
Abstract
Investigations on the phase behaviors and structural properties of mono-, di- and poly-valent metal carboxylates are reviewed with reference to developments in experimental and theoretical concepts surrounding their liquid crystalline properties. The main methods of structural investigation such as X-ray diffraction, infrared and 13C-NMR spectroscopies are examined in detail on the basis of common synthetic routes leading to the isolation of pure compounds. A detailed review of the thermal behaviors of several metal carboxylates is presented along with proposed theories and molecular models for odd–even alternation, chain length effects, phase structures and mesophase formation. Theories explaining the effects of metal ion radii and chain unsaturation are also discussed. Proposed degradation mechanisms resulting in the formation of various products and kinetic studies are also considered. Though this review highlights a number of investigations on the structural and phase properties of the mostly widely studied carboxylates, the results presented here strongly indicate that there is room for further studies on some of these systems.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Overview: metal carboxylates as metallomesogens
Metal carboxylates (soaps) are of different types based on the metal ion radius, hardness, softness, valency and alkyl chain structure. As a result, they exhibit diverse physicochemical properties; hence, they find extensive commercial applications in protective coating agents, paints, ink driers, polymer stabilizers, catalysts, waterproofing agents, lubricants, fuel additives and fungicides [1, 2]. Some of these compounds are also used in photothermogravimetry [3], as low angle calibrants for neutron diffraction instruments [4, 5] and in the manufacturing of pharmaceuticals [6–9]. However, the most exciting aspect of metal carboxylate chemistry stems from their ability to undergo mesomorphic phase transitions (metal containing liquid crystal) of various textures. In fact, from a historical perspective, metal carboxylates were the first metal containing liquid crystal compounds (metallomesogens), reported in 1855 by Heintz, for magnesium(II) tetradecanoate [10]. Similar observations were reported by Vörlander [11] in 1911 for several alkali metal carboxylates. Subsequently, a number of other mesomorphic metal carboxylates were successfully prepared and shown to exhibit thermotropic lamellar and columnar mesophases in their phase sequences [10, 12, 13]. Furthermore, some metal carboxylates formed smectic type mesophases which are of considerable commercial importance since they are often applied in liquid crystal display devices (LCDs) where they offer considerable advantages over conventional nematic and twisted nematic display devices; that is, they off shorter (faster) switch times, larger field of view and higher complexity. They are also more environmentally friendly, cheap and easier to synthesize relative to currently used liquid crystal compounds [14–16]. Hence, despite the advent of other forms of display mechanisms, such as light emitting diodes (LEDs), the technological relevance of liquid crystals remains significant [16]. However, the applicability of metal carboxylates in LCDs is limited since they form thermotropic liquid crystals (liquid crystals formed above ambient temperature). Therefore, considerable investigations have been carried out on their phase properties, resulting in several proposals [10–13] for the factors governing their phase transition behaviors. These studies also revealed that several metal carboxylates formed ionic melts which find application in ionic solvents and for the manufacture of portable energy storage devices such as lithium ion batteries which makes use of lithium salts [14–16]. The magnetic properties of cobalt(II) and manganese(II) n-alkanoates have been the center of investigations by Kambe and coworkers [17]. For instance, manganese(II) stearate was found to exhibit two-dimensional (2D) magnetic properties which was applicable in magnetic switches, hence, the possibility exists for their use in data storage devices. Additionally, metal carboxylates are generally insoluble in water but are usually soluble in various organic solvents in which they exhibit lyotropic mesomorphism, where the soap molecules exist as small aggregates of various shapes, classified as micelles [6, 18].
Theories explaining the structural and phase properties of these compounds are presently the center of a myriad of investigations. For example, a detailed theoretical study was carried out by the cancer research institute of Philadelphia on the three-dimensional (3D) binding of carboxylate moieties to different metals [19]. The relevance of the study stemmed from the fact that such groups were abundant in the active sites of several biological enzyme systems as part of glutamine and aspartate side chains that are active metal binders. However, despite the many uses of these compounds, most of the basic theoretical aspects of their physical and chemical properties remained largely underdeveloped up until the last two decades, where the majority of investigations were more meticulously carried out and focused on zinc(II), lead(II), copper(II), lithium(I), sodium(I), potassium(I), silver(I) and lanthanide(III) carboxylates. Such studies revealed a step-wise melting mechanism for these compounds, as indicated by the presence of several pre-melting transitions in their phase sequences [20, 31]. Unfortunately, though highly detailed thermodynamic explanations were proposed for some compounds, the majority of these studies resulted in no molecular and lattice structure–phase behavior correlation models. However, for lead(II) [21–27], copper(II) [28, 29], zinc(II) [30–32] and silver(I) [33–36] carboxylates where both crystal–crystal and mesomorphic transitions were observed in their phase sequences, a number of thermodynamic phase models were proposed. Several studies have also been carried on the thermal degradation of lead(II) [37, 38] and copper(II) [39, 40] carboxylates where their decomposition mechanisms, activation parameters and significant kinetic factors adduced. Theoretical investigations on the phase behavior of metal carboxylates have also been carried out by a number of authors via molecular modeling calculations such a density functional theory (DFT) [41, 42]. Considering the commercial importance of metal carboxylates, understanding their molecular structures and phase behaviors should be of considerable interest. Therefore, in this review attempts will be made to give a general overview of investigations on the structures and mesomorphic behaviors of the more widely studied metal carboxylates and the contributions of such reports to the current body of knowledge on the phase properties of metallomesogens.
Approach to synthesis
The synthesis of pure metal carboxylates presents a real but not insurmountable challenge. This involves simple reactions in which the acidic hydrogen of the alkanoic acid is replaced by a metal [20]. Such changes can be accomplished in several ways: the action of base (hydroxide or oxide) on fatty acids; double decomposition of the soluble salt of a fatty acid and the salt of a mineral acid or by saponification of glycerides or other esters of fatty acids with a base or metallic oxide [20]. These reactions hold true generally for mono- and di-valent soaps such as potassium, sodium, lithium, and lead carboxylates among others. On the other hand, preparation of trivalent and higher valence soaps appears to be particularly difficult. For example, there have been many attempts to utilize conventional methods, but these did not result in the formation of pure anhydrous compounds [20]. In many cases, soap hydrates as well as basic [M(OH) x (RO2)], and or acidic [(RCO2) n M(RO2H)], compounds were obtained. For example, studies involving the synthesis of aluminium(III) carboxylates resulted in the formation of both acidic and basic products [43–45]. After many attempts to produce the anhydrous tri-soap, it was demonstrated that it could be prepared only under anhydrous conditions [20, 46, 47]. Similarly, for chromium(III) stearate, the normal products have been shown to be a mixture of basic soaps and excess of acid [20, 48]. Various studies showed that basic chromium(III) carboxylates were easily prepared if hydroxyl species or water were present [49–51]. However, it has been shown that tri-soaps of chromium(III) could be prepared, with difficulty, only under anhydrous conditions [52]. In contrast, the hydrated trivalent lanthanide(III) soaps were readily prepared even in aqueous solution [20, 53, 54].
In light of these and other similar observations, the general methods for preparation of the carboxylates of the poly-valent metal ions include [20]: precipitation from aqueous solutions of sodium or potassium carboxylates by the addition of solutions of appropriate salts of the poly-valent metal; ligand exchange between the metal chloride or acetate and the long chain fatty acid in an appropriate anhydrous solvent, fusion of metal oxides, hydroxides or carbonates with the fatty acids and direct reaction of the metal and the fatty acid. The precipitation route is particularly simple, and whilst earliest studies involved, in general, reactions in aqueous solutions, it is usually more prudent to use alcohols or alcohol–water mixtures as solvents. The use of aprotic solvents, however, seemed to be more appropriate in these precipitation reactions as they generally produced anhydrous compounds. Upon precipitation, soaps were purified by recrystallization from organic solvents, such as ethanol, benzene or toluene or from neat fatty acids.
Molecular and lattice structure of metal carboxylates
Overview
As indicated before, the predominant liquid crystal phase exhibited by most metal carboxylates is the smectic phase, particularly types A and C; SmA and SmC [20]. This is because metal carboxylates possess layered structures in the solid state which is maintained on heating. The smectic phase is an example of a calamitic liquid crystal phase where in its structure the molecules possess more order than in the nematic (N) phase and are also arranged in layers which are preferentially aligned, on average, in a particular direction (called the director, n) [55]. Of the various types of smectic phases, two are more predominant, especially for metal carboxylates: the SmA, where the director is parallel to the layers’ normal and the molecules are arranged such that the average tilt angle is zero; the SmC, where the director and the molecules are tilted at an angle, θ, to the normal (Fig. 1) [56]. There are also the SmB, SmD, SmE, SmF and SmG phases. In each case, there is considerable fluidity and easy diffusion between the layers is possible though their viscosities are much higher than nematic phases [55, 57]. In general, these phases, arise from rod-like shaped molecules in which the axial part of the molecule is larger than the radial part.

Prevalent molecular arrangements of calamatic phases [58]
The rod-like molecular shape of some metal carboxylates can be said to derive from the basic structure of carboxylic acids, where they exist as dimers through interaction between hydrogen bonding which allows for the formation of a small core relative to the size of the peripheral part of hydrocarbon chains. This type of structure allows for a lattice in which the molecules have lateral intermolecular van der Waals interactions, responsible for stabilizing the layered phase structure, leading to a smectic phase [58].
Incorporation of a metal, interacting through the carboxylate groups, essentially maintains the rod-like structure with a small core and flexible peripheral parts leading to layered packing. The type of packing structure is dependent on (1) the nature of the metal and associated coordination geometry; (2) the type of carboxylate bonding mode and (3) the type of substituents on the carboxylate, viz. hydrocarbon chain length, branched hydrocarbon chain, etc. [18, 31, 58]. Invariably, the elongated molecules packed in a layered lattice must possess two characteristics: (1) positional order and (2) orientational order, which are responsible for the formation of a particular type of mesophase [55, 56]. Positional order is controlled by the basic molecular structure and shape, particularly from the size of the core relative to the molecular length and orientational order, which is strongly dependent on the extent of chain tilting. Certainly, changing the chain length is an important approach to inducing mesomorphism and it is widely understood that the longer the peripheral hydrocarbon chain length relative to the core, the more likely will a smectic phase be exhibited [20]. Not only do the metal ions offer rigidity at the center, which is important for positional order, they also maintain molecular polarizability along the molecular axis, giving rise to important intermolecular interactions responsible for mesophase formation [58].
Metal–carboxylate coordination and solid-state lattice structure
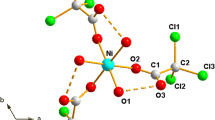
As can be inferred from the foregoing, the structural chemistry of metal carboxylates is quite fascinating because carboxyl groups can coordinate in different ways to the metal ion resulting in the formation of mono-, di- and polymeric forms of these compounds [59]. It has been shown in numerous reports that metal carboxylate coordination was central to their molecular packing and lattice structures. Therefore, various techniques have been employed to elucidate the structures of the room temperature solids [10, 20], though, for many long chain adducts few reports have been published on their molecular and lattice arrangement in the crystal system. This challenge arises because many compounds do not produce crystals of suitable morphology for single crystal X-ray analysis [12, 20, 60–62], that is, the crystals are usually twined or exist as thin needles that are very fragile and do not diffract well. This is due to their low molecular symmetry and flexibility of the polymethylene chains. Nevertheless, single crystal and powder X-ray diffraction (PXRD) studies in conjunction with infrared (IR), Raman, high-resolution solid-state nuclear magnetic resonance and UV/Vis absorption spectroscopies as well as but to a lesser extent, magnetic susceptibility and photoluminescence studies have allowed some advances in molecular structure determination [10, 18, 61, 63]. Notwithstanding the challenges in structural elucidation, a very useful approach that can allow for fairly precise postulations about the packing structure involves the use of composite data from PXRD, IR and molecular calculations. In recent times this approach, used by several authors [1–7, 20], has proven to be quite reliable since structures of metal carboxylates have fairly typical IR spectra (Fig. 2) and X-ray diffraction patterns arising from the nature of the carboxylate coordination, layered packing and chain conformation.

Typical infrared spectra for metal carboxylates, exemplified by zinc carboxylates [31]
Infrared spectroscopy
The use of vibrational spectroscopy in the study metal–carboxylate chemistry provides invaluable insights where metal–metal interactions, chain conformation and packing are concerned; hence, it is extensively used by a number of authors on various metal carboxylate systems [18, 45, 60, 64]. Typical IR spectra for metal carboxylates, exemplified by zinc(II) hexanoate and pentadecanoate (Fig. 2), were characterized by strong absorptions in the region of ca. 1,700–1,200 cm−1 and weak to medium bands between ca. 1,400 and 500 cm−1 [31, 46, 60]. The absence of hydroxyl absorption bands in the region of 3,500–3,300 cm−1 confirmed that the compounds were anhydrous as well as absence of a carbonyl absorption band in the region of 1,730 cm−1 and its replacement by absorptions in the region of 1,500, 1,400, 950, 580 and 540 cm−1 indicates that there was complete resonance in the C–O bonds of the carbonyl group as a result of coordination with the metal [18, 31].
The C–O bands were assigned as: asymmetric stretch, νas(COO) ~1,400–1,530 cm−1; symmetric stretch, νs(COO) ~1,410–1,390 cm−1; deformation, νd(C–C)COO ~940–960 cm−1; bending, ν δ (COO) ~745 cm−1; out of plane twisting, ν τ (COO) ~580 cm−1 and rocking, ν ρ (COO) ~550 cm−1. Splitting of C–O bands correlated with a lowering of COO symmetry and the relative strengths of the metal–carboxylate bond due to head group and side chain lateral van der Waals interactions [20, 31, 60]. For a homologous series, near invariance of the ρ(COO) absorption at around 550 cm−1 gives evidence of isostructures. Additionally, for triclinic or hexagonal chain packing, a single band for ρ(CH2), at around 720 cm−1, is expected, however, splitting of this band is indicative of orthorhombic or monoclinic packing [11, 65]. The nature of chain packing can be inferred from ρ(CH2) and δ(CH2) absorption bands with splitting indicating horizontal packing and if not, parallel packing is assumed [11]. Further, the progression of regularly spaced absorptions, in the region of 1,347–1,232 cm−1, arising from the wagging vibrations of methylene groups, are typical for aliphatic chains and indicate all-trans conformation of hydrocarbon chains [65].
In order to understand the importance of IR spectroscopy in obtaining structural information, it is useful to explain the points outlined in the foregoing with respect to coordination. To that end, Nakamoto and co-workers [66] stated that, site group or factor analysis, based on knowledge of the crystal structure, is desirable when taking into consideration the symmetry of the ligand, from the spectra obtained, in the crystalline state. For the most part, it was anticipated that the effects of coordination on ligand absorption were much stronger than the effects of the overall crystal field, hence, rigorous site group analysis may be ignored. Furthermore, by coordination, all the fundamentals (bands) were more or less shifted according to their modes of vibration. As the metal–ligand bonds become stronger, shifts to lower wavenumbers become greater. When coordination lowers the symmetry of the ligand, forbidden vibrations of the free ions are permitted, and degenerate vibrations became energetically distinct [64, 66]. Therefore, vibrational energy shifts, the magnitude of splitting and the intensity of the newly permitted bands are useful indicators of the effect of coordination. In addition, it is generally expected that emerging bands of the metal complex as well as those of the free ion, increase in intensity on formation of the metal complex. Such changes are affected by the type of metal carboxylate coordination mode(s), of which there are various possibilities (Fig. 3): chelating-type bidentate; Z, Z-type bridging bidentate; Z, E-type bridging bidentate; E, E-type bridging bidentate and bridging tridentate [66, 67].

