tris–HCl [pH 8], 120 mM EDTA [pH 8], 10% SDS) and 500 μL phenol] were added to the frozen samples. Cells were disrupted after thawing in the FastPrep®-24 Instrument at 4.0 m s−1 for 30 s, followed by two times 6.5 m s−1 for 40 s, with intermediate incubation on ice for 5 min. After centrifugation (14,165 g for 1 min, 20 °C), 750 µL phenol:chloroform:isoamyl alcohol (25:24:1 v/v) was added to 750 µL of the supernatant, thoroughly mixed and incubated on ice for 5 min, interrupted by thorough shaking every 90 s. Three repetitive cleaning cycles of centrifugation (17,650 g, 15 min, 4 °C), addition of an equal volume of chloroform:isoamyl alcohol (24:1) to the supernatant and incubation on ice (5 min, regular shaking) were applied to remove the phenol. In the meantime, 500 µL of a DEPC-treated PVPP suspension (Polyvinylpolypyrrolidone, Sigma-Aldrich, 0.5 g mL−1) were loaded on a spin column (Maxima Spin Column GF-F1 2 mL, GeneON), and centrifuged at 2150 g for 3 min. The flow-through was discarded and 500 µL of the nucleic acid extract were loaded on the spin column with PVPP. After centrifugation (2150 g, 3 min, 20 °C), 200 µL of ddH2O were added to the column and the centrifugation repeated. Of the flow-through, 500 µL were amended with 250 µL LiCl (8 M, Sigma-Aldrich) and incubated overnight at −20 °C. After ultracentrifugation (210,000 g, 30 min, 4 °C), the pellet was resuspended in 15 µL of RNAse-free ddH2O. Concentration and quality of the extract was assessed using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific Inc.).
DNA was separated from 2.5 µL of the nucleic acid extracts from litter after electrophoresis on agarose gels {0.8% (w/v) agarose dissolved in 1× TBE–buffer [88 mM Tris, 88 mM boric acid, 2.0% (v/v) 0.1 M EDTA, pH 8.0], supplemented with 0.005% of ethidium bromide}, at 100 V (400 mA) for 20 min. The parts containing large-sized DNA (≥ 10,000 bp) were excised and DNA was extracted from gel slices using the NucleoSpin® Gel and PCR Clean-up kit (Macherey–Nagel), according to the manufacturer’s instructions.
To compare community structures between living and decaying leaves, we extracted DNA from a subset of eight litter disks and eight surface-sterilized dried leaf disks (collected in 2014), collected in five positions in each of five plots (AEW05, HEW06, HEW11, SEW05 and SEW09), as detailed above. DNA from litter and leaf disks was extracted as detailed above for nucleic acid extraction from litter samples (Table S3).
To purify RNA, 2.5 µg of nucleic acids from litter were treated with 37.5 U of DNase I (New England BioLabs Inc.) in the corresponding buffer [10 mM Tris (pH 7.6), 2.5 mM MgCl2, 0.5 mM CaCl2] for 30 min at 37 °C. The reaction was stopped by dilution with ddH2O to a volume of 500 µL, and addition of 500 µL phenol:chloroform:isoamyl alcohol solution (25:24:1). Phenol was removed and RNA precipitated with LiCl as detailed above. The RNA pellet washed with 400 µL of 80% ethanol, ultracentrifuged again (210,000 g, 30 min, 4 °C) air dried for 10 min and resuspended in 9.5 µL of H2O. After quantification with the NanoDrop 1000 spectrophotometer, up to 100 ng of RNA was reverse transcribed using the ProtoScript® II First Strand cDNA Synthesis Kit (New England BioLabs Inc.), according to the manufacturer’s instructions. Negative controls (without addition of reverse transcriptase) were processed in parallel for all RNA extracts. Absence of (amplifiable) DNA contaminations in the RNA extracts was confirmed by consistently negative amplification results under the conditions detailed below.
Library preparation and sequencing
The fungal barcoding region (i.e. ITS rRNA gene region) (Schoch et al. 2012) was amplified for assessing the composition of the fungal assemblages. Preparation of the amplicon libraries comprised two sequential amplification steps to allow for multiplexing of multiple samples. The first PCR was performed with the fungal specific primer combination ITS1F (Gardes and Bruns 1993) and ITS4 (White et al. 1990) extended at the 5′-ends by TAG sequences and part of the sequencing primers (Table S4). The reactions contained 5.25 µL of ddH2O, 0.25 µL of each primer (10 µM), 0.5 µL of template and 6.25 µL of GoTaq® G2 Hot Start Colorless Master Mix (Promega). The amplification started at 95 °C for 3 min, followed by 33 cycles of 94 °C for 27 s, 57 °C for 60 s and 72 °C for 90 s, and ended after a final elongation step at 72 °C for 7 min. Of the Rapid PCR Cleanup Enzyme Set kit (New England Biolabs), 2 µL diluted 1:5 in ddH2O, were added to 5 µL of PCR product and incubated at 37 °C for 15 min followed by 80 °C for 15 min. The primers applied in the second PCR included sequences of the sequencing primer, indices and the P5 and P7 adapters for the Illumina sequencing (Table S4). The reactions included 6.5 µL of ddH2O, 0.5 µL of each primer (10 µM), 5 µL of purified PCR product and 12.5 µL of GoTaq® G2 Hot Start Colorless Master Mix. The amplification was achieved in 6 cycles of 94 °C for 27 s, 53 °C for 60 s and 72 °C for 90 s, between initial denaturation and final extension as detailed for the first PCR. The final PCR products were visualized in 0.8% agarose gels stained with ethidium bromide and ImageJ version 1.50b (https://imagej.nih.gov/ij) (Schneider et al. 2012) was used to quantify the amplified fragments. Similar concentrated PCR products were pooled equimolarily and purified with Dynabeads® Sequencing Clean-Up (Invitrogen™, LifeTechnologies) or with CleanPCR® Nucleic acid Clean up (CleanNA, GC biotech B.V.), according to the manufacturer’s instructions. Amplicons between 450 and 1500 bp in length were selected from purified pools using the Blue Pippin (Sage Science, United States). DNA concentration was assessed with a Qubit® 2.0 fluorometer (Life Technologies, Carlsbad, CA, USA) and amplicon size distribution using a Bioanalyzer 2100 (Agilent Technologies GmbH & Co. KG, Waldbronn, Germany). Sequencing was performed by the sequencing service of the Faculty of Biology at LMU Munich, using an Illumina MiSeq® sequencer (Illumina, Inc., San Diego, CA, USA) with 2 × 250 bp paired end sequencing (MiSeq Reagent Kit v3 Chemistry, Illumina, Inc., San Diego, CA, USA).
