
Abstract
In this study, we report the direct precipitation of nano-HA in the present of maltodextrins with the different dextrose equivalent (DE) values in the range of 10–30. Characterization of the obtained samples, using X-ray diffraction and Fourier transform infrared spectrophotometry, indicated that the presence of maltodextrins, with the different DE values, does not affect the phase composition and structure of the obtained composites. Morphology studies of the samples, using field emission scanning electron microscope and transmission electron microscope, revealed that maltodextrin has obvious effect on the size, shape, and morphology of hydroxyapatite nanoparticles. In particular, in studied DE range, maltodextrin DE 28–30 with dominant structure of debranched chain is the most preferable choice to obtain the composite with highly dispersed nanoparticles. In vitro assay on pre-osteoblast MC3T3-E1 cells demonstrated the ability of the composites to stimulate alkaline phosphatase activity and mineralization during differentiation of the cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In recent years, nano-hydroxyapatite (HA) has been widely used in biomedical applications as a material for damaged bones or teeth and other bone-related fields, scaffold materials, and drug delivery agents due to the fact that HA has a formula of Ca10(PO4)6(OH)2, which is similar to the mineral phase of natural bone and results in its excellent biocompatibility and high osteogenic potential (Klawitter and Hulbert 1971; Murugan and Ramakrishna 2007). Recently, an interesting research direction is to reinforce nano-HA in a polymer matrix, which is assumed to improve the mechanical and biological performance as well as to overcome the aggregation of HA nanoparticles. This aggregation itself is a key problem in the production of high-performance nanocomposite (Hule and Pochan 2007; Monika Supova 2009). For this purpose, natural polymers have received more attention because of their biocompatible and biodegradable properties. There are intensive studies which have been focused on the development of composites of HA with collagen (Yoruc and Aydinoglu 2015), chitosan (Yamaguchi et al. 2001; Chen et al. 2002), and alginate (Rajkumar et al. 2011). Collagen/HA composite materials have been tailored by the addition of different inorganic and organic components or synthesized with oriented structure to improve their morphology and properties. More recently, nanocomposite of HA/chitosan/alginate was prepared and the pore morphologies and mechanical properties of the composite scaffolds were determined, and their bioactivity and osteoconductivity were evaluated (Kim et al. 2015). In other study of Danilchenko et al. (2009), the porous chitosan–hydroxyapatite materials have shown good osteoconductive properties in in vivo experiments.
Among the natural polymers, starch and hydrolyzed-starch products are also widely used in food and pharmaceutical industries. In addition, their hydroxyl groups can play the role of nucleation sites for the formation of HA crystals, subsequently regulating their growth and arrangement. However, as far as we know, there are only a few literature data on the composites of HA and starch or its hydrolyzed products. Sadjadi, Meskinfam, and co-workers have synthesized HA via an in situ biomimetic process in the presence of wheat starch (Sadjadi et al. 2010) and gelatin/starch (Meskinfam et al. 2011, 2012). The result revealed that the shape and morphology of HA are influenced by the presence of biopolymers as a template. It leads to the formation of rod-like HA which is similar to this inorganic component in natural bone. Shakir et al. (2015) reported that they have synthesized successfully a nanocomposite system incorporating dextrin (its character was not informed) into nano-HA/chitin matrix via co-precipitation route for the application in bone tissue engineering.
The aims of this study are to prepare HA/maltodextrin composites and to determine the effect of dextrose equivalent value and chain structure of maltodextrin on the morphology and particle size of the obtained composites. The in vitro effect on the function of pre-osteoblastic MC3T3-E1 cells is investigated to evaluate the ability of the composites to stimulate osteoblast differentiation and mineralization. Maltodextrin is produced from starch hydrolysis and characterized by the degree of hydrolysis, expressed by the dextrose equivalent (DE) value. DE is defined as the percentage of reducing sugar calculated as dextrose (glucose) on a dry weight basis (Dziedzic and Kearsley 1995). DE value is the most common parameter used to characterize maltodextrins. Its theoretical value is inversely proportional to number average molecular weight; the higher the DE value, the shorter the glucose chains. It should be noted that maltodextrin (DE above about 10) has been more clearly identified and independent from plant resource (wheat, maize, cassava, etc.) in comparison to starch (Roller and Jones 1996; Dziedzic and Kearsley 1995).
