
Abstract
The transhydrogenation of pentane (P) and 1-hexyne (1HY) was investigated over 4% CrOx/Al2O3 and potassium-doped 4% CrOx/Al2O3 catalysts over a range of temperatures (523–773 K) with a 5:1 P:1HY ratio. Over the CrOx/Al2O3 catalyst, transhydrogenation clearly occurred at temperatures below 625 K where the yield of alkenes was higher for the co-fed system than for a combination of the individual yields. Due to the acidic nature of the alumina, many of the products were alkylated olefins and alkylated hydrocarbons formed by coincident alkylation and isomerisation. When pentane was added to a feed containing 1-hexyne, the extent of carbon deposition was reduced. By comparing transhydrogenation to limited hydrogen 1-hexyne hydrogenation at 623 K, it was shown that the processes of hydrogenation and transhydrogenation were different, with hydrogenation favouring alkanes, while transhydrogenation favoured alkenes. This may be because pentane dehydrogenation only releases two hydrogen atoms, which only allows 1-hexyne to hydrogenate to 1-hexene. Therefore, if the rate of alkene isomerisation and desorption is faster than that of pentane dehydrogenation, only alkenes will be observed. The latter proposal would suggest that the dehydrogenation/hydrogenation process is closely coupled and would be consistent with pentane influencing 1-hexyne surface chemistry. The effect of the potassium doping was to increase the yield of alkenes. The reason for this may be related to changes in the nature of the surface chromia species. The potassium also neutralised the acid sites on the alumina, reducing the extent of alkylation and hydrogenolysis, which suppressed the formation of other alkynes in the product mix.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The production of olefins has been of major industrial importance since World War II using catalysts to yield high octane aviation fuel [1]. Since then, the petrochemical industries have been constantly developing new processes for improved olefin production, involving new catalyst formulations and modifications to reactor and plant design. This effort has also been extended to accommodate different hydrocarbon feeds in order to maximise production [2]. Currently, olefins are produced via a number of processes in oil refining industries to successfully maximise the production. For example in the late 1980s, the Catofin™ process was designed for on-demand production of propylene and isobutylene using propane and isobutane, respectively [3,4,5].
The primary purpose of the transhydrogenation process historically has been to convert low-value cracked hydrocarbons into valued distillate products. Although transhydrogenation is not a new technology for production of olefins [6,7,8,9,10,11], there is scant scientific attention towards the invention, even though the invention has the potential to lift the equilibrium limitation of direct alkane dehydrogenation for the production of olefins by constantly removing the hydrogen produced through hydrogenation of an alkyne or alkadiene [12,13,14]. Hence, transhydrogenation combines the two processes (alkane dehydrogenation and alkyne hydrogenation) over a catalyst to form two alkenes. Alkane dehydrogenation is endothermic (typically around + 125 kJ mol−1) while alkyne/alkadiene hydrogenation is exothermic (typically around − 165 kJ mol−1); therefore, by coupling the endothermic dehydrogenation process with the exothermic hydrogenation process it is possible to generate a process, where the reaction conditions may be adjusted in order to produce a reaction that is net endothermic, net exothermic or thermally stable, which can simplify and reduce the cost involved in the process [11, 15]. For example, the transhydrogenation process between pentane and 1-hexyne is thermodynamically favoured at most temperatures.
The reaction sequence is thought to occur through dehydrogenation followed by hydrogenation with the hydrogen produced during the dehydrogenation used in the subsequent hydrogenation process:
At high temperatures, however, secondary reactions such as cracking and carbon deposition are also favoured; therefore, the reaction tends to get less selective as temperature is increased. The extent and nature of the carbon deposit will be related to the reactant and reaction temperature used.
The recent literature has revealed transhydrogenation reactions on various catalysts such as supported vanadia, chromia and doped chromia, most of which involved the use of LPG hydrocarbons. Jackson et al. [12,13,14] studied transhydrogenation of propane and propyne over chromia/alumina in a 1:1 ratio. The transhydrogenation reaction was shown to produce propene above the equilibrium value expected from propane dehydrogenation at 823 K. By 873 K, the conversion of propane was ~ 80%, but the yield of propene was low, which was suggested to be due to secondary reaction of the propene forming methane and carbon deposits. Transhydrogenation was also performed between propyne and butane over a 1% vanadia/θ-alumina catalyst at 823 K. Propyne and butane were co-fed, which resulted in an increase in conversion of propyne to propene compared to when it was fed singly over the catalyst. The transhydrogenation reaction was observed to deliver a ~ 72% increase in propene yield, while all the butane reacted was converted to butene isomers.
In this paper, the activity of a chromium oxide/alumina catalyst and a potassium-doped chromium oxide/alumina catalyst for the transhydrogenation of pentane/1-hexyne was evaluated primarily for the production of olefins and other valuable products. Most of the previous work has centred on the C-3 system, but in this work we were interested to see if transhydrogenation was applicable to a wider range of hydrocarbons. The C-6 hydrocarbons allowed us to study the behaviour of conjugated (2,4-hexadiene) and non-conjugated hexadiene (1,5-hexadiene) isomers in comparison with 1-hexyne. The information on these systems will be reported in a subsequent paper. Pentane was chosen as the alkane as it was also a liquid hydrocarbon and in principle could make a full range of unsaturated species. The ratio of alkane:alkyne was chosen as ~ 5:1. Due to the stronger adsorption of the alkyne relative to the alkane, it was decided to have an excess of alkane. The specific ratio was chosen for experimental convenience. A wide temperature range was chosen to fully probe the transhydrogenation reaction. At temperatures below 600 K, pentane dehydrogenation has an equilibrium position that gives < 1% pentene, whereas by 773 K the equilibrium value is ~ 20%. This would allow us to determine if pentane dehydrogenation was a controlling factor in transhydrogenation as the yields should follow the dehydrogenation equilibrium, even though the overall reaction is not equilibrium limited under these conditions.
Experimental
A 4% chromium oxide/alumina catalyst was prepared by incipient wetness impregnation using a γ-alumina support. The alumina support was pre-dried at 353 K for 16 h prior to impregnation. An aqueous solution of [NH4]2Cr2O7 (99 +% Aldrich) was used to prepare the catalyst with a 4% w/w loading. The γ-alumina support (surface area 208 m2 g−1, pore volume 0.52 ml g−1) was supplied by Johnson Matthey, UK. After impregnation, the catalyst was dried for 2 h at 353 K and then at 393 K overnight. A potassium-doped 4% chromium oxide/alumina catalyst was prepared by incipient wetness impregnation using the above catalyst. The catalyst was treated with a solution of KOH sufficient to generate a 10% loading of potassium. This was then dried at 353 K for 2 h and then at 393 K overnight. Finally, both samples were calcined at 873 K for 6 h. After calcination, the samples were ground using a mortar and pestle and sieved to a uniform particle size of 250–425 µm.
Thermo-gravimetric analysis was performed on the catalysts using a combined TGA/DSC SDT Q600 thermal analyser coupled to an ESS evolution mass spectrometer for evolved gas analysis. The samples (~ 10–30 mg) were heated from ambient temperature to 1273 K using a heating ramp of 15 K min−1 in 2% H2/N2 or 2% O2/Ar. Relevant mass fragments were followed by online mass spectrometry.
Elemental analysis was carried out using PerkinElmer analyst 400, atomic absorption spectrometer, with winlab 32. Typically, 0.1 g of catalyst was accurately measured to 0.001 g and digested with aqua regia reagent (1:3 HNO3/HCl). CHN analysis was performed by combustion using a CE-440 elemental analyser.
Colorimetric analysis was conducted to determine the oxidation state of the chromium on the catalyst (dry catalyst, un-calcined, calcined and the reduced catalyst). Cr(VI) reacts with diphenylcarbazide in acid solution to produce a red–violet colour of unknown composition which can be measured at 540 nm. The procedure is nearly specific for Cr(VI). The acid range is critical such that the final acidity must be between 0.7 and 1.3 M H+. The process was calibrated by preparing chromium standards before samples containing no more than 25 μg Cr were pipetted into a 50-ml volumetric flask, diluted to approximately 40 ml and mixed well. To this, 5 ml 0.5 M sulphuric acid and 1 ml diphenylcarbazide solution were added, made up to the mark and mixed well. This was then allowed to stand 5–10 min for colour development and the absorbance measured at 540 nm.
The activity and selectivity of the catalyst were determined using an all-glass, atmospheric, flow, microreactor (Fig. 1). The catalyst (0.5 g) was reduced in situ with pure hydrogen (40 ml min−1) by heating to 823 K and holding at this temperature for 2 h. After reduction was completed, the flow was switched to argon (40 ml min−1) and the system purged for 30 min.