Processing of sequencing data
The sequence reads were processed using the open-source software QIIME version 1.9.0 (Caporaso et al. 2010) as detailed by Röhl et al. (2017). Briefly, the TAG sequences were extracted and the reads were quality filtered (Phred > 19), allowing a maximum of one ambiguity base on each sequence. Reads were retained when at least 0.35% of the read consisted of consecutive high quality base calls and truncated after 19 consecutive low quality base calls. Reads were assigned to samples (i.e. demultiplexed) according to their indices and TAGs. ITS1 reads were extracted and trimmed at the 5′-end to comprise only the final 11 bp of the SSU rRNA gene region in addition. The pre-processed sequence data were deposited in the European Nucleotide Archive database (http://www.ebi.ac.uk/ena, accession numbers PRJEB19870 and PRJEB21700). Based on comparative analyses of clustering algorithms (Röhl et al. 2017), we decided to cluster the demultiplexed reads into operational taxonomic units (OTUs) using CD-HIT-OTU for Illumina reads version 0.0.1 (Fu et al. 2012), applying a similarity threshold of 97%. A table listing the read count per OTU and sample (Table S5) was generated and used for further analyses. Taxonomic affiliation for the representative sequence of each OTU was assigned using UNITE database version 7.0 (Kõljalg et al. 2013) as reference (Table S6). The samples considered to compare community structures between living and decaying leaves were independently processed (Tables S3, S7, S8).
Samples with less than 1000 reads were discarded. After processing, the original dataset included 610 fungal OTUs (represented by 16,571,285 quality-filtered sequence reads) in 388 successfully processed samples (Table S9). To account for potential errors due to tag switching (Carlsen et al. 2012; Sinha et al. 2017), we applied an abundance threshold for each OTU, only above which it was considered to be present in the respective sample (see Supplementary file S1 for details, Fig. S1). To filter potential erroneous detections, an equivalent to 1% of the total read count of each sample was subtracted from the read counts of each OTU in the respective sample. This trimmed dataset included 470 fungal OTUs (represented by 14,988,069 quality-filtered sequence reads) in the 388 samples (Table S10).
Statistical analyses and guild delimitation
Classification of OTUs to fungal guilds (endophytic, foliar and litter-indigenous fungi) reflects their detection in the respective sampling categories according to the trimmed dataset. Fungi detected in surface-sterilized samples were defined as endophytes (i.e. occurring inside the leaves), while fungi were considered as foliar when detected in non-surface sterilized leaves (i.e. occurring on the leaf surface and/or inside the leaves). Fungi detected only in litter samples were considered as indigenous litter fungi.
Sequencing depth of the original dataset did not correlate with OTU richness in samples of the foliar assemblages in 2014 and the DNA-derived litter communities (i.e. present fungi) in 2015 (Fig. S2). While comparisons of species richness are therefore reliable for these categories, those for the DNA-derived (i.e. present) endophytic and RNA-derived (i.e. active) litter fungi may be slightly biased. We consider the weak correlation (r2 < 0.07) between sequencing depth and OTU richness in these categories tolerable and therefore refrained from excessive data manipulation by rarefying (McMurdie and Holmes 2014). RNA-derived data were more strongly correlated to sequencing depth (r2 = 0.19), but the results were consistent with the DNA-derived data and the higher variation between samples did not compromise statistical significance. Significant positive correlations were not observed in the trimmed datasets, but applying the threshold resulted partly in significant negative correlations (Fig. S3).
The OTU tables generated from the original and trimmed datasets, respectively (Tables S5, S10) were imported into Primer7 version 7.0.11 (Clarke and Gorley 2015) and read counts were standardized per sample by the total number of reads. For analyses considering the relative read abundance, a Bray–Curtis similarity matrix was calculated. For analyses considering the OTU occurrence, the table was transformed into presence/absence data and a Jaccard similarity matrix was calculated. For each of the performed analyses, the same number of samples from each compared sampling category was considered per plot. The same samples were considered when DNA- and RNA-derived data was compared. Analyses of similarity (ANOSIM) were calculated using 10,000 permutations. Species richness (N), Shannon diversity (H′) and Pielou’s eveness (J′) were calculated using Primer7 DIVERSE methods. Area-proportional Euler Diagrams were generated using the BioVenn web application (Hulsen et al. 2008). Kruskal–Wallis test was performed using STATISTICA 64 version 12 (StatSoft, Inc.). All statistical tests were considered significant at P < 0.05.
Co-occurrence networks per sampling category were calculated based on the original dataset (Table S5) for each plot using CoNet version 1.1.beta (Faust et al. 2012). To compare community structures between living and decaying leaves the dataset derived from 8 discs was used (Table S7). Network inference was adapted from Faust et al. (2015), considering OTUs with a minimum occurrence in at least 3 samples from the respective plot. The obtained networks were visualized and processed as undirected networks in Cytoscape version 3.3.0 (Shannon et al. 2003), with only positive edges being considered for the analyses. The (local) clustering coefficient of a node (i.e. OTU) was considered as a measure of interconnectivity among its neighbours, where 0 represented no connections among neighbours and 1 denoted that every neighbour was connected to each other (Albert and Barabasi 2002).
Data accessibility
The sequence data are available from the European Nucleotide Archive (http://www.ebi.ac.uk/ena/) under study PRJEB19870 and PRJEB21700. Corresponding metadata are provided in Tables S2 and S3. All data generated or analysed during this study are included in this published article and its supplementary information files.
Results
Rare fungi distinguish foliar from endophyllous assemblages
To assess the impact of the host plant on composition of the initial litter decomposing community, we compared foliar and leaf-endophytic fungal assemblages, which are presumably separated by the host’s defence response. In the falling autumn leaves, we found 119 fungi; i.e. fungal OTUs, which represent clusters of highly similar barcoding sequences. Seventy-two of these fungi also occurred inside the leaves; i.e. as endophytes (Fig. 2a). The 47 fungi not detected inside leaves accounted for < 2% of the total foliar fungal assemblage (Fig. 2b) and were three times less frequent than fungi co-occurring endophytically (Fig. 2c). The 31 fungi detected exclusively in surface-sterilized, but not in untreated leaves occurred at similarly low abundance and frequency, indicating a technical bias due to undersampling. Only half (58%) of the endophytic fungi, however, were also detected at the same plot in the untreated foliar samples at the time of sampling (Fig. S5). The proportion of endophytic fungi with foliar occurrence at a certain plot increased significantly to 75 and 88%, when calculated in relation to the entirety of fungi found in all foliar samples from the corresponding region or the whole sampling area (i.e. Germany), respectively. All these trends were similar when the original dataset was analysed (Figs. S4, S6).