Evaluating the mode of coordination in metal carboxylate systems involves use of the magnitude of separation, ∆ν cm−1, between νa(COO) (carboxyl asymmetric) and νs(COO−) (carboxyl symmetric) stretching vibration bands [60, 63, 64, 66]. However, according to Edwards and co-workers [68] the use of ∆ν can be misleading, especially in cases of variable coordination modes, in some carboxylates, which make it difficult to assign, unambiguously, the correct coordination mode/s. Nevertheless, IR studies on a series of metal acetates, showed that for chelating bidentate coordination ∆ν was in the vicinity of 100 cm−1 or less but for bridging bidentate bonding the value was ca. 150 cm−1 [20, 60, 61, 68]. Additionally, studies on some metal haloacetates indicated that [20] a higher value of νas(COO−) relative to the values for the ionic soaps (Na+ and K+) was indicative of bridging coordination whilst lower values confirmed chelating coordination [60, 64]. Since monodentate coordination removes the equivalence of the C–O bonds resulting in a higher value for νas(COO−) and a lower value for νs(COO−), ∆νmon will be much greater than ∆νion (ca. 138 cm−1) [60]. Moreover, when COO− is unidentate, one of the C–O bonds should have enhanced double-bond character and should give rise to a high-wavenumber band [69] in the 1,590–1,650 cm−1 region of the spectrum. This approach has been utilized by many authors [31, 57, 61] and has resulted in fairly accurate descriptions of the metal–carboxylate coordinations for several compounds.
Clearly, for these compounds, molecular packing was a function of the type of carboxylate coordination; hence, various packing arrangements such as monolayers, bilayers, interdigitated bilayers were observed [20]. Other effects such as alternating orientation of the metal and hydrocarbon planes were observed. Typically, the hydrocarbon chains in the lamellar are packed in the fully extended all-trans conformation. This was proven by a number of authors [20, 27, 31] on the basis of IR data as outlined in the forgoing.
Structural information from powder X-ray diffraction
Typical room temperature X-ray powder diffraction patterns, for metal carboxylates (Fig. 4) are characterized by evenly spaced reflections from successive d planes in the order of: 1:1/2:1/3:…1/n characteristic of a lamellar structure [30, 31, 61–64]. Generally, the shorter chain compounds have evenly spaced reflections at low angles (2θ) but for the longer chain adducts a progression of evenly spaced low-intensity reflections are usually observed at, 20° ≤ 2θ ≤ 25° which becomes irregularly spaced at higher angles due to side chain packing interactions of the methylene hydrogen [61–63]. Indexation and refinement of the diffraction patterns are usually carried out to determine the unit cell parameters and hkl miller indices (typically, 00l, 0k0, h00) from which the d values were obtained [20, 31]. Such parameters were useful in presenting a picture of the molecular and lattice packing of these compounds. Further calculations carried out by several authors [18, 27, 31] have shown that comparison of the d-spacing, dexp, which represents the distance between successive metal planes, and the calculated molecular length, dL [dL = dC–H + (n − 1)dC–C sin 55° + dC–O + r x+M ], was indicative of whether the compounds were packed in a monolayer, bilayer or interdigitated bilayer arrangement within the lamellar.

Powder X-ray diffractograms of silver(I) carboxylates [70]
Monovalent alkanoates
The phase behaviors of monovalent metal alkanoates have been the subject of several investigations for a number of decades, especially those of lithium, sodium, potassium, thallium and silver. In the case of lithium carboxylates, some of the most comprehensive studies have been carried out by Shoeb et al. [71], Ferloni and Westrum [72], and Martinez Casado et al. [27], prior to which only the crystal structure of some hydrated lithium alkanoates (lithium formate monohydrate and lithium acetate dihydrate) have been determined [27].
Sodium(I) and potassium(I) carboxylates
Despite being extensively used as surfactants in detergents, the solid-state structures of sodium and potassium carboxylates have not been investigated to any appreciable extent. This is possibly due to their inability to form crystals of suitable morphology for X-ray single crystal diffraction, especially for the long chain homologues. Nonetheless, according to Kovalenko et al. [42], molecular modeling calculations, based on the isolated propanoate ion, were in agreement with experimental data. These results showed that the most probable coordination mode, for sodium carboxylates, was via chelating bidentate or monodentate bonding [42]. However, no molecular models were presented for these compounds, hence, very little is known about their three-dimensional lattice structures, intermolecular interactions and exact metal–carboxylate bonding. Nonetheless, fair amount of infrared analysis was carried out on these compounds, which gave a clear picture of the structure of the hydrocarbon chains. For example, Bui [73] in his studies of some short chain sodium n-alkanoates, with nc = 1–12, inclusive, showed that the intensities of the methyl and methylene group symmetric and asymmetric vibration bands were related to the number of methylene groups in the alkyl chain in agreement with previous reports on other metal carboxylate systems [30, 60–64]. In this study, it was suggested that the longer chain sodium compounds had greater ionic character relative to the shorter chain adducts. This suggestion is quite contradictory since it is well established in the literature [15–26] that shorter chain compounds were of higher melting points, indicating greater ionic charter. Furthermore, since ∆ν = 217–227 cm−1 [73] the clear inference was for ionic metal–carboxylate coordination, irrespective of chain length and not covalent as suggested. This assertion was supported by the presence of a UV absorption in the region of 186–187 nm corresponding the π–π* transition of C=O.
For potassium carboxylates, no accurate crystal structures have been reported that shows clearly, the type of metal–ligand bonding and their lattice packing. According to Lewis and Lomer [74] and Lomer [75] these compounds showed extensive chain length-dependent polymorphism; that is, compounds with 4–10 carbon atoms, inclusive, when crystallized at ambient temperature from alcohol formed a distinct phase, classified as A whilst those with nc = 12–18, inclusive, formed phase B. Furthermore, with the exception of the butanoate and hexanoate, all compounds transitioned to a third phase; C, on heating [75]. X-ray data, in agreement with data from Piper [41], showed that the compounds crystallized in a monoclinic crystal system with P21/a space group for the A polymorph; however, for polymorph B, the compounds were triclinic with P1 symmetry [75]. Though the structure for potassium decanoate A has been investigated by Lomer [75], no three-dimensional molecular models were proposed to explain its’ molecular and lattice packing. Nonetheless, these results showed that the molecules were tilted at ca. 57.9o with respect to the metal basal plane within 1.123 g cm−3 unit cells. Interestingly, UV absorption measurement carried out by Bui [73] showed two main transitions associated with the strong π–π* and low probability n–π* transitions of the C=O group. These transitions, observed in the range of 186–189 nm, were used to support the IR data in proposing that metal–carboxylate bonding was either via ionic or bridging coordination, though no detailed explanations were presented for such conclusions.
Lithium(I) carboxylates
Shoeb et al. [71] in their studies on a series of anhydrous lithium soaps (nc = LiC8…18) suggested, on the basis of infrared data, that the compounds were isostructural with ionic lithium–carboxylate interactions. However, the ionic character of the metal–carboxylate bonding increases with decreasing chain length as indicated by the observed decrease in melting point with increased chain length. Furthermore, PXRD data indicated that there was a change in the tilt angle of the hydrocarbon chains relative to the lithium basal planes with chain length [71]. However, this study was limited since there was no description of the metal carboxylate coordination, type of molecular packing and/or the effect of chain length on their lattice structures. Recently, more extensive structural studies were carried out by White et al. [76] and it was concluded, based on room temperature PXRD and IR data, that the ionic molecules were packed as lamellar bilayer structures in a triclinic crystal system composed of fully extended all-trans hydrocarbon chains. Interestingly, single crystal structures reported by Casado et al. [77] for lithium(I) propanoate and pentanoate showed carboxylate groups bound in a chelating bidentate mode to tetrahedral lithium(I) ions. These structures showed that the tilted hydrocarbon chains were fully extended and arranged as non-overlapping lamellar bilayer structures. Unit cell parameters presented by White and Ellis [113] for some long chain lithium(I) carboxylates, adduced from PXRD data, showed that the layers within the triclinic unit cells were chain length dependent. However, according to Casado et al. [77], the tilt angle of the alkyl chains, of LiC2 and LiC3, relative to the ionic layer was, on average, 60. The crystal structures (Fig. 5) showed that metal carboxylate bonding in the basal was such that a two-dimensional lithium polymeric structure was formed (coordination polymer). Frequency differences between asymmetric and symmetric COO stretching (∆ν = νas − νs), ∆ν ≈ 125 cm−1 vibrations confirmed bridging carboxylate coordination as indicated by PXRD. However, since ∆ν ~144 cm−1 for the longer chain adducts, as reported by White and Ellis [113], the clear inference was for change in head group coordination with chain length.

Single crystal structures for short chain lithium(I) n-alkanoates [77]
Along with infrared data, recent studies by Bui [73] suggested, based on UV absorption data, that lithium carboxylates (nc = 1–12, inclusive) were of a greater covalent character relative to other alkaline metal compounds. The spectra of these compounds were characterized by a single π–π* transition in the range of 192–188 nm. Surely, the wavelengths of these absorptions were related to the type of metal carboxylate interaction, however, no such analyses were presented in this study. Interesting, the Raman and infrared spectra were, for the most part, identical. However, the Raman spectra did not show clear splitting of the carboxyl asymmetric vibration. This indicated significant mechanical coupling with another vibrational mode, possibly the stretching of the C–C bond adjacent to the COO.
Silver(I) carboxylates
Perhaps the most similar metal–carboxylate analogues to the dimeric alkanoic acids are silver(I) carboxylates. For example, investigations by Bremmer et al. [78] showed that the hydrogen bonded protons of the carboxylic acid dimers were replaced by silver ions to form a Z, Z-type bridging bidentate coordination structure. However, since the coordination spheres of the Ag+ were not satisfied by the two carboxylates groups, additional ligands, typically neutral ones, can become coordinated to the metal centers, leading to polymeric structures serving as secondary building units for metal–organic frameworks. Not only are silver(I) carboxylates useful in this regard but the use of the behenate, stearate and/or a mixture in thermography and photothermography has attracted significant interest in their thermochemical properties [78]. Though these compounds were known to exist as lamellar structures, the exact 3D arrangement of the silver–carboxylate coordination and hydrocarbon packing for longer chain compounds were unknown [18, 79]. This was because they did not produce suitable crystals for X-ray single crystal analysis. Nonetheless, investigation by Tolochko et al. [80], on the structure of silver stearate, indicated based on EXAFS and PXRD, that the compounds were arranged as dimeric units, similar to that proposed by Bremmer et al. [78]. This data also showed that the Ag-to-Ag distance in the dimers was ca. 2.90 Å which was similar to the bond distance in metallic silver, indicating possible metal–metal interactions. A more extensive investigation on a homologous series of silver(I) carboxylates, AgC3–AgC24, inclusive, by Binnemans et al. [79] indicated that the fully extended hydrocarbon chains which were slightly tilted with respect to the metal plane, for these compounds, were arranged to form lamellar bilayer structures. Furthermore, the results showed that the inter-lamellar distance increased linearly with chain length, though the chains were proposed to be slightly interdigitated. Together with data from Tolochko et al. [80], Binnemans et al. [79] concluded that the structures for the series were as shown in Fig. 6, where the silver ions were bridged by carboxylate ligands to form eight-membered rings, held together by extensive head group intermolecular interactions to form infinite 2D sheets. Interestingly, since in the aforementioned eight-membered rings, the Ag–Ag distances were equivalent to those in metallic silver, coulombic interactions were expected to be significant. Indeed, such postulates were supported by DFT calculations of the electronic structure for the dimeric silver(I) acetate structure, reported by Olson et al. [81]. Furthermore, in this study, vibrational frequencies for the Ag–Ag bond were confirmed by experimental and theoretical Raman studies.

Lamellar (a) and head group (b) structures of Ag(I) n-alkanoates, proposed by Binnemans et al. [79]
In addition to their coordination structures, PXRD data, presented by Nelson and Ellis [70], confirmed that all silver carboxylates with nc 8–20, inclusive, were crystallized in a monoclinic crystal system with the fully extended all-trans hydrocarbon chains tilted ca. 75° with respect to the metal basal plane within the lamellar. Importantly, methyl groups from opposite layers in the bilayer do not overlap but were closely packed, resulting in methyl–methyl carbon–hydrogen van der Waals interactions [70]. This assertion was supported by molecular calculations and the presence of minimal reflections in the 20° ≤ 2θ ≤ 30° region of the powder diffractograms, associated with side chain van der Waals interactions. Extensive head group intermolecular interactions resulting in the formation of polymeric molecular layers, was also confirmed by infrared and solid-state 13C-NMR spectroscopies. This study culminated in a detailed structural model adequate in explaining the observed odd–even alternation in certain features that will be discussed in a following section [70].
The structures of other monovalent metal alkanoates, such as rubidium and caesium, have been investigated, though not in significant detail relative to other such compounds [73]. These salts, synthesized via fusion of the carboxylic acids with the metal carbonates, in anhydrous methanol, showed significant ionic character with decreasing chain length. Such assertions were made based on infrared data, which showed ∆ν in the range of 231–249 cm−1. Based on these observations ionic or bridging coordination was proposed. For caesium and rubidium carboxylates the UV absorption spectra showed a strong π–π* transition at ca. 200 nm and a weak n–π* transition at longer wavelength. Though no mention of possible structural similarities were mentioned by Bui, these data strongly indicated similar structural features for these compounds [73].
Divalent alkanoates
Though many metal carboxylato complexes were composed of ligands coordinated in a unidentate mode, there are frequent exceptions; for example, in some copper(II) carboxylate, metal ions were bridged by bidentate carboxylate ligands to form a robust cage-like 3D structures [42]. For lead(II) formate, adjacent lead ions were bridged by formate ligands to form an eight-coordinated 3D polymeric structure as was similarly, for a number of other short chain lead(II) carboxylates; some of which, showed preference for the eight- or seven-coordinate structure [44]. This was supported by Ellis and co-workers [20] who also proposed the same structure for the longer chain adducts.
Copper(II) carboxylates
Of the bivalent carboxylates, perhaps the most extensively studied and well understood structures are those of copper(II); hence, there have been little or no ambiguities about their structure relative to other metal carboxylate systems. This is partly due to the fairly easily predictable square planar coordination geometries, typical for copper complexes, as well as evidence adduced from single crystal X-ray diffraction studies, carried for a number of copper(II) carboxylates [20, 82–84]. These structures showed binuclear one-dimensional (1D) pseudo-polymeric chain or ladder structures, where each copper ion was surrounded by five oxygen atoms together with a single Cu–Cu bond shown in Fig. 7, for the octanoate [83]. Further, the square planar copper ions formed weak axial intermolecular interactions with one of the neighboring molecules resulting in columns arranged parallel to each other, to form columns as shown in Fig. 7b.