Block schematic of flow microreactor
Pentane (99 +% Aldrich) and 1-hexyne (97 +% Aldrich) were introduced to the argon carrier gas (30 ml min−1) using temperature controlled vaporisers. A molar feed ratio of 5.3:1 for the pentane:1-hexyne system was obtained using a temperature of 273 K for each vaporiser. The gas was then put through a static mixer before continuing to the reactor. The reaction was studied with each component individually and as a mixed feed, in the temperature range 523–773 K at atmospheric pressure. No hydrogen was co-fed in the process. The reactor temperature was controlled using a West controller and the reaction temperature monitored via a thermocouple placed in the catalyst bed. The weight hourly space velocity (WHSV) was 2.8 h−1 for pentane and 0.6 h−1 for 1-hexyne. The reaction products were condensed and collected in heptane at 253 K and then analysed by gas chromatography (Agilent series, FID detector) fitted with a 150-m Petrocol column. An online ESS mass spectrometer was also coupled to the reactor to continuously follow any non-condensed eluted products over the 2-h time period of the reactions. The conversion for each reactant feed was calculated as:
yield as:
Results
The alumina support and both catalysts (chromia/alumina and K-doped chromia/alumina) were analysed by XRD, and only the pattern for alumina was observed (Supplementary information). No chromia species were detected. This was expected as previous studies [16,17,18] have shown that crystalline chromium oxide is not observed at these loadings.
In order to determine the efficacy of the transhydrogenation process, the chromium oxide/alumina and potassium-doped chromium oxide/alumina catalysts were subjected to pentane and 1-hexyne individually and as a molar mixture of pentane:1-hexyne in the ratio (5.3:1) over the temperature range 523–773 K. The reactions were initially performed with the alumina support to evaluate its effect on the various reactants. The results were mainly cracking and alkylation products with ~ 50% and ~ 1% conversion at 623 K for the 1-hexyne and pentane, respectively. There was only a slight difference observed when 1-hexyne and pentane were co-fed over the alumina, with conversion of 1-hexyne still ~ 50%, but the pentane conversion was ~ 2% showing an increase of 1% compared to when it was run alone. The product distribution was very similar to that seen when 1-hexyne was run alone. This behaviour of alumina has been reported elsewhere in the literature [13, 14].
CrOx/Al2O3 catalyst
A standard TGA-TPR from ambient temperature to 1273 K on the chromium oxide/alumina gave a single weight loss/hydrogen uptake centred at ~ 650 K and is presented in Fig. 2.