Distribution of fungi to untreated (foliar) and surface-sterilized (endophytic) leaf samples according to the trimmed dataset. a Occurrence and b abundance of fungal OTUs in foliar and endophytic assemblages visualized by Euler-Diagrams. Abundance represents (standardized) read counts for each sampling category; i.e. the 41 co-occurring OTUs accounted for > 97% of the sequence reads. c Average frequency (i.e. proportion of colonized plots) of OTUs occurring exclusively in untreated (F), exclusively in surface-sterilized leaves (E) and co-occurring (FE) in both samples. Frequencies in foliar and endophytic assemblages are indicated separately and together for the co-occurring fungi
Assemblages of fungi (i.e. Operational Taxonomic Units, OTUs) were statistically indistinguishable between the foliar and endophyllous habitats (Fig. 3a, b) according to OTU occurrence (ANOSIM R = −0.011, P = 0.657) and abundance (R = 0.002, P = 0.397). Species richness and diversity were also statistically indistinguishable, but endophytic assemblages were more even than the corresponding foliar assemblages at the same plot (Fig. 3c–e). Considering the original dataset (i.e. all obtained sequence reads), foliar and endophytic assemblages differed significantly according to species richness and similarity of fungal composition (Fig. S7a–e).

Similarity among and structure of fungal assemblages according to the trimmed dataset. Average pairwise distance between fungal assemblages based on a abundance and b occurrence. Pairwise distances according to the ANOSIM R value (two-way nested ANOSIM) are ordinated by nMDS. Different letters denote significantly (ANOSIM P < 0.05) different communities. c Species richness (N), d Shannon diversity (H′) and e Pielou’s eveness (J′) of the fungal assamblages. Different letters denote significant differences (Kruskal–Wallis P < 0.05). Box plots indicate median values (squares), 25–75% range (boxes), non-outlier range (whiskers), outliers (circles) and extremes (asterisks). f Accumulation curve of observed species richness for each sampling category. Colours code fungal communities occurring in litter (L), and fungal assemblages in the phyllosphere (P) and inside leaves (i.e. with an endophytic occurrence) in 2014 (E14) and 2015 (E15)
Fungi occurring inside autumn leaves are more active in 1-year-old litter than indigenous litter fungi
Occurrence and contribution to the communities (relative abundance) was comparatively analysed for fungi in living and decaying leaves to estimate fungal turnover rates between the two habitats. Species overlaps were more pronounced according to the original dataset (Fig. S8a–d), but only presented for the trimmed dataset in the following: One-third (i.e. 46 out of 121) of the fungi inside autumn leaves also occurred in 1-year-old litter (Fig. 4a). A fifth of the 240 litter fungi were accordingly capable of colonizing the interior of living autumn leaves. The 46 litter fungi with endophytic occurrence in 2014 and/or 2015 accounted together for 42% of the fungal community in 1-year-old litter, even though they represented only 19% of the fungal OTUs. Five times more fungi were restricted to litter, i.e. presumably represent litter- or soil-indigenous fungi, but contributed only 58% to the fungal abundance in the community. The 47 litter fungi restricted to the leaf surface in 2014 accounted for < 6% of the litter community.

Contribution of fungi with endophytic occurrence to the litter community according to the trimmed dataset. a Number of litter fungi co-occurring (DNA-based) inside autumn leaves (i.e. with endophytic occurrence), and b contribution of the respective distribution groups to total fungal abundance in litter. c Number of active litter fungi (RNA) occurring inside autumn leaves (DNA). d Average contribution of individual litter fungi of the respective distribution groups to total activity in litter. Overlaps among categories indicate distribution patterns. Forty OTUs of non-endophytic foliar fungi contributed negligibly to the litter community (< 6%) and were treated as litter fungi for the visualization. Colours code OTU occurrence in litter (L) and inside leaves in 2014 (E14), 2015 (E15) and in both years (E1415)
To exclude the possibility that the formerly endophytic fungi were merely present as inactive stages in the 1-year-old litter, we targeted the barcoding region in the short-lived precursor rRNA in addition. This approach revealed 36 of the fungi with endophytic occurrence to be physiologically active in 1-year-old litter (Fig. 4c). They only represented 16% of the active fungi there, but their average activity clearly exceeded that of the other fungi in litter (Fig. 4d). Fungi detected endophytically in both years contributed on average > 4 times more to the overall fungal activity in litter than litter-indigenous fungi (Fig. 4d). Litter fungi detected as endophytes only in the year of litter sampling (i.e. 2015) were slightly more active in the litter than fungi occurring only as endophytes in the year before (i.e. 2014, Fig. 4d). Their activities were intermediate between soil-indigenous and fungi with recurring endophytic stage. Litter fungi with non-endophytic foliar occurrence contributed to the active community in litter as little as litter-indigenous fungi; i.e. 0.1% on average.
Fungi with endophytic occurrence are well embedded into the litter community
Despite the large number of fungi shared between autumn leaves and decaying litter, composition of the fungal communities colonizing 1-year-old litter in 2015 was clearly distinct from all phyllosphere assemblages (Fig. 3a, b). The litter communities were more diverse and richer in fungi and these were more evenly distributed than in the phyllosphere (Figs. 3c–f, S7c–f).
Co-occurrence networks revealed that fungal assemblages in leaves and litter are fundamentally differently structured: connections among fungi in the litter community exceeded those in the endophytic assemblages by 3.6 times (Fig. 5). Only in the litter community, were fungi with endophytic occurrence well connected among each other. The network clustering coefficients indicated that fungi with endophytic occurrence were consistently at least as well integrated into the litter community as fungi restricted to litter (Fig. 6). Integrating over all 22 plots, fungi with endophytic occurrence in one year were even significantly and slightly better embedded in the litter community than fungi restricted to litter (Kruskal–Wallis P < 0.05).

Interconnectedness of litter and endophytic fungi. Ratio between positive correlations in co-occurrence networks and number of detected OTUs per plot. Box plots indicate median values (squares), 25–75% range (boxes), non-outlier range (whiskers) and outliers (circles). Different letters denote significant (Kruskal–Wallis P < 0.05) differences between groups

Embeddedness of fungal OTUs with different distribution patterns in the communities in 1-year-old litter. Distributions of the network clustering coefficients are given for the OTUs of each distribution group according to Faust et al. (2015). Colours code OTUs with distribution patterns as specified in Fig. 3. The first letter of the plot ID codes the sampling region (A: Swabian Alb, H: Hainich-Dün, S: Schorfheide-Chorin) and the numerals specify the plots, as detailed in Table S2. Box plots indicate median values (squares), 25–75% range (boxes), non-outlier range (whiskers), outliers (circles) and extremes (asterisks). Significant differences (Kruskal–Wallis P < 0.05) between groups are indicated per plot by asterisks (*)
Detection of litter fungi inside autumn leaves increased along spatial scales
We ascribed the unexpectedly strong connection between leaf- and litter-inhabiting fungi to the extensive study area spanning three distant regions across Germany. To test this hypothesis, we analysed the distribution patterns across different spatial scales by assessing the contribution of fungi detected in falling leaves in autumn 2014 to the 1-year-old litter community in autumn 2015. Fungi detected just locally as endophytes contributed only to a limited degree to the litter community at the same plot, concerning fungal richness, abundance and activity (Figs. 7a–c, S9). This contribution increased more than 6, 3 and 19 times, respectively, when all fungi detected as endophytes in the corresponding region were defined as litter fungi with endophytic occurrence. Only by classifying all fungi as endophytic which occurred as such at any plot in the study area, previously endophytic fungi contributed as extensively to the litter community as detailed above (Fig. 3).