Materials and methods
Ca(OH)2, H3PO4, and C2H5OH 99.7% were extra pure, purchased from Merck. Maltodextrins were derived from the controlled hydrolysis of cassava starch using two enzymes of Amylex HT (Danisco US, Inc, Genencor Division) and Promozyme (Novozymes A/S, Denmark). The DE value was determined using Lane-Eynon titration, based on the reduction of CuSO4 in an alkaline tartrate solution (AOAC official method 923.09). The weight-averaged molecular weight (M w) was measured using Gel permeation chromatography (GPC, Agilent 1100HPLC using an ultrahydrogel 250 column, RI detector G1362A and pullulan (as) standard).
Concentration of enzymes and hydrolysis time were controlled in order to obtain three maltodextrins with different DE values in the range of 10-30 as follows:
Maltodextrin | P1 | P2 | P3 |
|---|---|---|---|
DE | 10–12 | 18–20 | 28–30 |
M w (g/mol) | 93,357 | 11,258 | 8025 |
Preparation of HA/maltodextrin composite
HA/maltodextrin composite samples were prepared by co-precipitation from Ca(OH)2 and H3PO4 in the presence of maltodextrin at room temperature. Ca(OH)2 suspension of 0.5 M was stirred in 30 min. Then, both aqueous solutions of 0.3 M H3PO4 and 5.0% maltodextrin were added gradually and simultaneously to the Ca(OH)2 suspension during vigorous stirring. When the addition of the solutions is finished, the mixture was stirred continuously for 3 h and left to stand for 24 h. The precipitation was filtered and washed with 1:1 ethanol/water mixture until reached pH 7 and then freeze dried for 24 h. Here, the molar ratio of Ca/P was 1.67 and maltodextrin content was about 25% in the composites.
A pure HA sample without maltodextrin was prepared in the same procedure.
Characterization methods of HA and HA/maltodextrin composites
X-ray diffraction (XRD) patterns were recorded on a D8 Advance Brucker using CuKα radiation. Infrared spectrophotometry (FTIR) was performed on Affinity-S1, Shimadzu. The morphology images were obtained from the field emission scanning electron microscope (FESEM, S4800-Hitachi) and transmission electron microscope (TEM, JEOL JEM-1010, sample was sonicated in distilled water for 30 min and deposited on carbon-coated copper grid). Thermogravimetric analysis (TGA) and differential thermal analysis (DTA) were carried out from room temperature to 600 °C in air at a heating rate of 10 °C/min on Setaram Labsys Evo thermal analyzer.
MC3T3-E1 cell assay
The mouse pre-osteoblastic cell line MC3T3-E1, obtained from the ATCC Cell Bank (Manassas, VA, USA), was cultured in α-modified minimal essential medium (α-MEM; Gibco BRL, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37 °C in 5% CO2 atmosphere. After two days, cells were subcultured using a EDTA–Tripsin 0.05% solution.
At 80% confluence, cells were cultured in the medium supplemented with 10 mM ß-glycerophosphate and 50 µg/ml ascorbic acid in a 96-well culture dish for differentiation. Studied samples, suspended in the culture medium at the different concentrations in the range of 4.0–0.16 µg/ml, were added into the culture wells. The medium was changed every 2–3 days.
After 7 days of differentiation, alkaline phosphatase (ALP) activity was determined using an ALP Assay Kit (Abcam, Cambridge, England) according to the manufacturer’s instructions.
Cell mineralization was evaluated by alizarin red S staining (0.04 M, Sigma-Aldrich, pH 4.4) after 15 days.
Experiments were repeated three times; data are expressed as the mean ± SD. Statistical analyses and significance were performed using GraphPad PRISM software version 4.0 (GraphPad Software, USA).
Results and discussion
Composite samples with maltodextrin of P1, P2, P3, and pure HA sample were denoted as HAP1, HAP2, HAP3, and HAp, respectively.
Phase structure and composition
XRD patterns of all samples are shown in Fig. 1. They are all similar and indicate the formation of phase structure of HA (compared to JCPDS file 00-024-0033) in the composites as well as the HAp sample. Broadening of the peaks is due to the small size of HA particles, especially in the composites.

XRD patterns of pure HAp and HA/maltodextrin composites
XRD results demonstrate that the presence of maltodextrins, with different DE values in the selected range, does not affect phase composition and structure of the obtained composites.
Figure 2 displays FTIR spectra of HAp, maltodextrin (representative sample of P3, the others are the same), and representative HA/maltodextrin composite (HAP3 sample).