TGA-TPR of chromium oxide/alumina catalyst
This is in keeping with the literature where reduction was detected at 643 K [19]. The catalyst was analysed by AAS analysis and colorimetric analysis to determine the amounts of Cr3+ and Cr6+ in the catalyst at various stages in the preparation. The results are presented in Table 1. Appreciable amounts of both Cr(III) and Cr(VI) oxidation states were observed at various treatment stages of the catalyst preparation. As expected, the dried catalyst contained a high percentage of Cr(VI) (3.21% c.f. the total loading 3.32%), while the reduced catalyst contained the lowest percentage of Cr(VI) at 0.22%.
These results confirm that the chromium in the catalyst has a mixed oxidation state even after reduction; therefore, the catalysts have been designated CrOx/alumina (and K-CrOx/alumina) to signify that the chromium species are non-stoichiometric oxides. This is in keeping with the literature [16, 18, 19].
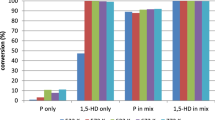
The reactant conversions were followed during the individually fed and co-fed reactions. An increase in the conversion of pentane was observed at all temperatures when pentane and 1-hexyne were co-fed compared to pentane dehydrogenation, while for 1-hexyne the conversion at the higher temperatures (> 573 K) showed a decrease when pentane was co-fed relative to 1-hexyne with no added pentane. The results are presented in Fig. 3.

Conversion comparison of pentane (P), 1-hexyne (1HY) and a 5.3:1 P:1HY mixture over CrOx/Al2O3
The product yields were calculated and are presented in Figs. 4, 5, 6, 7 and 8 (detailed analysis of the products is reported in Supplementary information, Tables S1–S5). As the temperature was decreased, the yield of olefin also decreased; however, the selectivity to olefins, which decreased as temperature decreased from 773 to 623 K, subsequently increased at the lower temperatures (573 K and 523 K).