Spatial links and differences of endophyllous and litter fungal assemblages. a–c Contribution of fungi with endophytic occurrence to the litter community in dependence on the spatial scale considered for categorization as endophytic fungus. Fungi were categorized as endophytes when occurring inside leaves at the same plot (local scale), in the corresponding region (regional) as the analysed litter and in any sample (national), respectively. Contribution to a OTU richness, b abundance (i.e. relative abundance of DNA-derived amplicons) and c activity (i.e. relative abundance of RNA-derived amplicons) is given for the 21 analysed plots, based on the trimmed dataset. Box plots indicate median values (squares), 25–75% range (boxes), non-outlier range (whiskers) and outliers (circles). Different letters denote significant differences (Kruskal–Wallis P < 0.05) among scales. d Magnitude (ANOSIM R) and significance (ANOSIM P) of differences in composition among plots (nested within regions) and regions is indicated for present fungal communities in litter in 2015 and for endophytic fungal assemblages in 2014, based on the original dataset. Asterisks indicate different levels of significance (***P < 0.0001; *P ≤ 0.01), “ns” indicates no significant differences (P > 0.05)
This indicated an interdependency of the fungal assemblages in leaves and communities in litter across larger spatial scales and accordingly a similar susceptibility to habitat conditions. Neither the fungal assemblages present in autumn leaves nor the active communities in litter were indeed influenced by forest management (ANOSIM R < 0.136, P > 0.143; Table S1). Only a weak impact of forest management on the communities present in litter was observed (ANOSIM R = 0.248, P = 0.041). Among regions, the litter communities differed much stronger in composition (ANOSIM R > 0.832, P < 0.0001) than fungal assemblages associated with autumn leaves did (R = 0.324, P = 0.007; Fig. 7d). Litter communities differed even among plots within regions (R > 0.402, P < 0.0001), while the corresponding endophytic assemblages were regionally indistinguishable (R = 0. 051 P = 0.088).
Discussion
Methodological constraints
Massive multiplexing of samples for metabarcoding is known to be prone to assignment of sequences to wrong samples due to “tag switching”, i.e. swapping of the sequences (i.e. tag and/or index sequences) which are introduced to designate samples (Carlsen et al. 2012; Sinha et al. 2017). To eliminate artefacts due to this tag switching, we applied an abundance threshold for accepting the presence of each OTU in each sample. Multiple lines of evidence (Supplementary File S1) indicate that tag switching effects are no longer relevant in the resulting trimmed dataset. However, the approach also eliminated 140 rare OTUs from the dataset. Because these OTUs were actually detected, the elimination was not restricted to false positive detections. This is also indicated by the correlation between read and OTU counts turning negative for some categories (Figs. S2, S3). This suggests a data processing artefact, because it is biologically implausible to detect fewer species (OTUs) by the expanding sampling effort (sequencing depth). This applies in particular if the sampling density is too low, as for the foliar communities (Fig. S3). Furthermore, the detected OTU richness in litter approached saturation according to the original dataset but still increased nearly linear according to the trimmed dataset (Figs. 3, S7). While OTU turnover between habitats was supported by both, the original and the trimmed dataset, it was generally more pronounced according to the original data (Figs. 2, 4, S4, S8). These results indicate that the magnitude of overlap between habitats is probably intermediate between those indicated by the corresponding original and trimmed datasets. We therefore regard the following discussion, which is based on the latter, as a conservative scenario.
Methodological biases are probably not the only reason to consider the discussed results as conservative. OTU turnover between foliar (Figs. S5, S6) and between endophytic assemblages and litter communities (Figs. 7, S9) consistently increased with the spatial scale considered. We therefore predict that an even more extensive sampling than already applied would reveal higher fungal turnover rates between the habitats; but the exact magnitude remains to be quantified.
Overlap of phyllosphere mycobiomes and fungal litter communities
At the time of budburst, leaves are virtually free of endophytes (Toti et al. 1993; Scholtysik et al. 2013) and distribution of the endophytes occurring later is highly localized within leaves (Espinosa-Garcia and Langenheim 1990; Lodge et al. 1996; Kinkel 1997; Duong et al. 2006; Johnston et al. 2006). This indicates that the majority of leaf-endophytes do not invade the leaves by extension from perennial (woody) organs. Fungi occurring endophytically are therefore supposed to predominantly invade leaves from the leaf surface (Cabral et al. 1993; Viret et al. 1994; Santamaria and Bayman 2005), and consequently occur first as epiphytes (Fig. 1). Differences between cultivatable epi- and endophytic assemblages (Santamaria and Bayman 2005; Osono 2008; Flessa and Rambold 2013; Zambell and White 2015) indicate that only part of the epiphytic fungi invades the leaves. This supports the idea that plants actively filter invading fungi (Vacher et al. 2016) and explain why plants growing at the same location harbour different microbiomes (Espinosa-Garcia and Langenheim 1990; Peršoh 2013). By the cultivation-independent approach we found that only rare and infrequent epiphytic fungi were restricted to the leaf surface, considering the whole extensive study area (Fig. 2a–c). This indicates the host plant only weakly filters invading fungi in autumn, which may be ascribed to decreasing defence capabilities in aging leaves (Balázs et al. 1973; Iwasa et al. 1996). These differences may be caused by methodological imbalances, such as cultivation biases, or biologically, such as by seasonal changes in leaf physiology. Identifying the actual cause will require assessment of the dynamics of leaf-endophytes throughout the growing season.
Endophytic fungi are supposed to rapidly sporulate and re-infect living leaves after leaf fall (Osono 2006). However, they have long been suspected to be involved in the decomposition of leaf litter (Hudson 1968; Promputtha et al. 2002, 2007, 2010; Suryanarayanan 2013; Saikkonen et al. 2015; Szink et al. 2016; Promputtha et al. 2017). On the other hand, soil fungi have been found earlier to constitute a major component of the endophytic fungal assemblage (Promputtha et al. 2007; Peršoh 2013). The close connection of both habitats is supported by the finding that a considerable proportion of endophytic fungi persists in litter for 1 year (Fig. 4a, b). This raises the question if there is a difference between endophytic fungi persisting in litter and litter fungi with transient endophytic occurrence. Colonizing the insides of leaves at the time of leaf-fall appears to be advantageous for the fungi in 1-year-old litter, because fungi with endophytic occurrence were more active in the litter than indigenous litter fungi without endophytic occurrence (Fig. 4c, d). While their activity inside the living leaves remains to be studied, endophytic fungi in general formed only unstructured loose assemblages there (Fig. 5). By contrast, litter fungi formed a strongly interconnected network with formerly endophytic fungi well integrated into the litter communities (Figs. 5, 6). Litter therefore appears to be their primary habitat and their occurrence in living leaves just a transient stage. The manifold interactions of endophytic fungi with plant hosts (for reviews see Hyde and Soytong 2008; Porras-Alfaro and Bayman 2011; Debbab et al. 2012; Hamilton et al. 2012; Hardoim et al. 2015; Berg et al. 2016; Vacher et al. 2016) are therefore probably mostly ascribable to fungal individuals and not to fungal communities.