FTIR spectra of pure HAp, maltodextrin P3, and HAP3 samples
Almost all of characteristic bands for HA and maltodextrin are present in the spectra of composite sample. Because of the presence of both HA and maltodextrin, some very weak bands were not observed and some bands become broaden or slightly shifted due to overlapping bands. In the previous publications on HA/polymer composites (Rajkumar et al. 2010; Wang et al. 2010), the interaction of HA and polymer, especially, the formation of bonds between Ca2+ ions and COO− or OH− groups of polymer has been proposed. However, also because of the overlap of main vibration bands of HA and maltodextrin, it is very difficult to observe probable change in FTIR spectra of their composite. PO4 3− group forms intensive absorption bands at 565 and 603 and at 1000–1100 cm−1 (Markovic et al. 2004), which were overlapped with the most characteristic bands of maltodextrin (Sun et al. 2013; Bartošová et al. 2013). An explicit peak at 3570 cm−1 is the characteristic of OH− group in HA molecule (Markovic et al. 2004). A wide band from 3600 to 3200 cm−1 and a band at about 1600 cm−1 are assigned to adsorbed water and stretching of the O-H bond of sugar (Souza et al. 2015). The weak intensity bands at 1410 and 1450 cm−1 are attributed to an impurity amount of CO3 2− in HA lattice (Markovic et al. 2004). Double peaks, near 2900 cm−1, are produced by the stretching of the C–H bond of the methyl group (Souza et al. 2015).
Maltodextrin content—thermal analysis result
Figure 3 shows the TGA-DTA curves of HAp, maltodextrin P3, and a representative composite (HAP3 sample). For HAP3 sample, DTA curve exhibits two exothermic peaks at 326 and 375 °C, associated with the weight loss of about 25%, corresponding to the thermal decomposition of maltodextrin. Meanwhile, for pure maltodextrin sample, the decomposition peaks are observed at the significantly higher temperature, 378 and 506 °C, respectively. The degradation temperature was shifted to lower temperatures indicating a decreased thermal stability, probably relating to the interaction between HA molecule and maltodextrin chain.

TGA/DTA curves of pure HAp, maltodextrin P3, and HAP3 samples
Maltodextrin content, determined approximately from the weight loss in temperature range of maltodextrin decomposition, was about 25 wt% composite as expected.
Morphology of the composites
SEM images of HAp and HA/maltodextrin composites are given in Fig. 4. It can be seen from the SEM image of HAp that pure HA particles are nearly round with a size of about 20–30 nm. However, they assembled into short chains, and then some chains stick together into clusters with a dimension of 30–60 nm in width and 80–100 nm in length. A similar morphology is also observed in the image of HAP1 but size of aggregates is smaller in HAp sample. In SEM image of HAP2, the agglomeration is reduced much more than HAP1; the particle chains were shorter and less linked together. The image of HAP3 sample presents a discrete dispersion of HA nanoparticles. Thus, the particle distribution was quite improved in HAP3 sample.

SEM images of a pure HAp and the composites of b HAP1, c HAP2, d HAP3
What is observed from SEM images indicates that template maltodextrin has affected the morphology of HA. Maltodextrins, in particular, with DE of 28–30 have given a rather uniform dispersion of HA particles. This is confirmed more clearly with TEM images of the composites (Fig. 5). There are rod aggregations with width of 30-50 nm and length up to 100 nm in the image of pure HA, corresponding to the clusters of particle chains in its SEM image. In HAP1 sample, the powder exhibits no serious aggregation. In the next image, the dispersion of nano-HA particles was improved further in HAP2 sample. In the TEM images of HAP1 and HAP2 composites, it can be seen nearly as spherical-shaped HA particles although they were still aggregated. In the image of HAP3, although HA particles were not entirely separated, they can be discerned rather clearly at the very small size, about 10–15 nm.

TEM images of a pure HAp and the composites of b HAP1, c HAP2, d HAP3
The main role of polymer in the composite formation is thought to offer nucleation sites and structural control on the morphology and size of nanoparticles, their distribution over the polymer matrix as well. As shown in some studies (Yin et al. 2011; Colovic et al. 2011), the concentration of carboxyl, hydroxyl, or amine groups in polymer molecule has a significant effect on speed and mechanism of HA nucleation. And then, the density of nucleation sites can also affect the size of particle aggregates and their morphology. In this study, maltodextrins were distinguished by chain length of molecule and branched structure, which are characterized by DE and M w values. Among used maltodextrins, P3 sample with DE 28–30 has the highest concentration of OH groups; the smallest particle aggregations were formed, possibly due to the largest number of nucleation sites. More importantly, by using Promozyme in starch hydrolysis, a debranching enzyme, maltodextrin P3 was intentionally obtained with almost linear molecular chain, while maltodextrin P1 and P2, using Amylex HT enzyme, possess linear and branched structure. This is also consistent to the determined result of their M w value that from P1 to P2, M w was much more decreased than those from P2 to P3. In HAP1 and HAP2 samples, the bulky structure of branched chain of maltodextrin P1 and P2 beneficials for the growth of nanoparticle aggregates. In HAP3, the narrow spaces of maltodextrin helical chains without branched side have protected HA nanoparticles from the agglomeration.