Yield of products at 773 K with feeds of pentane, hexyne and 5.3:1 pentane:hexyne feed over CrOx/alumina

Yield of products at 673 K with feeds of pentane, hexyne and 5.3:1 pentane:hexyne over CrOx/alumina

Yield of products at 623 K with feeds of pentane, hexyne and 5.3:1 pentane:hexyne over CrOx/alumina

Yield of products at 573 K with feeds of pentane, hexyne and 5.3:1 pentane:hexyne over CrOx/alumina

Yield of products at 523 K with feeds of pentane, hexyne and 5.3:1 pentane:hexyne over CrOx/alumina
Gaseous products were analysed by online mass spectrometry. In all cases, the evolution of hydrogen was observed at the start of the reaction but later declined, and after ~ 40 min no hydrogen was detected. Methane and ethene were observed to co-evolve with hydrogen but did not decline and were maintained to the end of the reaction. This was observed for reaction temperatures between 773 and 623 K, while only pulses of these gases were observed with 573 and 523 K temperatures (shown in Supplementary material, Figs. S1–S3). The results obtained at 773 K are presented in Figs. 9, 10 and 11.

Profile of the evolved gases over CrOx/Al2O3 using pentane only at 773 K

Profile of the evolved gases over CrOx/Al2O3 using 1-hexyne only at 773 K

Profile of the evolved gases over CrOx/Al2O3 using P/1HY at 773 K
After use, the catalysts were analysed to determine whether there had been any carbon laydown. The results are shown in Fig. 12, where it can be seen that the co-feed has a lower carbon deposition relative to 1-hexyne.

Carbon laydown on the spent CrOx/Al2O3 catalysts as a function of temperature
Hydrogenation of 1-hexyne was also performed to determine to compare the catalyst efficacy between hydrogenation and transhydrogenation. The reaction was performed using a limited hydrogen feed (2% H2/N2) in the ratio 2:1 hexyne:2% H2/N2. The results are shown in Fig. 13. Although there are some similarities in the yield of alkynes and alkadienes relative to transhydrogenation, there are significant differences, e.g. the yield of alkanes. Clearly, even in a hydrogen lean environment direct hydrogenation gives a significant alkane yield. This result may suggest that the hydrogen transfer in transhydrogenation does not simply go via an adsorbed hydrogen species.

Comparison of hydrogenation and transhydrogenation CrOx/alumina at 623 K
K-CrOx/Al2O3 catalyst
The catalyst was characterised by XRD and TPR. No XRD reflections associated with chromia were detected. TPR (shown in Supplementary material) gave hydrogen uptakes at 540 K and 760 K, which are consistent with the literature [20].
In general, increased conversions were obtained with pentane over the K-doped chromia catalyst compared to the non-doped catalyst. This was most marked with the pentane/1-hexyne co-feed, where higher conversions were obtained at all reaction temperatures compared to conversions obtained using the chromia catalyst. The results are presented in Fig. 14. The 1-hexyne conversions change very little between single feed and pentane co-feed.

Conversion comparison of P, 1HY and P/1HY mixed feeds over K-CrOx/Al2O3
The product distributions obtained over the doped catalyst had significant differences from those observed over the chromia catalyst. For example, 3-methylpentyne, one of the major products formed over the chromia catalyst, was not observed, and instead, an increased yield of 3-methyl-1-hexene and traces of benzene were observed. The distribution of the products (selectivity) changes with temperature and the % yield observed for individual products was also temperature dependent. The results are presented in Figs. 15, 16, 17, 18 and 19 (full detailed analysis is shown in Supplementary material, Tables S6–S10).

Yield of products at 773 K with feeds of pentane, hexyne and 5.3:1 pentane:hexyne feed over K-CrOx/alumina

Yield of products at 723 K with feeds of pentane, hexyne and 5.3:1 pentane:hexyne feed over K-CrOx/alumina

Yield of products at 623 K with feeds of pentane, hexyne and 5.3:1 pentane:hexyne feed over K-CrOx/alumina

Yield of products at 573 K with feeds of pentane, hexyne and 5.3:1 pentane:hexyne feed over K-CrOx/alumina

Yield of products at 523 K with feeds of pentane, hexyne and 5.3:1 pentane:hexyne feed over K-CrOx/alumina
The evolved gases observed with the doped catalyst are the same (H2, CH4 and C2H4) as observed over the chromia catalyst. However, with the pentane feed (Fig. 20), C2H4 was the major evolved gas. It was initially observed at ~ 10 min of the reaction, but later completely declined by ~ 60 min. Methane followed a similar pattern but with a much lower yield. This is in contrast to the gas evolution from the chromia catalyst. Hydrogen evolution was at a very low level, considerably reduced from that seen over the chromia catalyst. The results from 723 K are presented in Figs. 20, 21 and 22.