Previous studies suggested a lower overlap between fungi of leaves and litter and faster species turnover during early decomposition (Müller et al. 2001; Osono 2006; Voríšková and Baldrian 2013; Szink et al. 2016). These studies were mostly locally restricted, and agree largely with the conditions we observed for single plots. Fungi detected inside falling leaves at a certain plot contributed little to the corresponding local litter community, while those occurring anywhere in the study area as endophytes contributed considerably to each local litter community (Fig. 7a–c). As it appears implausible that the capability of litter fungi to invade living leaves is locally restricted, only a fraction of the transient endophytic occurrences was obviously detected at the time of sampling in a certain location. The species accumulation curves (Figs. 3, S7) indicate that compositional heterogeneity of endophytic fungal assemblages (Cordier et al. 2012a; Izuno et al. 2016) renders an undersampling at the local scale likely; even at the applied extensive sampling scale. In addition, the rapid changes in composition of endophytic assemblages (Suryanarayanan and Thennarasan 2004; Peršoh 2013) may have been only detected by sampling plots with different (micro)climatic conditions, thus representing different compositional stages of the phyllosphere microbiome at the sampling time. Leaves falling at a certain location and time may accordingly not include the entire spectrum of transient endophytic stages of the year’s litter fungi (Fig. 4a). Only litter accumulating over the leaf-falling period (Bray and Gorham 1964) may harbour the whole diversity of fungi assembling together with indigenous litter fungi into a locally structured litter community (Figs. 4a, c, 7d). This transition from endophytic assemblages to litter communities obviously involves rapid changes in the abundances of individual species (Voríšková and Baldrian 2013). Future analyses on this transition phase may provide valuable insights into the assembly of litter fungal communities and thus into the globally important decomposition process.
In this study, we compared for the first time fungal assemblages in falling and decaying leaves across extensive spatial and temporal scales. Fungal assemblages colonizing the phyllosphere were weakly structured, suggesting that the phyllosphere is colonized by a loose assemblage of fungi, which contribute to a well-structured community in decaying leaf litter. Assuming that the findings are generalizable for other tree species and developmental stages of leaves, this challenges our current understandings of the ecology of horizontally transmitted endophytic fungi and of litter decomposition processes. At the time of leaf-fall, endophytic fungi persisting in litter for less than 1 year represented only one part of the foliar microbiome (Fig. 4a). The other part became integrated into the litter community, indicating that they play a major role in early litter decomposition. It was shown that the initial decomposer community sustainably influences the assembly of successive decomposer communities and the decomposition process via priority effects (Cline and Zak 2015; Szink et al. 2016). Predictions of decomposition processes (Braakhekke et al. 2013) may therefore be refined by considering factors affecting composition of the endophytic assemblage during the leaf-falling period. Accounting for the immense spatiotemporal heterogeneity of leaf-endophytic fungal assemblages throughout the leaf-falling period (Fig. 7a–c) will be the major challenge for future experimental studies and modelling approaches.
Conclusion
A crucial function of the microbiome inside beech-leaves is apparently realized in dead leaves detached from the macroorganism. Decomposer communities originating from different tree individuals and species become intermixed on the forest floor. This renders delimitation of holobiont microbiomes aboveground as difficult as belowground. The close connection of plant individuals by their associated microbiomes challenges feasibility of the ‘plant holobiont’ concept in ecological field studies. Its striking relevance for nutrient (re)cycling, however, suggests an important role in ecology and evolution, i.e. in the adaptation of plants to changing environments (Zilber-Rosenberg and Rosenberg 2008).
References
Albert R, Barabasi A-L (2002) Statistical mechanics of complex networks. Rev Mod Phys 74(1):47–97. doi:10.1103/RevModPhys.74.47
Balázs E, Gáborjányi R, Király Z (1973) Leaf senescence and increased virus susceptibility in tobacco: the effect of abscisic acid. Physiol Plant Pathol 3(3):341–346. doi:10.1016/0048-4059(73)90005-2
Beiler KJ, Durall DM, Simard SW, Maxwell SA, Kretzer AM (2010) Architecture of the wood-wide web: Rhizopogon spp. genets link multiple Douglas-fir cohorts. New Phytol 185(2):543–553. doi:10.1111/j.1469-8137.2009.03069.x
Berg G, Rybakova D, Grube M, Köberl M (2016) The plant microbiome explored: implications for experimental botany. J Exp Bot 67(4):995–1002. doi:10.1093/jxb/erv466
Braakhekke MC, Wutzler T, Beer C, Kattge J, Schrumpf M, Ahrens B, Schöning I, Hoosbeek MR, Kruijt B, Kabat P, Reichstein M (2013) Modeling the vertical soil organic matter profile using Bayesian parameter estimation. Biogeosciences 10(1):399–420. doi:10.5194/bg-10-399-2013
Bray JR, Gorham E (1964) Litter production in forests of the world. In: Cragg JB (ed) Advances in ecological research, vol 2. Academic Press Inc., London, pp 101–157
Cabral D, Stone JK, Carroll GC (1993) The internal mycobiota of Juncus spp.: microscopic and cultural observations of infection patterns. Mycol Res 97(3):367–376
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5):335–336. doi:10.1038/nmeth.f.303
Carlsen T, Aas AB, Lindner D, Vrålstad T, Schumacher T, Kauserud H (2012) Don’t make a mista(g)ke: is tag switching an overlooked source of error in amplicon pyrosequencing studies? Fungal Ecol 5(6):747–749. doi:10.1016/j.funeco.2012.06.003
Clarke KR, Gorley RN (2015) PRIMER v7: user manual/tutorial. Primer E, Plymouth
Cline LC, Zak DR (2015) Initial colonization, community assembly and ecosystem function: fungal colonist traits and litter biochemistry mediate decay rate. Mol Ecol 24(19):5045–5058. doi:10.1111/mec.13361