Crystal structure of copper(II) octanoate (a) [83] and molecular for the columnar structures of copper(II) alkanoates (b)
Structural information for long chain copper(II) carboxylates which do not produce suitable crystals for X-ray analysis was obtained from electronic and infrared absorption spectroscopies [85]. Accordingly, the electronic absorption of the long chain adducts showed a band at 680 nm ascribed to the d–d transition of the copper(II) ion whilst the intense absorption at 300 nm was attributed to the ligand-to-metal charge transfer transition. The shoulder observed at ~370 nm indicated a metal-to-metal transition as a result of the metal–metal bond as was confirmed by magnetic susceptibility measurements [16, 58]. Additionally, IR data for all the compounds showed strong absorptions for the asymmetric and symmetric COO− stretching at ca. 1,590 and 1,410 cm−1, respectively, which gave ∆ν = 170 cm−1 confirming the bridging bidentate coordination as was predicted from PXRD data [18].
Zinc(II) carboxylates
Zinc(II) carboxylates have been synthesized in various polymorphic forms [20, 61, 62, 86–88], by a number of authors including Taylor [31]. Unfortunately some of these studies featured zinc(II) n-alkanoates synthesized in low purities and yields [20, 89, 90]; hence, discrepancies in the proposals for their structures and mesomorphic properties were evident. Additionally, though the structures for some compounds have been investigated the majority of studies were focused on the chain length homologues such as the acetate, butanoate, hexanoate and octanoate [87, 90–95]. Unfortunately, like most metal carboxylates, these compounds do not readily produce single crystals, consequently there had been no comprehensive proposals for the molecular structures and lattice packing for longer chain homologues. Though it was proposed that bidentate carboxylate coordination was most probable [18, 88, 90–96], an accurate picture of the molecular and lattice arrangement was not presented.
Investigations by Ishioka et al. [95, 96] involving the use of XAFS and IR spectroscopy, for ZnC18 and PXRD analysis involving ab initio Rietveld structure solutions, for ZnC11, ZnC12, and ZnC14, by Mesbah et al. [97] showed that the structures were arranged in layers with tetrahedral zinc ions, bridged by oxygen atoms of carboxylate groups coordinated in a bidentate mode. The calculations presented by Mesbah [97] were very useful, since proposed structures showed two crystallographically independent chains, for ZnC11; that is, hydrocarbon chains in one of the layers within the lamellar were packed parallel to the zinc plane whilst fully extended and those in the other layer were tilted at an angle of 3.96° to the metal planes. However, for ZnC12 and ZnC14, the chains in the layer were crystallographic equivalent. Hence, as expected the tetrahedral oxygen environment for the zinc ions was different from those for ZnC11 and ZnC12. For ZnC11, there were alternating ZnO4 tetrahedrons along the a-axis whilst for ZnC12 and ZnC14, this was not observed. Furthermore, the structure for ZnC11 was similar to single crystal data for the shorter chain adducts [97]. Based on these results it was concluded that for a homologous series there was a change in structure with chain length. For example, with all short chain compounds, with an odd number of carbons, an alternating bilayer arrangement was adopted whilst for the longer chain adducts, nc > 10, with an even number of carbons, the opposite was proposed [31].
Structural investigations carried out by Taylor and co-workers [31, 61, 62] on a homologous series of anhydrous zinc(II) butanoate to eicosanoate, inclusive, made use of several techniques such as IR spectroscopy, X-ray single crystal, PXRD and conoscopy in conjunction with density measurements and molecular calculations. These results confirmed that at room temperature the compounds were isostructural, where metal–carboxylate coordination was concerned; that is, metal–ligand bonding was via asymmetric Z, E-type (syn-anti) bridging bidentate mode. The hydrocarbon chains were all crystallized in the fully extended all-trans conformation and were tilted at an average angle of 60° with respect to the zinc basal plane within the lamellar. Interestingly, similar to Mesbah [97], the X-ray data indicated that the hydrocarbon chains for short chain adducts were packed differently from the long chain homologues. For example, a tail-to-tail bilayer arrangement was observed from the butanoate to the octanoate, inclusive, whilst an interdigitated bilayer arrangement was proposed for the longer chain adducts [31]. Though most of these observations were in agreement with Mesbah [97], no interdigitation of the hydrocarbon chains was proposed. Single crystal analysis of zinc(II) butanoate, pentanoate and hexanoate confirmed both the carboxylate coordination and lamellar bilayer arrangement of these short chain homologues. These results also showed that the compounds crystallized in the monoclinic crystal system with P2(1)/c, P1 (P21/a) and P1 (P21/c1) Bravais lattices, respectively [31, 88, 90–97]. The short chain homologues were composed of alternating bilayers between alternating zinc basal planes arising from crystallographically distinct zinc ions. Interestingly, powder data in agreement with the single crystal results indicated that the short chain length homologues had two molecules per unit cell (Z) but for long chain adducts Z = 1 [31]. For the butanoate, pentanoate and hexanoate, the complete structures consisted of 3D polymeric layered lattices in which component sheets were stacked in the direction of the longest unit cell side, similar to the structure proposed for the other homologues [96]. Interestingly, plots of interlayer (lamellar) spacing or unit cell long axis versus chain length were linear; however, there was an abrupt discontinuity between ZnC8 and ZnC9 as a result of changes in their molecular packing [31].
More accurate molecular calculations by Nelson et al. [98] considered zinc basal plane thickness and intermolecular distances between the hydrocarbon chains with in the lamellar; hence, it was shown (Fig. 8) that the hydrocarbon chains started to overlap for zinc compounds with nc ≥ 9; the degree of which increases with increasing chain length. Furthermore, measured densities and melting points, indicated differences in molecular packing for odd and even chain length compounds, attributed to geometries necessary for higher packing efficiencies and greater lattice stability. A more detailed discussion of the odd–even behavior including a more accurate description of the extent of packing was proposed by Nelson et al. [98] and will be discussed in a following section.

Lead(II) carboxylates
Though much of the early investigations on the structures of lead(II) carboxylates were focussed on the short chain length homologues, extensive studies have been carried out on the even chain adducts of various chain lengths. PXRD and solid-state NMR data confirmed lamellar structures, where the tilted hydrocarbon chains were crystallized in the fully extended all-trans configuration [20, 63]. However, as a consequence of the paucity of accurate structural data, for these compounds, both mono- and bi-layer arrangements were proposed for the lamellar packing of Pb(II) decanoate molecules [20]. Nonetheless, investigations by Harrison and Steel [99], on the crystal structure of anhydrous Pb(II) formate as well as the acetate trihydrate showed that the formate comprised a 3D polymeric network in which adjacent lead atoms were bridged by formate groups to form distorted eight-coordinated lead octahedrons. Interestingly, the data showed that the formate groups were dissimilar; that is, in some formate groups the C–O bonds (1.21 and 1.26 Å) were not equivalent. Though the high coordination number of the lead ions was surprising, other structures for the acetate trihydrate, pentafluorobenzoate and ethylene tetraacetate also showed similar coordination numbers and modes [20, 63].
Extensive IR, PXRD and microscopy studies for a homologous series of anhydrous Pb(II) carboxylates by Ellis and co-workers [20, 63], confirmed that head group bonding does not change with chain length in agreement with Mesubi [100]. Analysis of the IR data reported by Ellis et al. [63], showed that there was a non-equivalence of the CO bonds, hence, the Pb–O bonds were not identical in length, similar to the formate, where νa stretches were more intense than νs; an effect which increases with chain length. Interestingly, ∆ν values decrease with decreasing chain length suggesting unsymmetrical chelating bidentate coordination with possible carboxylate bridging, for the shorter chain compounds (PbC8…12). Furthermore, differences in the PXRD data resulted in the proposal, that there were differences in the molecular and lattice packing between short and long chain compounds; that is, for nc < 12, a bilayer arrangement of the hydrocarbon chains existed but for nc > 12, a monolayer arrangement was proposed. For example, marked differences in the high angle region, 20° ≤ 2θ ≤ 25°, of the powder diffractograms, associated with reflections from side chains packing interactions were observed [63]. However, despite the immense number of investigations carried out on these compounds, no detailed molecular models were proposed for them.
Lanthanide(III) alkanoates
Lanthanide(III) ions are known to take high coordination numbers such as 7–10 with the most popular coordination geometries being octa-coordinate square antiprism and the ennea-coordinate tricapped trigonal prism geometries [67]. As a consequence, lanthanide(III) carboxylates, exhibited diverse dimeric or linear polymeric structures, where the unit complexes were bridged by carboxylate ligands to form 3 or 2D structures as observed for some formates [18]. Furthermore, some structures were composed of many types of inter-metal ionic bridges (metal–metal bonds), which were different in some complexes; that is, they appeared as alternating with the polymeric chains [67, 101]. Some compounds showed simple bridges, where each pair of unit complex were linked by a single bidentate carboxylate group through both an E, E- and a Z, E-type bridging bidentate coordination. For example, trimeric scandium(III) formate was reported as octahedral, with three pairs of Z, Z-type carboxylate ions bridging three metal ions to form a 2D polymeric complex [67]. Similarly, scandium(III) chloroacetate was octahedral with metal ions bridged by three Z, Z-type carboxylate ions, respectively, to form a linear trimer [67]. Based on these results it is clear that some of the structures were quite complex and required a great deal of analytical sophistication for elucidation of their structures.
Though lanthanide carboxylates are widely used their anhydrous structures are not well known; that is, the majority of compounds isolated were hydrated. For example, lanthanide acetate was isolated as Ln(CH3COO)3·xH2O (x = 1, 1.5, 3, 4) [58]. Such complexes were easily obtained as crystals of suitable morphology for X-ray single crystal analysis from slow evaporation of aqueous solutions; hence, their coordination structures and molecular packing were well known. However, the structures for anhydrous acetates and longer chain compounds have not been extensively studied, up until the last 15 years when they were isolated as anhydrous powders. Hence, the majority of structural investigations on these adducts were carried out via PXRD and IR spectroscopy, which indicated that the compounds were crystallized as lamellar bilayer structures composed of 100 % all-trans methylene group conformers in the hydrocarbon chains, packed in either a triclinic or hexagonal lattice. For these compounds, the use of IR data was unsuccessful in distinguish between triclinic or hexagonal lattice. Nonetheless, IR analysis indicated that a symmetrical chelating bidentate coordination was present, since Δν = 120–133 cm−1 and the COO bands were shifted to lower frequencies relative to the ionic soaps [20].
Investigations by Binnemans et al. [59] on a series of neodymium(III) carboxylates (nc = 3–20, inclusive), indicated the presence of mesomorphic transitions in their phase sequence which showed significant structure dependence. The structure of the butyrate monohydrate was obtained from single crystal data but for the longer chain length homologues, PXRD and IR data were used to propose structural models. It was shown that, for this series, the structures consisted of ionic layers of neodymium ions, separated by bilayers of carboxylate anions anchored via Z, E-type bridging tridentate COO groups to the metal planes. Furthermore, the lamellar was composed of two crystallographically distinct neodymium ions, both having coordination number 9, allowing for a monocapped square antiprism geometry. In fact, the crystal structure for the butyrate (Fig. 9) showed Nd1 ions surrounded by two water molecules and five COO groups, four of which were bridging tridentate and the other bridging bidentate [59]. The Nd2 ions were coordinated by four bridging tridentate, one bridging bidentate and one chelating bidentate COO group. Based on these observations, the use of only IR data would be misleading since ∆ν of 125 cm−1 suggested only chelating bidentate COO coordination. The assumption was made that half the water molecules can be lost without a change in the carboxyl coordination mode of Nd1, however, the experimental results showed that there was a change in coordination number from 9 to 8 accommodated by the bridging bidentate COO group, transforming to the bridging tridentate mode [59].

Crystal structures of neodymium(III) butyrate monohydrate [59]
PXRD and IR investigations, by Hinz et al. [102], on a homologous series of praseodymium carboxylates gave similar results to that adduced from single crystal analysis, for lanthanum(III) butyrate monohydrate, previously reported [59]; hence, it was concluded that the structures for long chain lanthanide carboxylates were typical: a phenomenon possibly related to the lanthanide contraction effect. Binnemans et al. [57] isolated several lanthanide dodecanoates (Ln = Y, La, Ce–Lu; except Pm), as hemi- or mono-hydrates, however, all compounds were obtained in the anhydrous forms on heating and cooling from the melt. These compounds showed structures arranged in a lamellar bilayer arrangement, with alkyl chains in the all-trans conformation anchored to the metal basal plane, with respect to which they were tilted. Calculated and experimental values for the interlamallar spacing from PXRD did not decrease in magnitudes comparable to the decrease in metal ion radius and was suggested to be attributed to random deviations in chain flexibility (which affected the accuracy of the calculations, since 100 % all-trans conformation was assumed) and or experimental error [57]. It was proposed that lattice stability derives from the interplay between van der Waals forces, ascribed to the polymethylene chains and the electrostatic interactions associated with the Ln–OOC ionic layers. For the dodecanoates, it was assumed that if ionic radius decreases, then the bilayer was likely to contract resulting in increased interactions between the end methyl groups, thereby preventing the lattice from contracting too much. Hence, it was suggested that instead of a contraction of the overall lattice, there was an increase in the separation between the layers or tilting of the hydrocarbon chains which offsets any decrease in lanthanide ionic size [57, 58]. Therefore, the bilayer structure remained constant as evidenced from PXRD data. However, at increased temperatures, the repulsive electrostatic interactions which change with ionic radius became more prominent in controlling the lattice order since the van der Waals forces from the alkyl chains decreased significantly. Certainly, it would have been useful to calculate the thickness of the ionic layer as ion size changes. The structures of these compounds were assumed to be similar to that of Nd carboxylates based on observed similarities in their IR spectra and PXD patterns [57, 58].
Odd–even alternation
One of the challenges arising from a lack of crystal structures, for an extended homologous series, is the difficulty associated with modeling structural features responsible for odd–even alternation/variation in melting point, density and viscosity data, observed for several metal carboxylates [31, 61, 64, 70]. Similar to n-alkanes, n-alkanoic acids and other n-alkane derivatives, several metal carboxylates display this type of behavior, which was widely understood to be attributed to a difference in the packing of the hydrocarbon chains in the lattice. Though the phenomenon of odd–even alternation is well documented [20, 30, 31, 58], no plausible explanations were proposed to explain this effect until recently. Boese and co-workers [103–105], proposed the first comprehensive geometric model for odd–even variation in alkyl chain derivatives by studying the melting behaviors of n-alkanes and α,ω-alkanedithiols. This model was proposed based on X-ray data and involved representing the packed alkane chains in appropriate quadrilaterals which allowed for the revelation of void spaces in the packing structures [103–105]. The result was that the planes of the hydrocarbon skeletons, for the even chain alkanes, depicted parallelograms whilst the odd chain alkanes depicted trapezoids (Fig. 10). Based on these models, it was clear that in order to allow for the most efficient packing of geometric units, the approaching methyl groups adopted a staggered conformation resulting in columnar packing of the hydrocarbon units which were shifted with respect to each other. For even chain alkanes, intermolecular contact between the parallelograms at all sides resulted in fewer void spaces, whereas for the odd chain adducts only some parts of the molecules were efficiently packed; hence, more void spaces were present. Therefore, even chain alkanes had higher densities and melting points than their odd chain counterparts [103]. A similar approach was used to explain the melting point alternation in α,ω-alkanedithiols [103–105].