Profile of the evolved gases over K-CrOx/Al2O3 using P only at 723 K

Profile of the evolved gases over K-CrOx/Al2O3 using 1HY only at 723 K

Profile of the evolved gases over K-CrOx/Al2O3 using P/1HY at 723 K
After use, the catalysts were analysed to determine whether there had been any carbon laydown. The results are shown in Fig. 23, where it can be seen that overall the extent of carbon deposition is less than that found with the non-doped catalyst. The general trend is also similar in that the co-feed has a lower carbon deposition relative to 1-hexyne except at 523 K where the opposite is true.

Carbon laydown on the spent K-CrOx/Al2O3 catalyst as a function of temperature
Discussion
CrOx/alumina
On the basis of the characterisation results, the CrOx/alumina catalyst after reduction is similar to other chromia catalysts used for dehydrogenation reported in the literature [13, 16,17,18,19, 21]. The amount of Cr(VI) found in the sample is consistent with the literature for similar catalysts [16,17,18,19] as is the absence of any XRD detectable crystalline chromium oxide. At this loading of chromia, a mixture of surface species such as dimers and polymers of various lengths have been shown to be present by Raman spectroscopy [21].
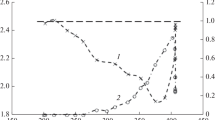
From the conversion data presented in Fig. 3 and the yield data presented in Figs. 4, 5, 6, 7 and 8, it is clear that when pentane and 1-hexyne are co-fed, they each alter the others reaction chemistry. Behaviour such as this has been observed previously with reactions between alkenes and alkynes [22] but not between alkane and alkyne. Examining the results when pentane was fed over the catalyst, the yield of olefins drops rapidly with temperature as expected from thermodynamics. At high temperatures (> 773 K), pentane can dehydrogenate to give pentenes; however, at lower temperatures the equilibrium position is such that only trace levels of olefin can be formed (~ 0.8% conversion at 600 K). Indeed, below 623 K pentane reactivity is very low. There is no such limitation for the hydrogenation of 1-hexyne to hexenes, but there must be a source of hydrogen. When 1-hexyne was passed over the catalyst at 773 K, a significant yield of alkene was observed and this can be associated with molecular breakdown of 1-hexyne, depositing carbon and liberating hydrogen that can be available for hydrogenation of other hexyne molecules. At lower temperatures, the extent of olefin formation from 1-hexyne is much less as carbon deposition decreases and the nature of the deposit will be hydrocarbonaceous, limiting available hydrogen. Therefore, it is easier to detect transhydrogenation at lower temperatures when other routes to olefins are less favoured. It should be remembered that reactions such as alkylation and isomerisation, as well as dehydrogenation and hydrogenation, are also taking place over the catalyst. The effect of transhydrogenation can be seen by examining the selectivity to alkenes (Fig. 24).