Bioactivity of the composites
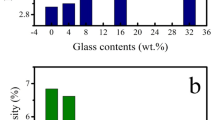
During differentiation, MC3T3-E1 cells exhibit a developmental sequence similar to osteoblasts in bone tissue, including production of ALP, processing of procollagens to collagens and the mineralization of the extracellular matrix (Quarles et al. 1992; Ogawa et al. 2007). Therefore, the influence of studied samples on the differentiation was evaluated by ALP activity and mineralization. The determined ALP activity (Fig. 6) demonstrates that in the presence of HAp or HAP composites, at the concentration of 4 µg/ml, ALP activity increased compared to negative control. It is more significant in cases of the composites. Especially, HAP3 sample showed an increase of nearly 30% relative to control.

Effect of pure HAp and the HA/maltodextrin composites on the ALP activity of MC3T3-E1. *p < 0.05 versus control
Similar to the ALP activity results, the osteoblast mineralization was improved by adding of HAP composites; HAp sample gave a negligible result (Fig. 7). Although this enhancement was not so large, probably due to low concentration of studied samples, an increasing trend of the mineralization from HAP1 to HAP3 can be seen. These results provide an evidence of the osteogenic potential of the composites.

Effect of pure HAp and the HA/maltodextrin composites on the mineralization of MC3T3-E1
Conclusion
Maltodextrins with the differences of DE value and molecular chain structure were prepared by the controlled hydrolysis of cassava starch using two enzymes of Amylex HT and Promozyme. Composites of HA and these maltodextrins were synthesized using the direct precipitation of nano-HA in the presence of the maltodextrins. SEM and TEM studies indicate that template maltodextrin has affected significantly the morphology of HA. In particular, in studied DE range, maltodextrin DE 28–30 with dominant structure of debranched chain is preferable choice to obtain the composite with highly dispersed nanoparticles. Furthermore, the osteogenic potential of the HAP composites was demonstrated by the stimulation of ALP activity and mineralization during the differentiation of pre-osteoblast MC3T3-E1 cells.
We acknowledge the financial support from the Ministry of Science and Technology Vietnam (No. KC.07.14/11-15).
References
Bartošová A, Soldán M, Sirotiak M, Blinová L, Michaliková A (2013) Application of FTIR-ATR spectroscopy for determination of glucose in hydrolysates of selected starches, Research papers, Faculty of materials science and technology in trnava Slovak university of technology in bratislava 21 (special number):116–121
Chen F, Wang Z-C, Lin CJ (2002) Preparation and characterization of nano-sized hydroxyapatiteparticles and hydroxyapatite/chitosan nano-composite for usein biomedical materials. Mater Lett 57:858–861
Čolović B, Marković D, Jokanović V (2011) Nucleation of biomimetic hydroxyapatite. Serb Dent J. doi:10.2298/SGS1101007C
Danilchenko SN, Kalinkevich OV, Lavrynenko SN et al (2009) Chitosan–hydroxyapatite composite biomaterials made by a one step co-precipitation method: preparation, characterization and in vivo tests. J Biol Phys Chem 9(3):119–126
Dziedzic, SZ, Kearsley, MW (1995) Handbook of starch hydrolysis products and their derivatives. Sringer Science + Business Media Dordrecht
Hule AH, Pochan DJ (2007) Polymer nanocomposites for biomedical applications. MRS Bull 32:354–358
Kim HL, Jung GY, Yoon JH, Han JS, Park YJ, Kim DG, Zhang M, Kim DJ (2015) Preparation and characterization of nano-sized hydroxyapatite/alginate/chitosan composite scaffolds for bone tissue engineering. Mater Sci Eng C Mater Biol Appl 54:20–25