For metal carboxylates, there has been no definitive proposal, adequately explaining odd–even alternation in melting point, density, IR and 13C-NMR, until recent studies by Taylor [31] and Nelson and Ellis [70] for the zinc(II) and silver(I) carboxylates, respectively. In these studies IR, 13C-NMR spectroscopic and PXRD data were used to provide evidence for the various lattice interactions. Molecular calculations were also used to provide evidence for the spacing between interacting groups within the molecular lattice. For example, long chain zinc(II) n-alkanoates, nc = 9–20, inclusive, showed odd–even variation in the methyl group, νas(CH3) vibration and the solid-state 13C-NMR chemical shift (δ) [31]. According to Taylor [31], the all-trans conformation of the hydrocarbon chains and chain interdigitation resulted in the CH3 groups of even chain homologues being in close proximity to the zinc ions as opposed to the odd chain homologues whose natural zig-zag structure resulted in the CH3 groups pointing along the zinc basal plane and was therefore, at a greater distance from the zinc ion. This model was correlated with observed odd–even variation in IR, 13C-NMR, density and melting point [31]. However, further analysis by Nelson et al. [98] lead to the proposal of more accurate molecular models for zinc(II) carboxylates. In these models short chain length compounds, nc < 9, did not show overlapping of the hydrocarbon chains, hence, methyl–methyl carbon–carbon interactions between the layers within the lamellar did not influence their molecular packing; therefore, odd–even alternation was not observed. However, for longer chain compounds, nc > 9, the chains formed overlapping instead of interdigitated bilayers. The extent of lamellar overlap increased with increasing chain length and as such, odd–even alternation observed in the νa(CH3) values were due to methyl–methylene interactions of chains from opposite layers within the lamellar [98]. This close approach of the methyl and methylene group carbons resulted in London dispersion forces that cause both carbons to become partial positively polarized; hence, partial negative charges were induced on the hydrogen atoms of the methyl and methylene groups, resulting in attraction between the hydrogen atoms of the alkyl chains from opposing layers. This resulted in reduced methyl C–H bond strength and frequencies as the vertical distance between the hydrocarbon planes within the lamellar decreased. Hence, these effects were greater for compounds that were more efficiently packed carboxylates [98].
In the case of silver(I) n-alkanoates [70], odd–even alternation was observed in νasCH3 absorptions, in addition to the νasCOO frequencies, which was never reported for any other metal–carboxylate system; that is, νasCOO values were consistently higher for the even chain compounds indicating greater metal–carboxylate interactions. For the methyl groups, stretching frequencies for even chain adducts occurred at higher frequencies, than for odd chain compounds, as a result of the relative difference in the closeness of approach of the hydrocarbon chains from different layers in the bilayer. This resulted in different extents of methyl–methyl and methyl–methylene and van der Waals interactions between the chains [70]. The chemical shifts for even chain length compounds were also consistently higher than for odd chain adducts; that is, they were more deshielded. By calculating the distance between hydrocarbon tails within a lamellar, Dbt = (dexp/sinr) − 2dL, Nelson and Ellis [70] showed that methyl–methyl carbon–hydrogen interactions between tail groups from opposite layers in the bilayer were clearly possible. Such interactions resulted in hydrogen atoms from closely packed tail groups becoming partial positively polarized by nearby methyl carbons from opposite layers within the lamellar, hence, the methyl carbons were partially negatively polarized; an effect which was a function of the vertical distance between tail group carbons. Consequently, tail group C–H bonds lengthened and weakened, as evidenced from the lower asymmetric methyl stretching frequencies and greater electron densities on methyl carbons for odd chain homologues. Odd–even alternation was also reported for the chemical shift, of the carboxyl carbon which is a first in metal carboxylate chemistry [70]. Based on this observation it was proposed that the carboxyl carbon for odd chain compounds had a greater electron density as a consequence of higher resonance in C–O bonds of carboxyl groups. However, though they did not discuss the basis for this difference in COO resonance for odd and even chain compounds, it may be assumed that the extent of metal–ligand interactions for odd and even chains may have influenced polarization along the chains, the way in which they were packed and also head group interactions. Perhaps the symmetry of the head groups was different for odd and even chain adducts [70].
Thermotropic phase transition behavior
Overview: mesophase structures
Metal carboxylates constitute a large class of metallomesogens, several of which have been reported on by various authors [20] and it was first observed by Luzzati and co-workers [116] in the1960s that alkali, alkaline earth and transition metal soaps exhibited non-linear liquid crystallinity. This investigation provided the foundation for modern understanding of self-assembly of amphiphilic systems and importantly, non-lamellar mesomorphism. Furthermore, formalized analysis of self-assembled systems in terms of molecular shape of constituent amphiphiles, is of fundamental importance in all fields involving molecular interfaces, from the study of synaptic endocytosis in neural transmission to the understanding of primitive cellular membranes. The study of the thermal behavior of these compounds allows for these applications in various fields of modern science.
Several phase structures including lamellae, discs, rods and ribbons in various 1D, 2D and 3D arrangements have been proposed for several metal carboxylates systems [20, 58, 106]. The phase behaviors of these carboxylates depend on both the metal and carboxylate chain length, but the assignment of many of the mesophases is often controversial and certain of the proposed structures must be treated with caution. The transitions are normally first order, although second order transitions have been suggested for several compounds such as cadmium(II) and barium(II) stearates [20, 58]. For many metal carboxylates, not only are the phase sequences dependent on alkyl chain length, but they are also dependent on lattice arrangement and metal–carboxylate interaction.
Monovalent carboxylates
Their interesting thermochemical properties and possible commercial applications have resulted in several decades of studies on the phase properties of metal carboxylate systems. For example, studies on the physicochemical properties of these compounds have shown that when sufficiently heated, the room temperature crystalline solid transforms into the isotropic melt via several intermediate steps, some of which were liquid crystalline for some metal carboxylates [107–110]. However, assignment and accurate characterization of these transitions have been controversial, hence, some of the proposed phase structures must be treated with caution. According to Skoda [111] the phase sequences for even chain lithium(I) carboxylates with nc = 14–18, inclusive, consisted of three transition ranges. These were classified as the polymorphic transition range at ca. 383 K adjacent to the higher temperature crystalline modification phases which transitioned into a mesomorphic ribbon phase at ca. 473 K followed by the isotropic melt in the region of ca. 503 K. A second order transition was also observed at 353 K for these compounds. Similar studies were carried out by Ferloni and Westrum [72] via DSC analysis, on lithium carboxylates with nc = 1–20, inclusive. Their phase sequence showed several crystal–crystal pre-melting transitions associated with changes in the hydrocarbon chain tilting and lamellar packing. The melts were associated with molten hydrocarbon chains anchored to unperturbed polar head groups [72]. According to Gallot and Skoulios [112], the phase sequences for lithium carboxylates with nc > 11 showed several pre-melting transitions, most of which were plastic crystal or lamellar phases, composed of ribbon-like structures, assembled in a rectangular centered mesh. Based on these results and POM studies, the following stepwise fusion mechanism was proposed: [112]
Lamellar crystal I ↔ lamellar crystal II ↔ plastic crystal ↔ melt.
Though several meta-stable phases associated with changes in van der Waals interactions in the lattice were observed for these compounds, the majority of transitions were reversible [113]. Furthermore, this was supported by White and Ellis [113] who showed that lithium(I) soaps with nc = 8–18, inclusive, exhibited enantiotropic phase behaviors. Their phase sequences were composed of only solid–solid transitions; the number of which increased with increasing chain length as a result of greater alkyl chain flexibility. For example, only one intermediate phase was observed for LiC10…12 [112, 113], when heated from the room temperature lamellar crystal to the melt:
Lamellar crystal I ↔ lamellar crystal II ↔ isotropic liquid and two for LiC14…18:
Lamellar crystal I ↔ lamellar crystal II ↔ plastic crystal ↔ isotropic liquid.
Since, super-cooling was observed for all transitions in the phase sequences, for all lithium carboxylates, it was concluded that reversion of the alkyl chains to the original lamellar arrangement was kinetically controlled [113]. This phenomenon, which was more pronounced for longer chain adducts, due to greater chain flexibility, confirmed that the observed transitions were solid–solid rather than liquid crystalline [114]. Calculated molar enthalpies (∆fusHm) and entropies (∆fusSm) of fusion, for lithium carboxylates, reported by several authors [111], showed reasonable agreement indicating that the compounds in these studies were structurally similar and were of high purity. Since plots of ∆fusHm and ∆fusSm versus nc showed no appreciable change with increasing chain length, for compounds with nc < 13, similar fusion mechanisms were proposed for these adducts [113, 114]. However, for longer chain adducts a linear relationship between these parameters and chain length was observed, confirming strong chain length dependence and different fusion mechanisms for short and long chain lithium carboxylates [113, 114]. Furthermore, since the plot of total molar enthalpy (∆totalHm) versus nc was linear with a slope less than the contribution per-methylene group (3.8 kJ mol−1) [115], for the fusion of n-alkanes, incomplete alkyl chain fusion was proposed for these compounds in the melt [113]. Phase behaviors for the shorter chain adducts, investigated by DSC and electrical conductivity measurements, revealed a single pre-melting transition at 549 K followed by the melt at 604 K for lithium propanoate [77]. For the pentanoate, fusion was observed at 576 K subsequent to two pre-melting transitions at ca. 206 and 325 K. Since the pre-melting transitions showed super-cooling of up to 5 K, it was concluded that the observed transitions were crystal–crystal rather than liquid crystalline [115]. Based on electrical conductivity data, the basis of these transitions was assigned to increased metal and carboxylate ion mobility as the compounds become increasingly molten with increased temperature [115]. Such conclusions were supported by variable temperature infrared data, which indicated increased methyl and methylene group rotation to yield conformational disordered states (con-dis) of various energies with increased temperature. This was further supported by the fact that the ∆H values, for the pre-melting transitions, were in the energy range for rotation of methylene groups [115]. Interestingly, there was a general consensus among authors where assignment of the phases for lithium(I) carboxylates were concerned.
Other alkali metal alkanoates have also been the subject of extensive investigations. For instance, Luzzati et al. [116] concluded, based on X-ray powder diffraction analysis, that the phase textures for anhydrous sodium carboxylates were composed of ribbon-shaped polar head group aggregates. These were dispersed in a sea of liquid hydrocarbon moieties that were arranged in a two-dimensional oblique or rectangular lattice [117]. In earlier studies [118–120] these phases were classified as sub-waxy, waxy, super-waxy and subnet, based on their lattice parameters. Interestingly, some of these phases were optically isotropic whilst others were birefringent [116]. According to Corkery [65], sodium(I) carboxylates exhibited both thermotropic and lyotropic mesomorphic transition behaviors. Their phase textures appeared as birefringent oily streaks for moist samples whilst for dry samples they were weakly birefringent at ambient temperature. In the region of ca. 393–403 K, a streaky feathery fan-like texture which deformed under a shear stress was detected by POM studies [65]. However, since this phase changed to spherulites when aged, the observed mesophases might have been due to the presence of impurities; hence, these phase textures must be treated with caution. Similarly, Sokolov [121] reported a fusion temperature of 571 K, for sodium(I) propanoate, in addition to four uncharacterized solid–solid transitions at ca. 560, 490, 468 and 350 K detected by a visual-polythermal technique. However, Baum et al. [122] obtained a fusion temperature of ca. 563 K via hot-stage polarizing microscopy for this same compound. Subsequent reports by Ubbelohde et al. [123] indicated a mesophase at 567 K which was above the fusion temperature of 566 K (∆fusHm = 8.8 kJ mol−1) adduced from DSC measurements. For this same compound, DSC analyses carried out by Ferloni et al. [124] showed a fusion temperature of 561 ± 1 K along with two uncharacterized solid–solid transitions, at ca. 498 and 475 K. Other reports have also been published on this compound; however, there was hardly any agreement where the transition temperatures, number and textures were concerned [125–127]. This is probably due to polymorphism, which is common for these compounds, and or the presence of impurities, since no C, H analyses results were presented in any of the aforementioned reports. Similar inconsistencies have been reported for long chain sodium n-alkanoates [128]. For instance, it was concluded, based on X-ray diffraction data, that sodium octadecanoate, eicosanoate and docosanoate formed a mesomorphic phase at the clearing point (after the melt). According to these reports a smectic I mesophase which transitioned into a smectic II phase was observed for the octadecanoate; however, for the longer chain adducts only a smectic II mesophase was formed at the clearing point [128]. These results are quite contradictory to the majority of results presented for metal carboxylates systems. For example, the assertion that there were post-melting transitions in the phase sequences for these compounds indicated inaccurate phase characterization. Such inconsistencies confirmed the need for more intensive investigations into the phase behaviors of sodium carboxylates from a theoretical and experimental perspective.
A study of the thermodynamic and transport parameters of some molten ionic salts showed variable thermal stabilities even in the absence of air, due to the great diversity of degradation pathways [129]. However, the presence of excess fatty acid retarded the decomposition rate by absorbing hydroxyl ions accumulated in the salts during synthesis. Melting volumes were studied and shown to follow a linear regression equation, ρ = ρ* − @(T − T*), where ρ and ρ* as well as T and T* were associated with the solid and liquid phases, respectively. The densities were measured using specially designed equipment. Interestingly, after a single heating–cooling cycle increased crystal volumes were observed for sodium butyrate, indicating the development of rifts in the crystals. Furthermore, based on DSC data it was concluded that in some compounds one or more of the observed pre-melting transitions were liquid crystalline. However, since no C, H-analysis or microscopy data were presented to prove the purities of these samples and to characterize the phases, the results presented should be accepted with caution. Electrical conductance measurements, carried out in a conventional Pyrex dipping cell, showed a general increase with increasing temperature. However, for sodium butyrate there were three points of steep increases, indicating significant changes in the mobility of the current carriers. For example, the steepest increase in conductance for sodium isobutyrate was observed at the melt. For the proposed liquid crystalline phase no significant change in conductance was observed, indicating that they were not real mesophases. This was confirmed by the high viscosities (~100 P in most cases) observed for them [129]. Interestingly, the viscosities of most compounds decrease to ~0.2 P at the clearing points, indicating significant changes in structure as they transition to the purely liquid phase. It was postulated that such changes might be associated with positional randomization of the anions and or cations as well as configurational changes due to thermal motion of various parts (–CH2–, –COO– and C–C) of the anion or cation and or the aggregation of ions [129].
Though several investigations have been carried out on the thermal behaviors of potassium carboxylates, no C, H-analysis results were presented for these compounds. Hence, the proposed phase sequences and corresponding thermochemical data must be viewed with caution. For instance, the phase sequences for potassium butanoate [124] showed five solid–solid transitions, at ca. 133, 464, 552, 626 and 677 K, inclusive of a liquid crystalline phase. For potassium propanoate two solid–solid transitions were observed, at ca. 255 and 353 K, prior to the isotropic melt at 638 K [130]. Dilatometric analysis of potassium acetate showed three transitions between room temperature and the melt at 577 K [131]. Variable temperature crystallographic analyses showed that for KC1 these transitions were associated with changes in crystal packing from orthorhombic to monoclinic: phase I (428 K) to phase II (349 K) [131]. These results indicated that the phase behaviors of these compounds were controlled by changes in alkyl chain packing with increased temperature. Investigations on the palmitates (hexadecanoate) of alkali metals from Li to Cs, inclusive, carried out by Vold and Vold [120], showed a total of three transitions at ca. 366, 462 and 484 K for lithium palmitate but for the potassium adduct, four transitions were observed at ca. 340, 403, 447 and 527 K. Dilatometric traces, collected between 273 and 423 K, showed two pre-melting transitions, at ca. 242 and 388 K for Rb and 234 and 368 K for Cs carboxylates. However, DSC traces for samples of these compounds, prepared by a different method, showed temperature values that were different from those adduced from dilatometry. This indicated possible contamination of the samples analyzed via dilatometry or that the second samples were different polymorphs of the compounds [120]. DSC traces for lithium palmitate showed four transitions, inclusive of the melt, whilst five transitions were observed for potassium and four for rubidium and caesium palmitates [120]. The transition at lowest temperature (374–340 K), for all compounds, was classified as an inter-crystal transition based on X-ray single crystal and powder diffraction analyses. These results were also used to classify the second transition, in the range of 400–470 K, as a waxy transition and subsequent phases, as sub-neat and neat in accord with Bolduan and coworkers [132]. Increased fusion temperatures observed on going from Li to Cs indicated that increased metal ion radius resulted in a more rigid carboxylate network [120]. Unfortunately, despite the myriad of investigations carried out on these compounds the underlying theories of their phase properties remains largely underdeveloped. Nonetheless, a study of the enthalpy of formation of the alkali metal salts was reported by Piedade and coworkers [133]. This investigation made use of some basic concepts of classical thermodynamics. Therefore, the molar enthalpies of formation, ∆fHm, for Li, Na, K, Rb and Cs acetates were calculated to be 714.4, 711, 722, 723 and 726 kJ mol−1, respectively [133]. These results indicated that the metal ion radius of metal carboxylate systems affected their packing energies; hence, the significant increase, ∆fHm, observed on going from Na to K indicated significant changes in lattice packing at this point due to changes in the metal ion radius. Indeed, calculated lattice energies using the Kapustinskii [134, 135] approximation: ∆latU0 = 1.079 × 105(νZ+Z−/(r+ + r−)), where ν is the number of ions and Z+ and Z− were the charges on them, showed decreasing values with metal ionic radius from 911 for CH3COOLi to 668 kJ mol−1 for CH3COOCs in agreement with Jenkins and Roobottom [136]. Recent studies by Bui [73], on some short chain monovalent carboxylates (MC1–C12, where M = Li, Na, K, Rb, Cs), confirmed the presence of several solid–solid pre-melting transitions in their phase sequences; the number of which increased with chain length. The calculated transition enthalpies for lithium compounds were in the range of 36–330 J/g. The onset of decomposition occurred in the range of 600–780 K. Interestingly, decomposition occurred at higher temperatures for the longer chain adducts, as did the fusion. This is quite contradictory to the results published by several authors; [20, 58, 113, 115, 130] that is, melting temperature decreases with increased chain length as the more weaker van der Waals forces becomes prevalent relative to head group electrostatic forces. Such discrepancies in the report by Bui [73] might be due to impurities in the samples as no elemental analysis data were presented to validate the purity of the compounds. For sodium carboxylate, the onset of decomposition occurred in the range of 670–750 K with weight losses in the range of 14–76 %. Interestingly, the percentage weight loss increased with increasing chain length. Unfortunately, no explanations were proposed for this observation except to say that the likely paralytic intermediate was Na2C2O4. Furthermore, though a number of pre-melting phases were observed in the phase sequences of these compounds, the number of such transitions was not in agreement with the literature. This is possibly due to polymorphic differences between the samples reported by Bui [73] and those in the literature or the presence of impurities in his samples [117–124]. Interestingly, though significant super-cooling was observed for the observed pre-melting phases, some of them were classified as liquid crystalline. Surly, this interpretation is incorrect, as liquid crystalline phases do not under cool. Furthermore, though an ionic mesogenicity rule was used to show that these compounds could form liquid crystal phases, metal carboxylates are not totally ionic; hence, the Buis’ phase postulates were inaccurate. Furthermore, though odd–even alternation was present in the melting point data, no mention of this feature was made by Bui [73] or explanations presented.
TGA measurement for potassium, rubidium and caesium carboxylates showed weight losses in the regions of 17–70 % (T = 860–750 K), 10–58 % (T = 834–760) and 12–50 % (T = 737–751), respectively [73]. These mass losses were associated with the loss of metal oxalate and an organic moiety such as a ketone. DSC data showed a greater number of transitions for K, Rb and Cs salts, compared to Li and Na compounds, indicating that some of the transitions observed might be associated with changes in the metal basal plane structure. For example, a total of seven transitions were observed for Rb octanoate and Cs heptanoate. However, for the smaller K ion, six transitions were observed for the decanoate.
The results of several investigations, carried out by various methods, on the phase behaviours of thallium(I) carboxylates (TlC1…..TlC n , n = 1, 2, 3…) have shown that they go through several pre-melting phases when heated from the room temperature solid to the melt, some which were observed at sub-ambient temperatures whilst the majority were super-ambient phases of various textures and morphologies, depending on the chain length [137–141]. For example, the phase sequence of thallium(I) hexanoate showed two first order transitions at ca. 204 and 208 K [139–141] in addition to several solid–solid phases and a single mesophase [142, 143]. For the decanoate [144] four solid–solid transitions were observed at ca. 233, 388, 306 and 327 K, followed by a mesophase at 405 K with ∆H and ∆S of 5.67 kJ mol−1 and 13.97 J mol−1 K−1, respectively, whilst the melt was observed at 484 K. Phase sequences for the longer chain thallium(I) n-alkanoates [138] from the tridecanoate to heptadecanoate showed several transitions, some of which were classified as mesomorphic based on their appearances. For instance, the phase sequence for the tridecanoate showed a single solid–solid transition at 365 K followed by the fusion at 468 K. For TlC15, a single solid–solid phase was observed at 353 K followed by a mesophase at 455 K and the melt at 488 K. Similarly, for TlC17 a single solid–solid phase followed by a mesophase and the melt were observed at 302, 421 and 502 K, respectively, whilst for TlC16 two solid–solid phases were observed followed by a mesophase and the fusion at ca. 398, 415, 425 and 500 K, respectively. Odd–even alternation in heat capacity Cp (at constant pressure) versus nc was observed; however, no model was proposed to explain this behavior. POM studies by Peltzl and Sackmann [145] revealed that the optical properties of thallium(I) carboxylates were chain length and temperature dependent. This was attributed to the rotation of CH2 groups about carbon–carbon bonds resulting in the formation of gauche conformers in the alkyl chains [138]. Thallium(I) carboxylate were also found to exhibit lyotropic mesomorphic phase behavior when combined with the parent acid [146–148]. Unfortunately, no detailed investigations have been carried out to adequately explain this phenomenon.
The thermophysics of dimeric silver(I) carboxylates (RCO2)2M2, is of commercial importance since they are applied in photothermogravimetry and other imaging techniques as a source of silver [35]. However, their interesting phase properties represent their most significant possible industrial application since they exhibit phase transitions of various textures when heated from the room temperature crystal to the melt. Some of their pre-melting phases have been described as liquid crystalline as well as solid–solid phases [79]. Mesophase to mesophase transitions have also been reported for several adducts. Furthermore, according to several authors [149–152], these compounds melted in the range of 383–393 K with decomposition, to yield silver nanoparticles for both short and long chain adducts. Investigations by Binnemans and coworkers [79], on a homologous series of silver n-alkanoates, nc = 4–26, inclusive, showed at least two smectic type mesophases in the phase sequences of several compounds. However, recent studies, though not refuting claims for the formation of mesomorphic phases, did not confirm more than one such phase for a single compound [70]. Furthermore, though there was a general consensus among authors, where the textures of the pre-melting phases were concerned, there were some discrepancies surrounding the number of transitions. For example, according to Binnemans [79], five pre-melting transitions at 376, 387, 400, 447 and 484 K (mesophase) followed by the melt at 508 K were observed for silver(I) n-dodecanoate. However, investigations by Nelson [70] showed that the phase sequence for this compound was composed of three pre-melting transitions at 374, 388 and 485 K followed by the melt at 510 K. Similar discrepancies were observed in the ∆H and ∆S values as well as the number of transitions in the phase sequences for the other homologues [70, 76]. Decomposition of these compounds at the melt was well documented; [20] hence, several proposals for their decomposition products and mechanism have been reported. For example, based on variable temperature infrared measurements (Fig. 11), Nelson [70] proposed the formation of an alkene which resulted from the decarboxylation of the dissociated dimeric silver carboxylates; that is, a mixture of carbon dioxide and carbon monoxide were released along with the metal oxide in accord with Binnemans [79].