Alkene selectivity in C5–C7 products from co-feed of pentane and 1-hexyne
Over the CrOx/alumina catalyst, the alkene selectivity and yield increase between 623 and 773 K as would be expected from thermodynamics (and kinetics). However, the alkene selectivity also increases as the temperature is decreased from 623 to 523 K. Indeed, the yield of alkenes is slightly greater at 573 K than 623 K (Figs. 6, 7). At 573 K, the yield of n-C6 alkenes is over seven times the yield obtained from 1-hexyne alone. This enhancement in alkene yield can be assigned to transhydrogenation between pentane and 1-hexyne, which has no thermodynamic limitations at these temperatures (ΔG = − 47 kJ mol−1). However, as stated above, transhydrogenation is not the only reaction taking place. Although the total alkene yield is observed to have generally increased during the transhydrogenation process, most of the alkenes are isomers of the primary target products as well as alkylated alkenes. Indeed, many of the alkylated products are alkenes, which suggest that in essence, rapid isomerisation and alkylation are occurring coincidentally with the transhydrogenation process, e.g. 3-methylpentyne and 1,4-hexadiene (both isomers of 1-hexyne) are common significant products from both the 1-hexyne feed and pentane/1-hexyne co-feed (Supplementary information). It is also noticeable that there is very little alkane formation.
Gas phase products are also produced with all three reaction feeds: pentane, 1-hexyne and pentane/1-hexyne mix (Figs. 9, 10, 11). Only hydrogen, methane and ethene were detected no C3 or C4 gases were observed. These three gas phase products are typical of cracking and hydrogenolysis. It is noticeable that the hydrogen evolution is not maintained, suggesting that the sites responsible for this reaction are deactivated during the course of the reaction. The results presented in Fig. 12 confirm that there is carbon deposition, which may result in the loss of activity. It is noticeable that the hydrogen evolution maximises at different times when 1-hexyne is the feed and when there is a pentane/1-hexyne co-feed. The co-feed evolution maximises ~ 10 min, whereas the 1-hexyne feed gives a maximum hydrogen evolution at ~ 20 min. The pentane feed system shows a broad evolution of hydrogen that would be consistent with an evolution at ~ 10 min and ~ 20 min. This suggests that there may be two sites with 1-hexyne only able to access one, whereas pentane may access both. This would invoke competitive adsorption, which would be a route by which pentane may influence 1-hexyne surface chemistry.
To better understand the transhydrogenation process, a limited hydrogen hydrogenation of 1-hexyne was performed over the CrOx/alumina catalyst (Fig. 13) and it is clear that hydrogenation and transhydrogenation are not similar processes. Hydrogenation, even with a limited hydrogen feed, produces a significant yield of n-alkanes. This catalyst is not optimised for alkene selectivity from hydrogenation and so produces the thermodynamically favoured alkane. In contrast, virtually no n-alkane is produced from the transhydrogenation reaction, and even the yield of iso-alkane is lowest during transhydrogenation, suggesting that the hydrogen transfer process is much more controlled than in hydrogenation. This has mechanistic implications for our understanding of transhydrogenation. The hydrogenation reaction reveals that if atomic hydrogen is present on the surface, then alkane is the favoured product. For the transhydrogenation reaction to favour alkenes, it must control the hydrogen supply or there are two types of hydrogen on the surface. It has been shown using isotopic labelling that different hydrogen populations do exist on the surface [13]. Therefore, the difference between transhydrogenation and hydrogenation may relate to 1-hexyne sampling different hydrogen populations. The other possibility is that as pentane dehydrogenation only releases two hydrogen atoms, this only allows 1-hexyne to hydrogenate to 1-hexene. Therefore, if the rate of alkene isomerisation and desorption is faster than that of pentane dehydrogenation, only alkenes will be observed as there is never free hydrogen for further hydrogenation.
K-CrOx/alumina