Coince A, Caël O, Bach C, Lengellé J, Cruaud C, Gavory F, Morin E, Murat C, Marçais B, Buée M (2013) Below-ground fine-scale distribution and soil versus fine root detection of fungal and soil oomycete communities in a French beech forest. Fungal Ecol 6(3):223–235. doi:10.1016/j.funeco.2013.01.002
Collado J, Platas G, González I, Peláez F (1999) Geographical and seasonal influences on the distribution of fungal endophytes in Quercus ilex. New Phytol 144(3):525–532. doi:10.1046/j.1469-8137.1999.00533.x
Cordier T, Robin C, Capdevielle X, Desprez-Loustau M-L, Vacher C (2012a) Spatial variability of phyllosphere fungal assemblages: genetic distance predominates over geographic distance in a European beech stand (Fagus sylvatica). Fungal Ecol 5(5):509–520. doi:10.1016/j.funeco.2011.12.004
Cordier T, Robin C, Capdevielle X, Fabreguettes O, Desprez-Loustau M-L, Vacher C (2012b) The composition of phyllosphere fungal assemblages of European beech (Fagus sylvatica) varies significantly along an elevation gradient. New Phytol 196(2):510–519. doi:10.1111/j.1469-8137.2012.04284.x
Debbab A, Aly AH, Proksch P (2012) Endophytes and associated marine derived fungi—ecological and chemical perspectives. Fungal Divers 57(1):45–83. doi:10.1007/s13225-012-0191-8
Dickie IA, Alexander I, Lennon S, Öpik M, Selosse M-A, van der Heijden MGA, Martin FM (2015) Evolving insights to understanding mycorrhizas. New Phytol 205(4):1369–1374. doi:10.1111/nph.13290
Duong LM, Jeewon R, Lumyong S, Hyde KD (2006) DGGE coupled with ribosomal DNA gene phylogenies reveal uncharacterized fungal endophytes. Fungal Divers 23:121–138
Espinosa-Garcia FJ, Langenheim JH (1990) The endophytic fungal community in leaves of a coastal redwood population diversity and spatial patterns. New Phytol 116(1):89–97. doi:10.1111/j.1469-8137.1990.tb00513.x
Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, Huttenhower C (2012) Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol 8(7):e1002606. doi:10.1371/journal.pcbi.1002606
Faust K, Lima-Mendez G, Lerat J-S, Sathirapongsasuti JF, Knight R, Huttenhower C, Lenaerts T, Raes J (2015) Cross-biome comparison of microbial association networks. Front Microbiol 6:1200. doi:10.3389/fmicb.2015.01200
Fischer M, Bossdorf O, Gockel S, Hänsel F, Hemp A, Hessenmöller D, Korte G, Nieschulze J, Pfeiffer S, Prati D, Renner S, Schöning I, Schumacher U, Wells K, Buscot F, Kalko EK, Linsenmair KE, Schulze E-D, Weisser WW (2010) Implementing large-scale and long-term functional biodiversity research: the biodiversity exploratories. Basic Appl Ecol 11(6):473–485. doi:10.1016/j.baae.2010.07.009
Fisher PJ, Petrini O, Petrini LE, Sutton BC (1994) Fungal endophytes from the leaves and twigs of Quercus ilex L. from England, Majorca and Switzerland. New Phytol 127(1):133–137. doi:10.1111/j.1469-8137.1994.tb04267.x
Flessa F, Rambold G (2013) Diversity of the Capnocheirides rhododendri-dominated fungal community in the phyllosphere of Rhododendron ferrugineum L. Nova Hedw. 97(1):19–53. doi:10.1127/0029-5035/2013/0110
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28(23):3150–3152. doi:10.1093/bioinformatics/bts565
Fukasawa Y, Osono T, Takeda H (2009) Effects of attack of saprobic fungi on twig litter decomposition by endophytic fungi. Ecol Res 24(5):1067–1073. doi:10.1007/s11284-009-0582-9
Gao X-X, Zhou H, Xu D-Y, Yu C-H, Chen Y-Q, Qu L-H (2005) High diversity of endophytic fungi from the pharmaceutical plant, Heterosmilax japonica Kunth revealed by cultivation-independent approach. FEMS Microbiol Lett 249(2):255–266. doi:10.1016/j.femsle.2005.06.017
Gardes M, Bruns TD (1993) ITS primers with enhanced specificity for basidiomycetes—application to the identification of mycorrhizae and rusts. Mol Ecol 2(2):113–118
Guo LD, Hyde KD, Liew EC (2001) Detection and taxonomic placement of endophytic fungi within frond tissues of Livistona chinensis based on rDNA sequences. Mol Phylogenet Evol 20(1):1–13. doi:10.1006/mpev.2001.0942
Guo LD, Huang GR, Wang Y, He WH, Zheng WH, Hyde KD (2003) Molecular identification of white morphotype strains of endophytic fungi from Pinus tabulaeformis. Mycol Res 107(6):680–688. doi:10.1017/S0953756203007834
Hamilton CE, Gundel PE, Helander M, Saikkonen K (2012) Endophytic mediation of reactive oxygen species and antioxidant activity in plants: a review. Fungal Divers 54(1):1–10. doi:10.1007/s13225-012-0158-9
Hardoim PR, van Overbeek LS, Berg G, Pirttilä AM, Compant S, Campisano A, Döring M, Sessitsch A (2015) The Hidden World within Plants: ecological and Evolutionary Considerations for Defining Functioning of Microbial Endophytes. Microbiol Mol Biol Rev 79(3):293–320. doi:10.1128/MMBR.00050-14
Hawksworth DL (1991) The fungal dimension of biodiversity: magnitude, significance, and conservation. Mycol Res 95(6):641–655
Hudson HJ (1968) The Ecology of Fungi on Plant Remains Above the Soil. New Phytol 67(4):837–874. doi:10.1111/j.1469-8137.1968.tb06399.x
Hulsen T, de Vlieg J, Alkema W (2008) BioVenn—a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9:488. doi:10.1186/1471-2164-9-488
Hyde KD, Soytong K (2008) The fungal endophyte dilemma. Fungal Divers 33:163–173
Iwasa Y, Kubo T, van Dam N, de Jong TJ (1996) Optimal level of chemical defense decreasing with leaf age. Theor Popul Biol 50(2):124–148. doi:10.1006/tpbi.1996.0026
Izuno A, Tanabe AS, Toju H, Yamasaki M, Indrioko S, Isagi Y (2016) Structure of phyllosphere fungal communities in a tropical dipterocarp plantation: a massively parallel next-generation sequencing analysis. Mycoscience 57(3):171–180. doi:10.1016/j.myc.2015.12.005
Johnston PR, Sutherland PW, Joshee S (2006) Visualising endophytic fungi within leaves by detection of (1 → 3)-β-d-glucans in fungal cell walls. Mycologist 20(4):159–162. doi:10.1016/j.mycol.2006.10.003
Jumpponen A, Jones KL (2010) Seasonally dynamic fungal communities in the Quercus macrocarpa phyllosphere differ between urban and nonurban environments. New Phytol 186(2):496–513. doi:10.1111/j.1469-8137.2010.03197.x
Khan AL, Al-Harrasi A, Al-Rawahi A, Al-Farsi Z, Al-Mamari A, Waqas M, Asaf S, Elyassi A, Mabood F, Shin J-H, Lee I-J (2016) Endophytic fungi from frankincense tree improves host growth and produces extracellular enzymes and indole acetic acid. PLoS ONE 11(6):e0158207. doi:10.1371/journal.pone.0158207