Changes in infrared spectra for silver eicosanoate with increased temperature [70]
Iweg and Hecht [153] posited that the decomposition of silver(I) butanoate produced butanoic acid, carbon dioxide, carbon and the metal by way of several elementary reactions. Similar results were reported for silver acetate, however, molecular hydrogen was also proposed as a decomposition product [154]. For the longer chain adducts, metallic silver, carbon dioxide and an alkyl radical, which subsequently combined with another such radical, were proposed [149] in agreement with Akanni [20].
Divalent carboxylates
Phase transition behaviors for long and short chain lead(II) carboxylates in both the solid (neat) and solution state have been investigated extensively [20–26]. For example, though, sub-ambient solid-state DSC analyses down to 180 K, revealed no additional transitions, super-ambient scans revealed a single intermediate transition for compounds with nc < 10 whilst for nc = 10 two such transitions were observed in the ranges of 337–382 and 343–379 K. The phase at ca. 355 K, showed extensive super-cooling and was, therefore, characterized as crystal–crystal rather than liquid crystalline [23]. However, the birefringence and texture of the phase centered at 361 K indicated a SmC mesophase. Increased volume observed on transition into this phase [25], was indicative of significant disorder. However, solid-state [201] Pb-NMR measurements [25, 155] suggested that little or no perturbation in metal–carboxylate bonding and by extension the entire metal basal plane, occurred on going from the solid to the melt. These results indicated that the phase transitions, especially the melt, were associated with disordered alkyl chains anchored to unperturbed metal basal plane aggregates. This assertion was confirmed by transverse 1H-NMR relaxation studies which revealed that for lead(II) decanoate the sample was partly amorphous at ambient temperature, however, the percentage amorphous content increased with increasing temperature, without reaching the polar head group, as a result of progressive activation of conformational disorder in the polymethylene chains.
Though the formation of a SmA mesophase was first proposed for PbC10 [21–27], it has been agreed that only a single SmC phase with fan-like texture was formed for this compound. For the shorter chain adducts, nc = 2–5, inclusive, the formation of ionic melts and various glass phases have been proposed [20, 27]. For instance, on heating lead(II) pentanoate, from the room temperature crystal to the melt, two endotherms were observed at 328 and 356 K ascribed to a solid–solid phase transition followed by the isotropic melt. Interestingly, slow cooling of the sample, at 5 K min−1, showed only the crystallization transition whilst reheating showed a new transition at 322.9 K, classified as a glass phase [27]. These results indicated significant changes in the morphology of the sample on heating. However, these changes were temporary since aged samples (stored for 60 days at ambient temperature) showed total reversion to their original thermal behaviors. Phase sequences for lead(II) acetate and propanoate were almost identical; that is, only a single transition corresponding to the fusion was observed for both compounds [20]. Rapid cooling (20 K min−1) of these samples resulted in the formation of a glass phase. However, on second heating two glass transitions were observed prior to the fusion [115]. The thermal behavior of PbC4 was similar to that for PbC2 and PbC3, however, there were some noteworthy differences. For example, on heating, only a single endotherm ascribed to the fusion was observed for this compound. Interestingly, on cooling, the thermograms showed significant dependence on the time elapsed before reheating; that is, when the thermogram was recorded immediately after cooling, a single glass transition was observed but when aged, for a time in excess of 10 h., at room temperature, a new solid–solid transition was observed. These results indicated that the phase behaviors of lead(II) n-alkanoates were kinetically controlled and chain length dependent. Though very little investigations have been carried out on the unsaturated analogues, Adeosun and coworkers [156] showed that in contrast to lead(II) n-octadecanoate the mono-unsaturated adduct melted, in a single step, at a lower temperature. However, the alkyldiynoates which have been investigated for possible polymerization to poly(diacetylene) showed fairly ordered SmC phases at ambient temperature [157]. Such results indicated that the formation of room temperature liquid crystals were most likely for carboxylate with unsaturated hydrocarbon chains.
Thermal degradation mechanisms and kinetics for lead(II) n-alkanoates investigated by Ellis [37, 38] and Baraldi [158] via infrared emission spectroscopy, DTA and TGA showed that decomposition of the compounds, in the range of 465–640 K, occurred via a two step process which resulted in an overall mass drop of 59 % [38]. The major decomposition products, for PbC10, characterized by infrared spectroscopy, NMR measurements and elemental analyses, were nanodecan-10-one and lead(II) oxide along with small amounts of CO2 and CO gases [38]. However, according to Baraldi [158] decomposition of lead(II) formate resulted in the formation of the intermediate metal carbonate which subsequently decomposed to yield a mixture of the raw metal and metal oxide. These results also indicated that thermal degradation took place at a higher temperature in air than under inert atmosphere [158]. The decomposition kinetics for PbC10 investigated via thermogravimetric-mass spectrometry in conjunction with gas–liquid chromatography [158] indicated that the rate of decomposition was controlled by a diffusion mechanism at a contracting interface or by the remaining amount of reactant. Based on the popular Freeman and Carroll [159] model it was shown that the decomposition of PbC10 followed zero order kinetics with activation energy of 42.05 ± 2.3 kJ mol−1 in accord with Rasheed and Bhode [160]. Furthermore, it was proposed that the decomposition of the PbC10 took place via a free radical mechanism. Though it was expected that the thermal degradation mechanism for this compound would have been identical to that for the longer chain adducts, the observed differences for the dodecanoate, tetradecanoate and octadecanoate indicated slightly different degradation pathways [37]. For example, decomposition of PbC12 and PbC14 took place via two rapid steps in the range of 690–730 K, terminating after a total mass drop 62 and 65 %, respectively. However, for the octadecanoate, decomposition occurred via a single step resulting in an overall mass drop of 71 %. Based on these results, it was concluded that one of the major decomposition products for all compounds was lead(II) oxide [37]. The kinetic data for the decomposition of these compounds were analyzed through application of the modified Freeman–Carroll model [161], expressed as: ∆log (dw/dt) = n ∆log Wr − (∆E/2.303R) ∆(1/T) where dw/dt was the rate of reaction, n was the order of reaction, ∆E was the activation energy, R was the gas constant and T the absolute temperature. These results indicated that, similar to the degradation mechanism for PbC10, the decomposition process for PbC12, PbC13 and PbC18 were zero order and were controlled by a diffusion mechanism with overall activation energies of 629, 103 and 139.31 kJ mol−1, respectively [37]. Since the activation energies were different, it was concluded that there was a systematic relationship between chain length and the energy requirements for degradation of these compounds.
The monotropic mesomorphic transitions for copper(II) carboxylates of various chain lengths, have been characterized by POM, X-ray diffraction [65, 162–164], DTA, DSC [85], viscosity measurement [165], dilatometry [166] and variable temperature infrared spectroscopy [167], among other methods [168]. According to Giroud-Godquin et al. [164], the phase sequence for copper(II) dodecanoate consisted of a single discotic meophase at 380 K (∆H = 17.1 kcal mol−1) which was stable up to 523 K. However, phase sequences for the longer chain adducts, nc = 10–18, inclusive, showed a single lamellar crystal to columnar mesophase transition in the region of 383–393 K, followed by the isotropic melt at ~473 K, after which the samples decomposed [168]. EXAFS studies revealed that the metal basal plane remained virtually unchanged both in the lamellar crystal and mesomorphic phase [169]. However, variable temperature infrared analyses showed an increase in the stretching frequencies of the CH2 group as a result of disordering of the alkyl chains in the mesophase [167] in agreement with dilatometric and 1H-NMR studies [170]. These results confirmed that the mesophase was composed of partially disordered alkyl chains anchored to unperturbed metal basal plane aggregates. Interestingly, high temperature X-ray powder diffraction for two polymorphs of copper(II) decanoate indicated that for these compounds polymorphism does not play a direct role in their liquid crystalline properties [171–173]. For instance, since the d-spacing for both samples in the disordered columnar mesophase were ca. 15.6 Å and showed an identical increase of 1.2 %, their liquid crystal phases were concluded to be of the same structure.
Though most investigations on copper(II) carboxylates were focused on straight chain compounds, studies on the copper complex of (n-C9H12)2CHCH2CO2H revealed the formation of a columnar mesophase at 348 K [28]. Furthermore, copper(II) complexes where the terminal group (methyl or tail group) had been replaced by various groups such as a benzene ring, 1, 2-ditholane or 4-cyclohexyl phenyl moieties have also been investigated [29]. For copper(II) 1, 2-ditholane and 4-cyclohexyl phenyl substituted adducts, only a crystal to melting transition was observed. However, both copper(II) phenyl-propanoate and 13-henicosenoate showed a single mesomorphic transition at 428 and 336 K, respectively. Based on these results it was concluded that the tail groups played an important role in the phase behaviors of these compounds [28].
Though the thermal degradation of copper(II) carboxylates is to this day largely under investigated. Studies by Van My and coworkers [174] showed that the thermal degradation of short chain copper(II) carboxylates produced metallic copper under inert atmosphere whilst the oxide was produced in air [39, 40] along with the parent alkanoic acid and or the α-olefin in some cases [175]. However, no comprehensive mechanism or kinetic models were proposed for their degradation behaviors.
The structures and phase textures for zinc(II) n-alkanoates, when heated from the room temperature lamellar solid to the isotropic melt, have been investigated by several authors [30, 31, 58]. For example, recent studies by Ellis and Taylor [30, 31] on a homologous series of anhydrous zinc(II) carboxylates, [(CH3(CH2) n CO2)4Zn2], from the butanoate to eicosanoate, inclusive, revealed a single SmC transition in the phase sequences for the heptadecanoate to the eicosanoate, inclusive. The broken fan textures were observed under plane-polarized light, in the range of ca. 398–378 K (Fig. 12), on cooling from the isotropic melt, in agreement with Roberts and Friberg [32]. For the other homologues only solid–solid transitions were observed, in accord with Konkoly-Thege et al. [176].