The addition of potassium to catalysts is a standard method to change activity, change selectivity and reduce by-product formation [23]. Detailed spectroscopic and mechanistic analysis of CrOx/alumina has shown that the addition of potassium does not affect alkane dehydrogenation but significantly affects side reactions [21]. Therefore, the addition of potassium has a noticeable effect on product selectivity. For example, when pentane and 1-hexyne are co-fed at 623 K, the yield of alkenes was 30%, whereas over the CrOx/alumina catalyst under the same conditions the alkene yield was only 9%. Most strikingly, no alkynes are formed over the K-CrOx/alumina, specifically 3-methylpentyne, which was a major product via structural isomerisation over CrOx/alumina. Much of this change can be understood as a neutralisation of the acid sites on the alumina; indeed, it has been shown in a number of reports that the role of potassium is to remove acid sites [20, 21, 24]. Nevertheless, there is still considerable alkylation with significant yields of iso-alkanes (including methylcyclohexane) although this could be base catalysed [25, 26]. Alkylated alkenes such as 3-methyl-2-pentene and 3-methyl-1-hexene were also major products. Figure 24 shows the alkene selectivity as a function of temperature, and it can be seen that the alkene selectivity does not significantly change with temperature over the K-CrOx/alumina in contrast to the CrOx/alumina catalyst. This lack of variation in alkene selectivity across the temperature range would be consistent with transhydrogenation and is consistent with the detailed product analysis (Supplementary information), where hexene isomers are specifically enhanced. It is also clear that the K-CrOx/alumina catalyst gives an enhanced yield of alkenes compared to CrOx/alumina. A possible explanation for this difference may be related to the fact that addition of potassium to CrOx/alumina is known to change the nature of the surface chromia species [20]. In CrOx/alumina, at least five surface chromia species have been identified [16]; when potassium is added to a CrOx/alumina, the surface chromia species alter tending to form isolated and oligomeric chromia [20]. It is possible that these species facilitate transhydrogenation giving an increased alkene yield.
As with the CrOx/alumina catalyst, light gases were evolved from the reaction of the three feeds over K-CrOx/alumina (Figs. 20, 21, 22). However, the manner of their evolution is markedly different. For all three feeds, methane and ethene are evolved as pulses, typically over 0.3 h, whereas hydrogen is evolved over most of the time on stream. However, when the gas evolutions are examined in more detail, further differences between the two catalysts are found. For all the feeds, the major gas produced is ethene. This is in contrast to the gas evolution over the CrOx/alumina, where hydrogen and ethene were always the main gaseous products. The short pulse of the hydrocarbon gases produced over the K-CrOx/alumina suggests that a small number of sites catalysing cracking reactions such as
were rapidly deactivated. The extent of carbon deposition over the K-CrOx/alumina catalyst is noticeably less than over the CrOx/alumina (Fig. 23). This is in keeping with the literature [12, 18, 19] where potassium doping reduces the extent of carbon deposition. The pentane/1-hexyne co-feed shows lower carbon laydown than expected from a sum of each species individually, except for the reaction at 523 K where the extent of carbon laydown is similar to the sum of the individual feeds. Interestingly, if the effect of pentane on 1-hexyne surface chemistry was due to competitive adsorption, it may be expected that at lower temperatures where the adsorption of pentane is limited, the effect would be less and this would explain why at 523 K the carbon deposition is purely the sum of the pentane deposition and 1-hexyne deposition.
Conclusions
The transhydrogenation of pentane and 1-hexyne was investigated over CrOx/Al2O3 and K-CrOx/Al2O3 catalysts. Over the CrOx/Al2O3 catalyst, transhydrogenation clearly occurred at temperatures below 625 K, where the yield of alkenes was higher for the co-fed system than for a combination of the individual yields. Due to the acidic nature of the alumina, many of the products were alkylated olefins and alkylated hydrocarbons due to coincident alkylation and isomerisation. Pentane was shown to affect the chemistry of 1-hexyne over and above the transhydrogenation reaction, e.g. the extent of carbon deposition was reduced. By comparing transhydrogenation to limited hydrogen 1-hexyne hydrogenation at 623 K, it was shown that the processes of hydrogenation and transhydrogenation were different, with hydrogenation favouring alkanes, while transhydrogenation favoured alkenes. It was suggested that this may be due to different hydrogen pools that influence the final product or that as pentane dehydrogenation only releases two hydrogen atoms, this only allows 1-hexyne to hydrogenate to 1-hexene. Therefore, if the rate of alkene isomerisation and desorption is faster than that of pentane dehydrogenation, only alkenes will be observed. The latter proposal would suggest that the dehydrogenation/hydrogenation process is closely coupled and would be consistent with pentane influencing 1-hexyne surface chemistry. The effect of potassium doping was to increase the yield of alkenes. The reason for this may be related to changes in the nature of the surface chromia species suggesting site specificity for transhydrogenation. The potassium also neutralised the acid sites on the alumina, reducing the extent of alkylation and hydrogenolysis, which suppressed the formation of other alkynes in the product mix.