Kinkel LL (1997) Microbial population dynamics on leaves. Annu Rev Phytopathol 35:327–347. doi:10.1146/annurev.phyto.35.1.327
Koide K, Osono T, Takeda H (2005) Colonization and lignin decomposition of Camellia japonica leaf litter by endophytic fungi. Mycoscience 46(5):280–286. doi:10.1007/S10267-005-0247-7
Kõljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AFS, Bahram M, Bates ST, Bruns TD, Bengtsson-Palme J, Callaghan TM, Douglas B, Drenkhan T, Eberhardt U, Duenas M, Grebenc T, Griffith GW, Hartmann M, Kirk PM, Kohout P, Larsson E, Lindahl BD, Lucking R, Martin MP, Matheny PB, Nguyen NH, Niskanen T, Oja J, Peay KG, Peintner U, Peterson M, Poldmaa K, Saag L, Saar I, Schussler A, Scott JA, Senes C, Smith ME, Suija A, Taylor DL, Telleria MT, Weiss M, Larsson K-H (2013) Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol 22(21):5271–5277. doi:10.1111/mec.12481
Kumaresan V, Suryanarayanan TS (2002) Endophyte assemblages in young, mature and senescent leaves of Rhizophora apiculata: evidence for the role of endophytes in mangrove litter degradation. Fungal Divers 9:81–91
Lodge DJ, Fisher PJ, Sutton BC (1996) Endophytic Fungi of Manilkara bidentata Leaves in Puerto Rico. Mycologia 88(5):733. doi:10.2307/3760967
Margulis L, Fester R (1991) Symbiosis as a source of evolutionary innovation: speciation and morphogenesis. MIT Press, Cambridge
McMurdie PJ, Holmes S (2014) Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol 10(4):e1003531. doi:10.1371/journal.pcbi.1003531
Müller MM, Valjakka R, Suokko A, Hantula J (2001) Diversity of endophytic fungi of single Norway spruce needles and their role as pioneer decomposers. Mol Ecol 10:1801–1810
Okane I, Nakagiri A, Ito T (1998) Endophytic fungi in leaves of ericaceous plants. Can J Bot 76(4):657–663. doi:10.1139/cjb-76-4-657
Osono T (2006) Role of phyllosphere fungi of forest trees in the development of decomposer fungal communities and decomposition processes of leaf litter. Can J Microbiol 52(8):701–716. doi:10.1139/w06-023
Osono T (2008) Endophytic and epiphytic phyllosphere fungi of Camellia japonica: seasonal and leaf age-dependent variations. Mycologia 100(3):387–391. doi:10.3852/07-110R1
Osono T, Takeda H (1999) Decomposing ability of interior and surface fungal colonizers of beech leaves with reference to lignin decomposition. Eur J Soil Biol 35(2):51–56. doi:10.1016/S1164-5563(99)00112-0
Paulin MM, Nicolaisen MH, Jacobsen CS, Gimsing AL, Sørensen J, Bælum J (2013) Improving Griffith’s protocol for co-extraction of microbial DNA and RNA in adsorptive soils. Soil Biol Biochem 63:37–49. doi:10.1016/j.soilbio.2013.02.007
Peñuelas J, Rico L, Ogaya R, Jump AS, Terradas J (2012) Summer season and long-term drought increase the richness of bacteria and fungi in the foliar phyllosphere of Quercus ilex in a mixed Mediterranean forest. Plant Biol 14(4):565–575. doi:10.1111/j.1438-8677.2011.00532.x
Peršoh D (2013) Factors shaping community structure of endophytic fungi–evidence from the Pinus-Viscum-system. Fungal Divers 60(1):55–69. doi:10.1007/s13225-013-0225-x
Peršoh D (2015) Plant-associated fungal communities in the light of meta’omics. Fungal Divers 75(1):1–25. doi:10.1007/s13225-015-0334-9
Peršoh D, Theuerl S, Buscot F, Rambold G (2008) Towards a universally adaptable method for quantitative extraction of high-purity nucleic acids from soil. J Microbiol Methods 75(1):19–24. doi:10.1016/j.mimet.2008.04.009
Peršoh D, Melcher M, Flessa F, Rambold G (2010) First fungal community analyses of endophytic ascomycetes associated with Viscum album ssp austriacum and its host Pinus sylvestris. Fungal Biol 114(7):585–596. doi:10.1016/j.funbio.2010.04.009
Porras-Alfaro A, Bayman P (2011) Hidden fungi, emergent properties: endophytes and microbiomes. Annu Rev Phytopathol 49:291–315. doi:10.1146/annurev-phyto-080508-081831
Promputtha I, Lumyong S, Lumyong P, McKenzie EHC, Hyde KD (2002) Fungal succession on senescent leaves of Manglietia garrettii in Suthep-Pui National Park, northern Thailand. Fungal Divers 10:89–100
Promputtha I, Jeewon R, Lumyong S, McKenzie EHC, Hyde KD (2005) Ribosomal DNA fingerprinting in the identification of non sporulating endophytes from Magnolia liliifera (Magnoliaceae). Fungal Divers 20:167–186
Promputtha I, Lumyong S, Dhanasekaran V, McKenzie EHC, Hyde KD, Jeewon R (2007) A phylogenetic evaluation of whether endophytes become saprotrophs at host senescence. Microb Ecol 53(4):579–590. doi:10.1007/s00248-006-9117-x
Promputtha I, Hyde KD, McKenzie EHC, Peberdy JF, Lumyong S (2010) Can leaf degrading enzymes provide evidence that endophytic fungi becoming saprobes? Fungal Divers 41(1):89–99. doi:10.1007/s13225-010-0024-6
Promputtha I, McKenzie EH, Tennakoon DS, Lumyong S, Hyde KD (2017) Succession and Natural Occurrence of Saprobic Fungi on Leaves of Magnolia liliifera in a Tropical Forest. Cryptogam Mycol 38(2):213–225. doi:10.7872/crym/v38.iss2.2017.213
Purahong W, Hyde KD (2011) Effects of fungal endophytes on grass and non-grass litter decomposition rates. Fungal Divers 47(1):1–7. doi:10.1007/s13225-010-0083-8
Rodriguez RJ, White JF, Arnold AE, Redman RS (2009) Fungal endophytes: diversity and functional roles. New Phytol 182(2):314–330. doi:10.1111/j.1469-8137.2009.02773.x
Röhl O, Peršoh D, Mittelbach M, Elbrecht V, Brachmann A, Nuy J, Boenigk J, Leese F, Begerow D (2017) Distinct sensitivity of fungal freshwater guilds to water quality. Mycol Prog 16(2):155–169. doi:10.1007/s11557-016-1261-1
Rohwer F, Seguritan V, Azam F, Knowlton N (2002) Diversity and distribution of coral-associated bacteria. Mar Ecol Prog Ser 243:1–10. doi:10.3354/meps243001
Rosenberg E, Zilber-Rosenberg I (2016) Microbes drive evolution of animals and plants: the hologenome concept. MBio 7(2):e01395. doi:10.1128/mBio.01395-15
Saikkonen K (2007) Forest structure and fungal endophytes. Fungal Biol Rev 21(2–3):67–74. doi:10.1016/j.fbr.2007.05.001
Saikkonen K, Mikola J, Helander M (2015) Endophytic phyllosphere fungi and nutrient cycling in terrestrial ecosystems. Curr Sci 109:121–126