SmC phase textures for a ZnC17, b ZnC18, c ZnC19 and ZnC20 [31]
These transitions were associated with disordering of the alkyl chains due to methylene group rotation as a result of reduced van der Waals interactions caused by increased temperature in the lattice [177] and not the presence of impurities as previously suggested [178]. Furthermore, increased ∆TotHm and ∆TotSm values with increasing chain length were proposed to be a result of increased van der Waals interactions [32, 100]. Odd–even alternation observed in plots of ∆TotHm and ∆TotSm versus nc was attributed to different packing arrangements and intermolecular interactions in the lattice [30, 31]. Using XAFS data from Ishioka et al. [95]. Taylor [31] proposed that melting of the compounds proceeded via dissociation equilibrium:
This suggested that the isotropic liquid was a mixture of dissociated dimer and monomers as well as relatively small amounts of carboxylate and zinc ions.
Studies of unsaturated zinc(II) carboxylates such as the cis and trans isomers of zinc(II) 9-octadecenoate and 9, 12-octadecadenoates via DTA revealed a single transition associated with the fusion but at a lower temperature than for the corresponding saturated adducts. For other cis–trans isomers, the cis adduct was of lower melting point as a result of more reduced van der Waals interactions in the lattice [20]. These results also showed that the presence of an additional double bond resulted in further lowering of the fusion temperature for these compounds. Recent studies by Nelson et al. [98] on two geometric isomeric unsaturated zinc(II) carboxylates (zinc(II) 9-undecynoate and 10-undecynoate), consisting of a single triple bond in the alkyl chains, showed significantly different phase properties. For instance, though the compounds were of identical chain lengths, a SmC type mesophase was observed of zinc(II) 10-undecynoate at 344 K followed by the fusion at 350 K whilst only the fusion, at 429 K, was observed for the other isomer. Furthermore, ageing (60 days) of these compounds resulted in a 50 % increase in their transition enthalpies. Such observations along with extensive super-cooling of the crystallization transition were due to slow molecular equilibration as the hydrocarbon chains rearrange to their original room temperature lamellar order [98]. Unfortunately, reports on the thermal degradation of zinc(II) carboxylates are sparse; however, studies by Dollimore and Tonge [179] revealed that the major products of their decomposition were H2O, CO, CO2 and H2 along with the metal oxide. Other investigations have revealed the formation of a mixture of ketones, CO2, ZnCO3 and ZnO [180, 181].
The thermal behaviors of mercury(II) carboxylates have been investigated by X-ray diffraction [182, 183], DTA [107, 184], DSC analyses and POM [20]. Though it was initially suggested that even chain mercury(II) carboxylates [20] from the octanoate to octadecanoate, inclusive, showed smectic type mesomorphic transitions in their phase sequences, more recent studies by Ellis [183] revealed only solid–solid pre-melting transitions. Based on these studies it was concluded that the melt consisted of disordered alkyl chains. Unfortunately, though these compounds exhibited step-wise melting, not many investigations have been carried out on them. Hence, much of the theoretical and experimental aspects of their phase properties remain largely uninvestigated to the best of our knowledge. Nonetheless, thermal degradation studies on these compounds revealed the formation of a mixture composed of the carboxylic acid, and the corresponding α-olefin as a result of decarboxylation to release carbon dioxide and the raw metal [185].
Phase sequences for even chain cadmium(II) carboxylates of various chain lengths, investigated by X-ray diffraction [186], dilatometry [188] and DTA [108, 185, 187], showed a mesophase which consisted of infinite cylindrical micelles at ca. 373 K previous to the melt at ca. 503 K. However, these results were questioned by Konkoly-Thege et al. [176] on the basis of DTA studies which indicated that the results of previous investigations were due to impurities in the samples. Nonetheless, it was suggested that cadmium(II) carboxylates, from the dodecanoate to octadecanoate, formed a single mesophase ~10–15 K below the highly viscous melt, observed in the region of 373–393 K, which was composed of infinite cylindrical aggregates similar to those proposed for the mesophase [20]. For the decanoate a single SmA mesophase was reported by Mirnaya and coworkers [188]. Since, calculated total molar enthalpies and entropies of fusion were much less than for the corresponding lead(II) and zinc(II) n-alkanoates [20], incomplete alkyl fusion was concluded for the melt. However, increased conformational disorder in the alkyl chains with increasing temperature was indicated by variable temperature infrared measurements, DSC and Raman spectroscopy [176, 178, 179]. Such results confirmed that in the melt the hydrocarbon chains became molten without perturbation of the metal basal plane in accord with suggestions by Luzzati [116]. Interestingly, the hydration numbers of these compounds were found to be an influential factor where their phase behaviors were concerned.
Though no degradation or kinetic models have been proposed for these compounds it was shown by Yakerson [189] that the decomposition of Cd(CH3COO)2·2H2O was a multi-step process; that is, both moles of water were lost at ~423 K which was followed immediately by endothermic decomposition at a reasonable rate to yield the metal oxide and carbon dioxide by way of the meta-stable metal carbonate intermediate [190].
Polyvalent carboxylates
Lanthanide carboxylates
The thermochemical properties of various di- and trivalent lanthanide alkanoates [191, 192] have been investigated by DSC, [60, 193] TGA [60], X-ray powder diffraction [194] and POM [60]. The thermal behaviors of cerium(III) carboxylates [60] with nc = 8–12, inclusive, were shown to be markedly different from those with nc > 12; that is, the melting point for the shorter chain adducts were higher than for the long chain homologues. Such results indicated that cerium(III) carboxylates were significantly ionic at short chain lengths. The phase sequence for cerium(III) alkanoates [193] with nc ≥ 10, showed a single lamellar crystal to mesophase transition in the vicinity of 351 K followed by the fusion which was quite gradual [60]. However, since super-cooling was observed for all transitions, for these compounds, it was concluded that the pre-melting phases previously classified as liquid crystalline were not well formed; hence, they were classified as disordered smectic mesophases based on POM studies [60]. However, according to Binnemans and coworkers [193] the phase sequence for cerium(III) decanoate was composed of a single SmA mesophase at 379 K followed by the isotropic melt at ca. 411 K. Enthalpy values calculated for the first heating cycle showed a 10–15 % decrease during second heating; hence, incomplete fusion was concluded in support of the pseudo-isotropic melt, indicated by POM studies [60]. Total molar enthalpies (∆TotHm) for conversion from the lamellar crystal phase to the isotropic melt showed a linear relationship with chain which confirming that the main theoretical contributors to this parameter were: [195] ∆TotHm = ∆Uconf + ∆UvdW + ∆Uo + p∆V, where ∆Uconf and ∆UvdW were energy variations associated with intra-molecular conformational disorder such as gauche-trans isomerism and van der Waals interactions, respectively. Other energy contributions, such as head group electrostatic interactions, were represented by ∆Uo. Since changes in molecular volume were negligible, p∆V ≈ 0. This expression was used by Marques and coworkers [60] to explain the phase behaviors of some even chain Ce(III) carboxylates. Interestingly, though the decomposition of these compounds occurred at relatively low temperatures, in the region of 523–573 K, no comprehensive investigations on the kinetics of this process have been reported.
Partially hydrated praseodymium(III) n-alkanoates, Pr(C n H2n+1COO)3]. xH2O (x = 0.5–1), showed at least a single SmA-type mesomorphic transition, for all compounds with nc = 5–20, inclusive. These conclusions were based on POM and variable temperature X-ray studies [102]. However, since super-cooling was reported for these phases, such conclusions must be viewed with caution. Furthermore, according to Binnemans and coworkers [102] the melting temperatures for these compounds increase with increasing chain length, contrary to the results of previous studies. The effect of chain length on phase behavior is highlighted by the formation of two mesophases, for all compounds from the pentanoate to the octadecanoate whilst for the longer chain adducts, nc = 9–20, inclusive, only a single liquid crystalline phase was observed in the region of 358–403 K. The low temperature mesophase in the region of 385–363 K, for all compounds, showed no appreciable change in d-spacing and was classified as a lamellar mesophase in which molecular rotation was restricted due to the nature of the metal–carboxylate coordination geometry [102]. Such an observation represented a sharp contrast to the SmA phase, where reduction in d-spacing was quite marked.
Neodymium(III) n-alkanoates form the butyrate to eicosanoate, inclusive, isolated as the hemi- and monohydrates, showed two mesophases for compounds with nc < 15, inclusive. However, for the longer chain adducts only a single endotherm ascribed to the fusion was observed [59, 196]. Similar to Pr(III) [102], the high viscosity low temperature mesophase, in the region of 350–363 K, was labeled as an “M” phase whilst the other pre-melting transition, in the region of 365–374 K, was classified as SmA mesophase followed by the melt in the range of 388–439 K [59]. Since on cooling, the observed transitions were birefringent only under increased pressure, it was concluded that the molecules were aligned homeotropically (with alkyl chains perpendicular to the cover slip). Furthermore, X-ray powder diffraction studies on the mesomorphic transitions for these compounds showed a decrease in the d-spacing which was indicative of gauche–trans isomerism in the alkyl chains and possibly chain kinking. Based on these results it was concluded that the low temperature solid to mesophase transition was caused by partial melting of the hydrocarbon chains without changes in their tilt angle whilst for the SmA phase they were totally molten. Such descriptions of the phase structures for these compounds were nearly opposite to those for Li(I) [201–204], Ag(I) [70, 78, 79], Pb(II) [20] and Zn(II) [20, 30, 31, 65] among other metal carboxylate systems, adduced from X-ray diffraction, variable temperature 1H and 13C-NMR and variable temperature IR data. For all neodymium(III) carboxylates the melts were due to destruction of the ionic layers and the formation of an ionic salt melt [196]. However, since no electrical conductance data were presented to validate such claims, these results must be treated with caution.
Europium (Cn−1H2n−1CO2)3Eu, n = 14, 16, 18, 20, investigated by Li and coworkers [194] showed mesomorphic-like phase behavior in the temperature range of 373–398 K. However, observed super-cooling indicated that these transitions were solid–solid rather than liquid crystalline in agreement with Binnemans and coworkers [59]. Though several transitions were observed for these compounds on heating, the cooling traces showed fewer phases. This indicated that some of the phases observed on heating were metastable or that the mechanism of cooling caused them to coalesce [59, 196]. Furthermore, X-ray data indicated that heating resulted in gradual melting of the alkyl chains to yield states consisting of varying degrees of gauche conformers [193]. Therefore, the observed phases were mainly controlled by alkyl chain structure and packing. Unfortunately, despite the importance of these heavy metals, very little investigations have been carried out on the thermal behaviors of their unsaturated and branched chain alkanoate salts. Furthermore, detailed searches of the literature revealed no data for the thermal degradation kinetics and or mechanism for these compounds. Nonetheless, Binnemans and coworkers [196], reported that the decanoates for metals from lanthanum(III) to lutetium(III), inclusive, between one and two pre-melting transitions were observed. DSC thermograms for compounds with Ln = Sm–Lu, inclusive and Y, showed a single peak corresponding to the melt whilst for LaIII, CeIII, PrIII and NdIII an additional endotherm was observed, ascribed to a SmA mesophase. Super-cooling observed for these phases indicated the absence of mesomorphic transitions in their phase sequence [59]. Interestingly, the melting point decreased from La to Lu indicating that the phase properties of these compounds were dependent on the properties of the metal ion.
Carboxylates with metal–metal bonds
Subsequent to the discovery that copper(II) alkanoates were dimeric, several other di-metal carboxylates were prepared. Of these, the first were rhodium(II) carboxylates, which also had a binuclear structure similar to copper(II), but with a metal–metal double bond [197]. Giroud-Godquin et al. [168] showed that like copper(II) alkanoates, the rhodium(II) complexes formed columnar mesophases on heating from the room temperature solid to the melt. These transitions occurred between 338 and 428 K, corresponding to a transition from the crystalline phase to a mesomorphic state followed by decomposition above 493 K. It was also demonstrated that a reverse transition (mesophase to crystal) occurred at slightly lower temperature which was attributed to slow nucleation and crystal growth kinetics. Not surprisingly, as with many carboxylates, the change in phase on heating was accompanied by disordering of the aliphatic chains in the lamellar at room temperature, as evidenced from PXRD data, however, the columns are maintained by the rigid order of the Rh cores. Evidence from Raman spectroscopy showed that the Rh=Rh bond was sensitive to increased temperature but EXAFS measurements showed that the core structure is maintained in the mesophases. 13C-NMR spectral data suggested that the di-rhodium core underwent a fast axial bond exchange in the mesophase [197].
Di-ruthenium carboxylates (RuIIRuII) containing Ru=Ru double bond as well as the mixed-valent (RuIIRuIII) species formed columnar mesophases [197]. It was suggested, based on PXRD and EXAFS data, that the RuIIRuIII complexes had the same molecular organization as the copper alkanoates. Not surprisingly, branching and unsaturation of the carboxylates lead to a decrease in the melting points and the presence of two types of mesophases: hexagonal columnar and lamellar columnar. In the case of the RuIIRuIII carboxylates, magnetic susceptibility measurement as a function of temperature showed a sharp change in magnetic moment at the solid to mesophase transition, suggesting that there is some distortion of the core, similar to the di-rhodium compounds. These RuIIRuIII species which are cations, counterbalanced by X− anions (X− includes Cl− and RCOO−), to give Ru2(RCOO)4X, carboxylates, are such that the anion had a strong influence on the mesomorphism; those compounds with nc = 5–9, 12, 16 and X− = Cl−, are not mesomorphic whereas, those with X− = RCOO−, exhibit a columnar mesophase on heating. It was shown that packing between the chloride and carboxylate ions created void spaces between the dimers and as such, the use of bulky anions or a combination of bulky carboxylates with a small anion, effected a mesophase as they filled the interdimer space [20]. The molecular model proposed for the mesophase is shown in Fig. 13 in the case for the propanoate, Ru2(prop)5; (a), (b) show the polymeric dimer anion chain, and (c) the columnar hexagonal structure of the mesophase, built up by four polymeric chains.