References
Bhasin MM, McCain JH, Vora BV, Imai T, Pujadó PR (2001) Dehydrogenation and oxydehydrogenation of paraffins to olefins. Appl Catal A 221:397–419
Garba MD, Galadima A (2015) Efficiencies of green chemistry metrics in the activities of petroleum refinery process. Int Sci Invest J 4:65–87
Wiess AH (1970) The manufacture of propylene. In: Spillane LJ, Leftin HP (eds) Refining petroleum for chemicals. ACS, Washington, pp 153–178
Craig RG, Spence DC (2007) Catalytic dehydrogenation of liquefied petroleum gas by the houdry catofin and catadiene processes. In: Meyers RA (ed) Handbook of petroleum refining processes. McGraw-Hill Education, New York
Bipin VV, Pujado PR (2005) Catalytic dehydrogenation. In: Lee S (ed) Encyclopedia of chemical processing. CRC Press, Boca Raton, pp 379–395
Dessau RM, Partridge RD (1995) Production of Olefins by Transhydrogenation. WIPO Patent. Mobil Oil Corporation, Fairfax, VA
Andreas SBA, Neil DE, Thomas E, Damon S (2009) Transhydrogenation processes. WIPO Patent. SI Group, inc. (US/US), Schenectady, NY and five others
Gough A, Turner SK, Mercer J, Stitt, EH (1994) Process for cracking and transhydrogenation of hydrocarbon feedstock. WIPO Patent. Imperial Chemical Industries PLC, London (GB)
Arthur RJL, Aldridge C, Seine M (1966) Process for forming olefins by hydrogen transfer. US Patent. Exxon Research Engineering Co
Parris DA, Nunawading U (1985) Process for the manufacture of methyl t-butyl ether US Patent. Imperial Chemical Industries Australia Limited, Melbourne
Turner SK, Gough A (1997) Process for producing a branched chain olefin by isomerization and transhydrogenation. US Patent. Institut Francais Du Petrole (Rueil-Malmaison Cedex, FR)
Jackson SD, Matheson IM, Webb G (2003) Carbon deposition during transhydrogenation over chromia catalysts. Am Chem Soc Div Pet Chem 49:50–53
Jackson SD, Matheson IM, Webb G (2005) An isotopic study of the transhydrogenation of propyne with propane over a potassium-doped chromia/alumina catalyst. Appl Catal A 289:16–21
Wigzell F, Rugmini S, Jackson SD (2015) Transhydrogenation of propyne with butane over a vanadia/θ-alumina catalyst. Appl Petrochem Res 7:9–21
Garba MD, Jackson SD (2016) Catalytic upgrading of refinery cracked products by transhydrogenation: a review. Appl Petrochem Res 7:1–8
Fridman VZ, Xing R, Severance M (2016) Investigating the CrOx/Al2O3 dehydrogenation catalyst model: I. identification and stability evaluation of the Cr species on the fresh and equilibrated catalysts. Appl Catal A 523:39–53
De Rossi S, Caselleto MP, Ferraris G, Cimino A, Minelli G (1998) Chromia/zirconia catalysts with Cr content exceeding a monolayer. Appl Catal A 167:257–270
Jackson SD, Stitt EH (2003) Propane dehydrogenation over chromia catalysts: micro-catalysis, deactivation, macro-kinetics, and reactor modelling. Curr Top Catal 3:245–265
Edussuriya M, Jackson SD, Rugmini S (2010) Deactivation and regeneration of chromia and vanadia catalysts in alkane dehydrogenation. Curr Top Catal 9:15–24
Rombi E, Cutrufello MG, Solinas V, De Rossi S, Ferraris G, Pistone A (2003) Effects of potassium addition on the acidity and reducibility of chromia/alumina dehydrogenation catalysts. Appl Catal A 251:255–266
Jackson SD, Matheson IM, Naeye ML, Stair PC, Sullivan VS, Watson SR, Webb G (2000) Production of alkenes over chromia catalysts: effect of potassium on reaction sites and mechanism. In: Corma A, Fierro JLG, Melo FV, Mendioroz S (eds) Studies in surface science and catalysis. Elsevier, Amsterdam, pp 2213–2218
Canning AS, Jackson SD, Monaghan A, Wright T (2006) C-5 alkene hydrogenation: effect of competitive reactions on activity and selectivity. Catal Today 116:22–29
Moss WD (1983) Alkali doping in heterogeneous catalysis. Catal Rev 25:591–637
Liu D, Bai P, Wu P, Han D, Chai Y, Yan Z (2015) Surface chemistry and catalytic performance of chromia/alumina catalysts derived from different potassium impregnation sequences. Appl Surf Sci 351:250–259
Closson RD, Napolitano JP, Ecke GG, Kolka AJ (1957) Base-catalyzed alkylation with olefins. J Org Chem 22:646–649
Qafisheh N, Mukhopadhyay S, Joshi AV, Sasson Y, Chuah G-K, Jaenicke S (2007) Potassium phosphate as a high-performance solid base in phase-transfer-catalyzed alkylation reactions. Ind Eng Chem Res 46:3016–3023
Acknowledgements
The authors would like to fully acknowledge the funding provided, for one of us (MDG), by the Petroleum Technology Development Funds (PTDF) through the Government of Nigeria and the University of Glasgow for the placement.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Garba, M.D., Jackson, S.D. Transhydrogenation of pentane and 1-hexyne over CrOx/Al2O3 and potassium-doped CrOx/Al2O3 catalysts. Appl Petrochem Res 9, 113–125 (2019). https://doi.org/10.1007/s13203-019-0231-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-019-0231-3