Klawitter JJ, Hulbert SF (1971) Application of porous ceramics for the attachment of load bearing internal orthopedic applications. J. Biomed Mater Res Symp 5(6):161–229
Markovic M, Fowler BO, Tung MS (2004) Preparation and comprehensive characterization of a calcium hydroxyapatite reference material. J Res Natl Inst Stand Technol 109(6):553–568
Meskinfam M, Sadjadi MS, Jazdarreh H (2011) Biomimetic preparation of nano hydroxyapatite in Gelatin-starchmatrix. Int Sch Sci Res Innov 5(4):294–297
Meskinfam M, Jokaria F, Giahia M, Jazdarreh H (2012), In vitro bioactivity and biocompatibility evaluation of nano hydroxyapatite-gealtin-starch biocomposites. In: 4th Inter. Conf. on Nanostructures (ICNS4), Kish Island, I.R. Iran 12–14 March
Murugan R, Ramakrishna S (2007) Development of cell-responsive nanophase hydroxyapatite for tissue engineering. Am J Biochem Biotechnol 3(3):118–124
Ogawa N, Yamaguchi T, Yano S, Yamauchi M, Yamamoto M, Sugimoto T (2007) The combination of high glucose and advanced glycation end-products (AGEs) inhibits the mineralization of osteoblastic MC3T3-E1 cells through glucose-induced increase in the receptor for AGEs. Horm Metab Res 39:871–875
Quarles LD, Yohay DA, Lever LW, Caton R, Wenstrup RJ (1992) Distinct proliferative and differentiated stages of murine MC3T3-E1 cells in culture: an in vitro model of osteoblast development. J Bone Miner Res 7(6):683–692
Rajkumar M, Meenakshisundaram N, Rajendran V (2011) Development of nanocomposites based on hydroxyapatite/sodium alginate: synthesis and characterisation. Mater Charact 62(5):469–479
Rajkumar M, Sundaram NM, Rajendran V (2010) In-situ preparation of hydroxyapatite nanorod embedded poly (vinyl alcohol) composite and its characterization. Int J Eng Sci Technol 2(6):2437–2444
Roller S, Jones SA (1996) Handbook of fat replacers. In: Roller S (ed) Starch-derived fat mimetics: maltodextrins. CRC Press, Boca Raton, pp 99–118
Sadjadi MS, Meskinfam M, Jazdarreh H (2010) Hydroxyapatite—starch nano biocomposites synthesis and characterization. Int J Nano Dim 1(1):57–63
Shakir M, Jolly R, Khan MS, Iram NE (2015) Nano-Hydroxyapatite/dextrin/chitin nanocomposite system for bone tissue engineering. Int J Chem Mol Eng 2(9):774
Souza VB, Thomazini M, Carvalho Balieiro JC, Fávaro-Trindade CS (2015) Effect of spray drying on the physicochemical properties and color stability of the powdered pigment obtained from vinification byproducts ofthe Bordo grape (Vitis labrusca). Food Bioprod Process 93:39–50
Sun P, Yang H, Wang Y, Liu K, Xu Y (2013) Lipase-catalyzed synthesis and characterization of stearic acid dextrin ester. Res Health Nutr 1(1):7–11
Supova M (2009) Problem of hydroxyapatite dispersionin polymer matrices: a review. J Mater Sci Mater Med 20:1201–1213. doi:10.1007/s10856-009-3696-2
Wang J, Yu F, Qu L, Meng X, Wen G (2010) Study of synthesis of nano-hydroxyapatite using a silk fibroin template. Mater, Biomed. doi:10.1088/1748-6041/5/4/041002
Yamaguchi I, Tokuchi K, Fukuzaki H, Koyama Y, Takakuda K, Monma H, Tanaka J (2001) Preparation and microstructure analysis of chitosan/hydroxyapatite nanocomposites. J Biomed Mater Res 55(1):20–27
Yin N, Chen S, Ouyang Y, Tang L, Yang J, Wang H (2011) Biomimetic mineralization synthesis of hydroxyapatite bacterial cellulose nanocomposites. Prog Nat Sci Mater Int 21:472–477
Yoruc ABH, Aydinoglu AK (2015) Synthesis of hydroxyapatite/collagen (HA/COL) composite powder using a novel precipitation technique. Acta Phys Pol, A 127(4):1264–1267
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Phan, B.T.N., Nguyen, H.T., Đao, H.Q. et al. Synthesis and characterization of nano-hydroxyapatite in maltodextrin matrix. Appl Nanosci 7, 1–7 (2017). https://doi.org/10.1007/s13204-016-0541-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13204-016-0541-z