Schematic representation for the mesophase of mixed-valent di-ruthenium pentacarboxylates [198]
Extensive studies by Chisholm and coworkers [199] showed that carboxylates containing Mo–Mo and Cr–Cr quadruple bonds also exhibited columnar phases. The molybdenum alkanoates were mesomorphic from nc = 4–10 and the melting points were fairly constant in the region of 425 and 381 K, for the series, except for nc = 4 and 5. The clearing temperatures on the other hand decreased linearly with chain length, with no mesophases occurring above the decanoate. Similar phase behavior was observed for the di-chromium complexes, where they show crystal to liquid crystal transition but decompose at about 473 K. For the tungsten carboxylates, no mesophases were observed. These differences in phase transitions were surprising, especially considering that their structures were similar, even to those of the copper, rhodium and ruthenium di-carboxylates. It was proposed that since the phase behavior of these are almost identical to those of copper(II) alkanoates, similar dimeric interactions, axial metal–oxygen intermolecular interactions were preserved and responsible for the formation and stabilization of these phases. In fact, analysis of structural parameters of the compounds show that metal–oxygen intermolecular bond lengths were in the order of 2.33, 2.65 and 2.67 Å, for Cr, Mo, and W, respectively. Furthermore, it was also proposed using data from PXRD and 13C-NMR that the axial bonds are kinetically labile, being readily broken and formed, allowing for a dynamic behavior, as depicted through the columnar packing [199].
Other metal carboxylates
Though most investigations have been focused on the saturated carboxylates of Li(I), Na(I), K(I), Tl(I), Ag(I), Pb(II), Zn(II) and Cu(II), other systems have been studied but to a lesser extent. For example, the phase sequences for barium(II) and calcium(II) octadecanoates (stearate) showed more than one second order transition in addition to several first order phase changes [200]. Other alkaline earth metal carboxylates have also been investigated by various methods [201–206] and the results reviewed elsewhere [207]. These investigations revealed several intermediate transitions in their phase sequences, when heated from the room temperature crystal to the isotropic melts, some of which were classified as mesomorphic phases based on X-ray diffraction studies. They were classified as lamellae, discs, rods and ribbons, arranged in 1D, 2D and 3D networks [20]. However, based on X-ray diffraction studies in conjunction with other results, it was suggested that the observed transitions, for alkaline earth carboxylates, did not represent liquid crystal phases in a classical sense [208]. Further, Raman spectral and rheological measurements on calcium stearate as a function of temperature showed marked changes in yield stress, elastic moduli and viscosity, at ca. 398 K, confirming transition to a mesophase [209]. These results also showed that fusion temperatures increase from Mg to Ba, as expected. For aluminium(III) carboxylates, though several studies showed at least two transitions at ca. 373 and 438 K, in most cases the samples were either basic or hydrated [210]. Hence, these results must be treated with caution. Manganese(II) carboxylates, from the octanoate to hexadecanoate, showed a single intermediate phase, characterized as mesomorphic based on electrical conductivity and DTA measurements [103, 184, 211]. This phase was composed of infinite cylindrical micelles similar to Cd(II) carboxylates. However, for Mn(II) octadecanoate, two such transitions were observed. Though several endotherms were reportedly observed in the phase sequences of iron(III) dodecanoate and octadecanoate, in the region of 330–378 K, detailed and timely searches of the literature revealed no proposals for their phase structures [103, 212]. For cobalt(II) stearate, magnetic susceptibility and DTA measurements showed that the transitions observed at 372 and 381 K were associated with a solid–solid followed by a solid to plastic mesophase transition, respectively [17]. However, on cooling, an additional mesomorphic transition was observed in the region of 317–327 K. Such changes were due to perturbation in head group bonding. Phase sequences for rhodium(II) [164, 213, 214] and ruthenium [164, 215] carboxylates were similar to Cu(II) n-alkanoates, forming columnar mesophases consisting of infinite cylindrical micelles of binuclear metal carboxylate aggregates.
Binary metal carboxylate systems
Though the majority of investigations on the structures and phase behaviors of metal carboxylates were focused on pure compounds, in recent times, several studies have been carried out by various authors on the phase properties of binary carboxylate systems [20, 37, 38, 48, 55–59]. For example, studies by Spegt and Skoulios [206] showed that at high temperatures, a mixture of calcium and strontium octadecanoates was homogeneously arranged in a hexagonal phase. However, X-ray diffraction studies revealed that this phase was heterogeneous for a mixture of magnesium and calcium octadecanoates, corresponding to the phase behaviors of the individual carboxylates [201, 202]. Similarly, significant studies have been carried out by Hagino [216] on the phase properties of some binary even chain sodium carboxylate system, from the dodecanoate to octadecanoate, inclusive. However, though the phase diagrams for these systems have been presented, the composition and distribution of the components of the mixture were not discussed. Phase diagrams for binary systems of lead(II), zinc(II), cadmium(II), manganese(II) and mercury(II) octadecanoates have been presented by Adeosun and Ellis [217]. These investigations revealed that, though lead(II) octadecanoate showed an SmG phase when heated and the zinc(II) adduct melted directly, the addition of <0.2 mol fraction (x) of zinc octadecanoate to the lead(II) sample did not result in the formation of a new phase (Fig. 14a). However, when x > 0.2, a new phase was formed which was structurally different from the G phase and was labeled G2. However, when the dopant was changed to cadmium octadecanoate a new mesophase formed when x > 70 %. No such change was observed when the dopant was manganese octadecanoate [217].

Phase diagrams for various binary metal carboxylate systems [217]
Interestingly, when cadmium(II) octadecanoate was doped with the corresponding zinc(II) carboxylate, no new phase changes were observed up to 0.8 mol fraction of the dopant; however, at higher mole fractions there was a direct transition to the melt (Fig. 14b) [217]. The simple phase sequence of manganese octadecanoate, crystal to mesophase to melt, showed an additional phase on the low temperature side of the DTA curve, upon addition of the corresponding zinc adduct (Fig. 14c), which was resistant to a shear stress. Doping of lead(II) octadecanoate with the corresponding Hg(II) adduct resulted in the formation of a new laminar G phase when 0.4 < x < 0.9 as shown in Fig. 14d. However, at higher mole fractions, only the fusion was observed, in contrast to an earlier study [210] which showed a single pre-melting phase. Such an observation was due to pre-melting effects which occurred during thorough mixing of the samples. Mercury(II) octadecanoate have been mixed with other octadecanoates of various metals such as Mn(II), Cd(II) and Zn(II). Interestingly, the phase sequences for all systems were different indicating different interactions of the components in the mixture [217]. However, the solid-state structures of these mixtures were not investigated; hence, the exact interactions and packing dynamics of the components in lattice are unknown.
Phase behaviors of some binary lanthanide(III) carboxylate systems, La x Ln1–x(C11H23COO)3, where lanthanum dodecanoate was doped or used as a dopont with other lanthanide (Ln) dodecanoates, such as Nd, Eu, Ho, Tb and Yb, have been investigated by Binnemans and coworkers [218]. These results indicated that for systems of Eu and La, where the mole fraction of La was greater than or equal to 0.04, a SmA mesophase was observed at temperatures that showed no relationship with the percentage of the non-mesomorphic europium and lanthanum dodecanoates. This was due to the stabilizing effects of the larger La ion which also increased the melting and clearing temperatures of the substrate. For the Nd–Eu systems, decreased Nd mesophase stability was observed with increased Eu concentration. This effect was not predicted by the additivity law Tcl = T1 + x(T2 − T1) [219] where Tcl represents the clearing point of the mixture, T1 and T2 were the clearing temperature for the individual components and x was the mole fraction of the dopant. This was due to competition between the metal basal plane and alkyl chain fusion; that is, electrostatic and van der Waals forces. Phase diagrams for La x Tb1−x(C11Ho23COO)3, La x Ho1−x(C11H23COO)3 and La x Yb1−x(C11H23COO)3 systems showed that addition of the mesomorphic La(III) dodecanoate to the non-mesomorphic dodecanoates of Tb(III), Ho(III) and Yb(III) resulted in the formation of a liquid crystalline phase in each case. However, the fraction of La(III) dodecanoate required for mesophase formation was much greater for La–Yb than for the Eu–La system [218]. Based on these results it was concluded that meophase formation in doped lanthanide(III) carboxylate systems was strongly dependent on metal ion radius which provided a stabilizing effect for mesophase.
Binary mixtures of lithium(I) and thallium(I) carboxylates underwent both eutectic and peritectic reactions especially for the Li(I) and Tl(I) propanoates(C3) where a new compound, Li2Tl(C3)3, was formed [220]. This new compound showed a single solid–solid transition at ca. 368 K [∆H = 4.9 kJ mol−1 of Li2Tl(C3)3 or 1.6 kJ mol−1 of mixture] followed by the melt at 512 K. For the Li2Tl(C5)3 system formed from Li(I) and Tl(I) pentanoates (C5) a single solid–solid phase transition and an incongruent melt, similar to the peritectic fusion of Li2Tl(C3)3, was observed. Interestingly, these results also showed that doping of TlC5 with xLiC5, where x = 0 − 0.56, resulted in an extension of the mesophase stability from 35 K in the pure TlC5 to 110 K in the eutectic mixture of LiC5:TlC5, 0.19:0.81 ratio. Based on POM studies, this transition was identified as a SmA mesophase (Fig. 15) consisting of the typical focal conic domains of a neat phase [220].