Santamaria J, Bayman P (2005) Fungal epiphytes and endophytes of coffee leaves (Coffea arabica). Microb Ecol 50(1):1–8. doi:10.1007/s00248-004-0002-1
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to ImageJ: 25 years of image analysis. Nat Methods 9(7):671–675. doi:10.1038/nmeth.2089
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci USA 109(16):6241–6246. doi:10.1073/pnas.1117018109
Scholtysik A, Unterseher M, Otto P, Wirth C (2013) Spatio-temporal dynamics of endophyte diversity in the canopy of European ash (Fraxinus excelsior). Mycol Progress 12(2):291–304. doi:10.1007/s11557-012-0835-9
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–2504. doi:10.1101/gr.1239303
Shirouzu T, Hirose D, Fukasawa Y, Tokumasu S (2009) Fungal succession associated with the decay of leaves of an evergreen oak, Quercus myrsinaefolia. Fungal Divers 34:87–109
Simard SW, Perry DA, Jones MD, Myrold DD, Durall DM, Molina R (1997) Net transfer of carbon between ectomycorrhizal tree species in the field. Nature 388(6642):579–582
Sinha R, Stanley G, Gulati GS, Ezran C, Travaglini KJ, Wei E, Chan CKF, Nabhan AN, Su T, Morganti RM, Conley SD, Chaib H, Red-Horse K, Longaker MT, Snyder MP, Krasnow MA, Weissman IL (2017) Index switching causes “spreading-of-signal” among multiplexed samples in illumina HiSeq 4000. DNA. doi:10.1101/125724
Smith GR, Finlay RD, Stenlid J, Vasaitis R, Menkis A (2017) Growing evidence for facultative biotrophy in saprotrophic fungi: data from microcosm tests with 201 species of wood-decay basidiomycetes. New Phytol 215(2):747–755. doi:10.1111/nph.14551
Sun X, Ding Q, Hyde KD, Guo LD (2012) Community structure and preference of endophytic fungi of three woody plants in a mixed forest. Fungal Ecol 5(5):624–632. doi:10.1016/j.funeco.2012.04.001
Suryanarayanan TS (2011) Diversity of fungal endophytes in tropical trees. In: Pirttilä AM, Frank AC (eds) Endophytes of forest trees, vol 80. Springer, Dordrecht, pp 67–80
Suryanarayanan TS (2013) Endophyte research: going beyond isolation and metabolite documentation. Fungal Ecol 6(6):561–568. doi:10.1016/j.funeco.2013.09.007
Suryanarayanan TS, Thennarasan S (2004) Temporal variation in endophyte assemblages of Plumeria rubra leaves. Fungal Divers 15:197–204
Suryanarayanan TS, Venkatesan G, Murali TS (2003) Endophytic fungal communities in leaves of tropical forest trees: diversity and distribution patterns. Curr Sci 85:489–493
Szink I, Davis EL, Ricks KD, Koide RT (2016) New evidence for broad trophic status of leaf endophytic fungi of Quercus gambelii. Fungal Ecol 22:2–9. doi:10.1016/j.funeco.2016.04.003
Toti L, Viret O, Horat G, Petrini O (1993) Detection of the endophyte Discula umbrinella in buds and twigs of Fagus sylvatica. For Pathol 23(3):147–152. doi:10.1111/j.1439-0329.1993.tb00954.x
Vacher C, Hampe A, Porté AJ, Sauer U, Compant S, Morris CE (2016) The phyllosphere: microbial jungle at the plant-climate interface. Annu Rev Ecol Evol Syst 47(1):1–24. doi:10.1146/annurev-ecolsys-121415-032238
Viret O, Toti L, Chapela IH, Petrini O (1994) The role of the extracellular sheath in recognition and attachment of conidia of Discula umbrinella (Berk. & Br.) Morelet to the host surface. New Phytol 127(1):123–131. doi:10.1111/j.1469-8137.1994.tb04266.x
Voríšková J, Baldrian P (2013) Fungal community on decomposing leaf litter undergoes rapid successional changes. ISME J 7(3):477–486. doi:10.1038/ismej.2012.116
Weig AR, Peršoh D, Werner S, Betzlbacher A, Rambold G (2013) Diagnostic assessment of mycodiversity in environmental samples by fungal ITS1 rDNA length polymorphism. Mycol Prog 12(4):719–725. doi:10.1007/s11557-012-0883-1
White TJ, Burns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis M, Gelfand D, Sninsky J, White T (eds) PCR protocols: a guide to methods and applications. Academic Press, Cambridge, pp 315–322
Yuan Z, Chen L (2014) The role of endophytic fungal individuals and communities in the decomposition of Pinus massoniana needle litter. PLoS ONE 9(8):e105911. doi:10.1371/journal.pone.0105911
Zambell CB, White JF (2015) In the forest vine Smilax rotundifolia, fungal epiphytes show site-wide spatial correlation, while endophytes show evidence of niche partitioning. Fungal Divers 75(1):279–297. doi:10.1007/s13225-014-0316-3
Zilber-Rosenberg I, Rosenberg E (2008) Role of microorganisms in the evolution of animals and plants: the hologenome theory of evolution. FEMS Microbiol Rev 32(5):723–735. doi:10.1111/j.1574-6976.2008.00123.x
Acknowledgements
We thank all the members of the Geobotany working group (Bochum University) for their assistance during sample collection, nucleic acid extractions and library preparation and Miguel Romero Torres for surface sterilization. We also thank the managers of the three Exploratories, Kirsten Reichel-Jung, Katrin Lorenzen, Martin Gorke and all former managers for their work in maintaining the plot and project infrastructure; Christiane Fischer and Jule Mangels for giving support through the central office, Michael Owonibi for managing the central data base, and Markus Fischer, Eduard Linsenmair, Dominik Hessenmöller, Daniel Prati, Ingo Schöning, François Buscot, Ernst-Detlef Schulze, Wolfgang W. Weisser and the late Elisabeth Kalko for their role in setting up the Biodiversity Exploratories project. The work has been funded by the DFG Priority Program 1374 “Infrastructure-Biodiversity-Exploratories” (BE 2201/16-1 and PE 1673/5-1). Field work permits were issued by the responsible state environmental offices of Baden-Württemberg, Thüringen, and Brandenburg (according to § 72 BbgNatSchG).
Author information
Authors and Affiliations
Contributions
The study was designed by DB and DP; DP, MG and student helpers performed the sampling; MG conducted the laboratory work; AB supported preparation of the amplicon libraries and performed the sequencing; MG and DP analysed the data and wrote the manuscript. All authors discussed and contributed to the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Guerreiro, M.A., Brachmann, A., Begerow, D. et al. Transient leaf endophytes are the most active fungi in 1-year-old beech leaf litter. Fungal Diversity 89, 237–251 (2018). https://doi.org/10.1007/s13225-017-0390-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13225-017-0390-4