Smectic fan-like texture for the SmA phase of Li2Tl(C5)3 [220]
Phase diagrams for binary systems of sodium and barium(II) n-butanoates have been presented by Ferloni et al. [221]. These results indicated that barium butanoate exhibited a stepwise melting behavior, transitioning through a single pre-melting phase at ca. 519 K before melting at ca. 589 K. However, addition of sodium(I) butanoate (NaC4) resulted in significant changes in the phase sequence on heating and cooling. For example, on heating, a eutectic point was observed at ca. 425 K where the mole fraction of NaC4 was 0.62. Interestingly, the phase sequences for the different mixtures showed a SmA mesophase at 0.58 < x < 1.00, identified by POM studies. Two metatectic points were observed at 445 and 480 K where x = 0.47 and 0.20, respectively. These were associated with interactions of a solid phase, an unspecified mesophase (M) and an isotropic liquid phase [220]. Unfortunately, no structural models were presented to account for the interactions between the components of these mixtures. Nonetheless, studies on various other binary carboxylate systems [222] have been reported as well as metal carboxylate systems doped with other compounds [223–226]. However, no studies on the catalytic properties, degradation kinetics or mechanism have been reported for these systems. This indicates that there is significant room for further investigations in this area and possible extension in ternary and quaternary systems.
Conclusion and outlook
Over the last century, several investigations on the chemistry of metal carboxylates have been carried out by a number of authors. These studies were carried out manly via DSC, TGA, EXAFS, FT-IR, spin decoupled solid-state 13C- and 1H-NMR spectroscopy, dilatometry, POM, etc. Such studies were focused on the structure and thermal behaviors of mono-, di- and poly-valent metal carboxylates of saturated, unsaturated, branched and straight chain carboxylic acids. Interestingly, the majority of results showed that the type of metal carboxylate bonding observed in these compounds were dependent on metal ion properties such a radius, hardness, softness and valence. The structure of the carboxylate ion also played an important role in the symmetry of the metal–ligand coordination sphere. Hence, though sodium and silver were of the same ionic radius, when bound by carboxylate groups of the same structure, their observed bonding types were different. Similarly, the head group symmetry and phase behaviors of zinc(II) 9-undecynoate was different from that of zinc(II) 10-undecynoate. This indicated that metal carboxylate bonding was not controlled only by metal ion radius. The coordination structure for zinc(II), Pb(II) and Cu(II) decanoates were all different despite being composed of metal ions with the same oxidation state. This showed that metal–carboxylate coordination is not directly related to the charge on the metal ion. Odd–even alternation observed for long chain zinc(II), lithium(I) and silver(I) carboxylates were due to differences in molecular packing between, the more asymmetric odd chain adducts and their even chain counterparts. Since this behavior was also observed in the methyl group chemical shift and unusually the carboxyl chemical shift, for silver(I) compounds, differences in electron density distribution was indicated for odd and even and short and long chain compounds. Unfortunately, theories providing insights into the three-dimensional structures of these compounds from PXRD data are sparse. Such theories would be highly useful in understanding the structures of these compounds since they do not readily produce crystals of suitable morphology for single crystal diffraction. Furthermore, since it was established that the phase behaviors of these compounds were dependent on molecular packing which was a function of metal–carboxylate bonding, structural theories, for these compounds, could allow for the designing of new ionic liquids.
Furthermore, despite the numerous pre-melting transitions observed, for several compounds, complete and quantitative theoretical understanding of the structural features responsible for these phases remain underdeveloped for most of them. Such theories would allow for phase manipulation and possibly energy storage in the structures of these compounds. Being able to control their phase behaviors could also allow for their application in dye sensitized solar cells [227, 228] as electrolytes or even as ordered ionic solvent systems for controlling reaction conditions on a molecular scale. Therefore, future work in this field should place significant emphasis on the theory of the phase and structural properties of these compounds.
References
Buono FJ, Feldman ML (1979) Kirk-Othmer, encyclopedia of chemical technology. In: Mark HF, Othmer DF, Overberger CG, Seaborg GT (eds) 3rd edn, vol 8. Wiley, New York, p 34
Bossert RG (1950) Ultrasonic study of molecular interactions and compressibility behaviour of strontium soaps in chloroform-propylene glycol mixture. J Chem Educ 27:10
Huang TC, Toraya H, Blanton TN, Wu Y (1993) X-ray powder diffraction analysis of silver behenate, a possible low-angle diffraction standard. J Appl Crystallogr 26:180
Matthews FW, Warren GG, Michell JH (1950) Derivatives of fatty acids identification by X-ray diffraction powder patterns. Anal Chem 22:514
Keiderling U, Gilles R, Wiedenmann A (1999) Application of silver behenate powder for wavelength calibration of a SANS instrument—a comprehensive study of experimental setup variations and data processing techniques. J Appl Crystallogr 32:456
Engels T, von Rybinski W (1998) Liquid crystalline surfactant phases in chemical applications. J Mater Chem 8:1313–1320
Bonekamp K, Hegemann B, Jonas J (2007) Polarization microscopy of short chain sodium carboxylate mesophases. Mol Cryst Liq Cryst 87:13–28
Mehrotra KN, Kachhwaha R, Singh M (1983) Studies on beryllium soaps. Infrared absorption spectra and thermogravimetric analysis. Thermochim Acta 62:179–185
Burrows HD (1990) The structure, dynamics and equilibrium properties of colloidal systems. Kluwer Academic Publ., Netherlands, pp 415–426
Donnio B, Bruce DW (1999) Liquid crystal II: structure and bonding. Springer, New York, pp 193–247
Ohta K, Shibuya T, Ando M (2006) Flying-seed-like liquid crystals. J Mater Chem 16:3635–3639
Lee SJ, Han SW, Choi HJ, Kim K (2002) Structure and thermal behavior of a layered silver carboxylate. Phys Chem B 106:2892–2900
Klose G, Volke F, Hentschel M, Mops A (1983) Molecular motions and mesophases in anhydrous thallium (I) soaps studied by 1H- and 205TL-NMR. The 205TL chemical shift tensor. Mol Cryst Liq Cryst 90:245–254
Wilkes JS (2002) A short history of ionic liquids-from molten salts to neoteric solvents. Green Chem 4(2):73
Seddon KR (1998) Molten salt chemistry forum. In: Wendt H (ed) Proceedings of the 5th international conference on molten salt chemistry and technology, vol 53, pp 5–6
Welton T (1999) Room-temperature ionic liquids. Solvents for synthesis and catalysis. Chem Rev 99:2071
Kambe H, Ozawa T, Onoue M, Igarashi S (1962) Physicochemical studies on cobalt salts of higher fatty acids. VI. Some observations on thermal transitions of cobalt soaps, by differential thermal analysis, thermogravimetry, and magnetic measurement. Bull Chem Soc Jpn 35:81
Akanni MS, Burrows HD, Ellis HA, Asongwed DN, Babalola HB, Ojo PO (1984) Solution behavior of some divalent metal carboxylates in organic solvents. J Chem Technol Biotechnol 34A:127–135
Carrell CJ, Carrell HL, Erlebacher J, Glusker JP (1988) Structural aspects of metal ion carboxylate interactions. J Am Chem Soc 110:8651–8656
Akanni MS, Okoh EK, Burrows HD, Ellis HA (1992) The thermal behaviour of divalent and higher valent metal soaps: review. Thermochim Acta 208:1–41
Adeosun SO, Sime S (1976) Properties of molten carboxylates. Part 4: a quantitative differential thermal analysis study of melting and mesophase formation in some lead(II) carboxylates. J Thermochim Acta 17:351
Adeosun SO, Sime SJ (1978) The properties of molten carboxylates: part 7. Odd–even variations in melting and mesophase formation in the lead(III) carboxylates. Thermochim Acta 27:319
Ellis HA (1986) Thermotropic phase transitions in some lead(II) carboxylates. Mol Cryst Liq Cryst 139:281–290
Amorim da Costa AM, Burrows HD, Geraldes CFGC, Teixeira-Dias JJC, Bazium CG, Gullion D, Skoulios A, Blackmore E, Tiddy GJT (1986) Mesophase formation in lead(II) decanoate. Liq Cryst 1:215
Bazuin CG, Guillon D, Skoulios A, Amorim da Costa AM, Burrows HD, Geraldes CFGC, Teixeira-Dias JJC, Blackmore E, Tiddy GJT (1988) Thermotropic polymorphism in liquid-crystalline lead(II) alkanoates. Liq Cryst 3:1655
Ellis HA (1997) An X-ray and microscopic investigation of the thermal phases of lead(II) decanoate. Mol Cryst Liq Cryst 308:111–120
Martinez Casado FJ, Ramos Riesco M, Sanchez Arenas A, Garcia Perez MV, Redondo MI, Lopez-Andres S, Garrido L, Cheda JAR (2008) A novel rotator glass in lead(II) pentanoate: calorimetric and spectroscopic study. J Phys Chem B 112:16601–16609
Attard GS, Cullum PR (1990) Thermotropic mesomorphism in a branched chain copper(II) carboxylate and its pyrazine complex. Liq Cryst 8:299
Akopova OB, Shabyshev LS, Bobrov VI (1995) Synthesis, structure, and discotic mesomorphism of the new series of copper carboxylates. Russ Chem Bull 44(7):1210–1214
Taylor RA, Ellis HA (2009) Polymorphism, crystal–crystal and liquid crystalline thermotropic transition behaviour of even chain length zinc(II) n-alkanoates. Liq Cryst 36(3):257–268
Taylor RA, Ellis HA (2013) Zinc carboxylate liquid crystals: solid state structures and thermotropic phase transition behaviour of a homologous series of zinc(II) n-alkanoates. LAP Lambert Academic Publishing
Roberts K, Friberg S (1969) Phase transitions of adsorbed carboxylic acids on zinc oxide and of zinc soaps. Infrared and X-ray diffraction investigations. Kolloid Z Z Polym 230:357
Zou CF, Sahyun MRV, Levy B, Serpone N (1996) Mechanisms of latent image formation in photothermographic silver imaging media. J Imaging Sci Technol 40:94
Sahyun MRV (1998) Thermally developable photographic materials (TDPM): a review of the state-of-the-art in mechanistic understanding. J Imaging Sci Technol 42:23
Geuens I, Vanwelkenhuysen I (1999) Physical characterization of silver behenate as a tool for the development of thermographic and photothermographic materials. J Imaging Sci Technol 43:521
Zavlin PM, Batrakov AN, Velinzon PZ, Gaft SI, Kuznetsov LL (1999) Thermally developed photographic materials based on silver organic salts. J Imaging Sci Technol 43:540
Ellis HA (1981) Kinetics and reaction mechanism for the thermal decomposition of some even chain lead(II) carboxylates. Thermochim Acta 47:261–270
Ellis HA, Okoh EK (1982) Thermal decomposition kinetics of lead(II) decanoate. J Chem Soc Perkin Trans 2:1497–1501
Galwey AK, Jamiesson DM, Brown ME (1974) Thermal decomposition of three crystalline modifications of anhydrous copper(II) formate. J Phys Chem 78:2664
Reichert C, Fung DKC, Lin DCK, Westmore JB (1968) Thermal decomposition of copper(II) carboxylates: mass spectra of binuclear copper(I) carboxylates. Chem Commun 1094–1095. doi:10.1039/C19680001094
Piper SH (1929) An X-ray examination of some salts of the fatty acids. J Chem Soc 234–239. doi:10.1039/JR9290000234
Kovalenko VI, Gayfutdinova DN, Furer VL (2002) Ionic layers of potassium and sodium stearates at liquid-crystal transitions according to the data from infra-red spectroscopy. Physicochem Investig 2(6):37
Glazer J, McRoberts TS, Schulman JH (1950) The preparation of a stable aluminium dodecanoate (laurate) with no gelling properties in hydrocarbons. J Chem Soc 426:2082–2083
Smith GH, Pomeroy HH, McGee CG, Mysels KJ (1053) Preparation of aluminium di-soaps. J Am Chem Soc 1948:70
Scott FA, Goldenson J, Wiberley SE, Bauer WH (1954) Spectra of aluminium soaps and soap–hydrocarbon gels. J Phys Chem 58:61–64
Mehrotra RC, Pande KC (1956) Studies in aluminium soaps—I aluminium tri-soaps. J Inorg Nucl Chem 2:60–65
Mehrotra RC, Rai AK (1962) Studies in heavy metal soaps. Synthesis of chloride soaps of aluminium. J Indian Chem Soc 39:1–4
Wood JA, Seddon AB (1981) Identification of the chromium salt of stearic acid. Thermochim Acta 45:365–368
Eshel M, Bino A, Felner I, Johnston DC, Luban M, Miller LL (2000) Polynuclear chromium(III) carboxylates. 1. Synthesis, structure, and magnetic properties of an octanuclear complex with a ring structure. Inorg Chem 39:1376–1380
Eshel M, Bino A (2002) Polynuclear chromium(III) carboxylates. 3. Cyclic and cubane type hexachromium acetates. Inorg Chim Acta 329:45–50
Atkinson IM, Benelli C, Murrie M, Parsons S, Winpenny REP (1999) Turning up the heat: synthesis of octanuclear chromium(III) carboxylates. Chem Commun 285–286. doi:10.1039/A808304E
Rai AK, Parashar GK (1979) Thermogravimetric analysis of some higher carboxylate derivatives of chromium(III). Thermochim Acta 29:175–179
Misra SN, Misra TN, Mehrotra RC (1963) Organic salts of lanthanide elements—I. Preparation of tri-acylates of lanthanum and cerium(III) from aqueous solutions. J Inorg Nucl Chem 25:195–199
Misra SN, Misra TN, Mehrotra RC (1963) Organic salts of lanthanide elements—II: preparation of lanthanum and cerium(III) acylates from a non-aqueous medium. J Inorg Nucl Chem 25:201–203
Demus D, Richter L (1978) Textures of liquid crystals. Verlag Chemie, New York
De Vries A, Ekachai A, Spielberg N (1979) Why the molecules are tilted in all smectic a phases, and how the layer thickness can be used to measure orientational disorder. Mol Cryst Liq Cryst 49:143–452
Binnemans K, Görller-Walrand C (2002) Lanthanide-containing liquid crystals and surfactants. Chem Rev 102:2303–2345
Binnemans K (2005) Ionic liquid crystals. Chem Rev 105:4148–4204
Binnemans K, Jongen L, Bromant C, Hinz D, Meyer G (2000) Structure and mesomorphism of neodymium(III) alkanoates. Inorg Chem 39:5938–5945
Marques EF, Burrows HD, da Graca Miguel M (1998) The structure and thermal behaviour of some long chain cerium(III) carboxylates. J Chem Soc Faraday Trans 94(12):1729–1736
Taylor RA, Ellis HA, Maragh PT, White NAS (2006) The room temperature structures of anhydrous zinc(II) hexanoate and pentadecanoate. J Mol Struct 787:113–120
Taylor RA, Ellis HA (2007) Room temperature molecular and lattice structures of a homologous series of anhydrous zinc(II) n-alkanoate. Spectrochim Acta 68:99–107
Ellis HA, White NA, Hassan I, Ahmad R (2002) A room temperature structure for anhydrous lead(II) decanoate. J Mol Struct 642:71–76
Ellis HA, White NAS, Taylor RA, Maragh PT (2005) Infrared, X-ray and microscopic studies on the room temperature structure of anhydrous lead(II) n-alkanoates. J Mol Struct 738:205–210
Corkery RW (2004) A variation on Luzzati’s soap phases. Room temperature thermotropic liquid crystals. Phys Chem Chem Phys 6:1534–1546
Nakamoto K, Fujita J, Tanaka S, Kobayashi M (1957) Infrared spectra of metallic complexes. IV. Comparison of the infrared spectra of unidentate and bidentate metallic complexes. J Am Chem Soc 79:4904–4908
Ouchi A, Suzuki Y, Ohki Y, Koizumi Y (1988) Structure of rare earth carboxylates in dimeric and polymeric forms. Coord Chem Rev 92:29–43
Edwards DA, Hayward RN (1968) Transition metal acetates. Can J Chem 46:3443–3446
Cotton FA, Wilkinson G (1980) Advanced inorganic chemistry: a comprehensive text, 4th edn, vol 170. Wiley-Interscience, New York, pp 58–602
Nelson PN, Ellis HA (2012) Structural, odd–even chain alternation and thermal investigations of a homologous series of anhydrous silver(I) n-alkanoates. Dalton Trans 41:2632
Shoeb ZE, Hammad SM, Yousef AA (1999) Oleochemicals I: studies on the preparation and the structure of lithium soaps. Grasas Aceites 50:426–434
Ferloni P, Westrum EF Jr (1992) Thermodynamic properties of lithium n-alkanoates. Pure Appl Chem 64(1):73–78
Bui LH (2013) Alkali metal C1–C12 n-alkanoates. Masters of Science in Engineering Thesis, University of Alberta
Lewis ELV, Lomer TR (1969) The refinement of the crystal structure of potassium caprate (form A). Acta Cryst B25:702
Lomer TR (1952) The unit-cell dimensions of potassium soaps. Acta Cryst 5:11
White NAS, Ellis HA, Nelson PN, Maragh PT (2011) Thermal and odd–even behaviour in a homologous series of lithium n-alkanoates. J Chem Thermodyn 43:584–590
Casado FJM, Riesco MR, Perez MVG, Redondo MI, Lopez-Andres S, Cheda JAR (2009) Structural and thermodynamic study on short metal alkanoates: lithium propanoate and pentanoate. J Phys Chem B 113:2896–12902
Bremmer L, Burgard MD, Rodger CS, Swearingen JK, Rath NP (2001) Silver(I) carboxylates: versatile inorganic analogs of carboxylic acids for supramolecular network formation. Chem Commun 2468–2469. doi:10.1039/B108448H
Binnemans K, Deun RV, Thijs B, Vanwelkenhuysen I, Geuens I (2004) Structure and mesomorphism of silver alkanoates. Chem Mater 16(10):2021–2027
Tolochko BP, Chernov SV, Nikitenko SG, Whitcomb DR (1998) Nucl Instrum Methods Phys Res 405:428
Olson L, Whitcomb DR, Rajeswaran M, Blanton TN, Stwertka BJ (2006) The simple yet elusive crystal structure of silver acetate and the role of the Ag–Ag bond in the formation of silver nanoparticles during the thermally induced reduction of silver carboxylates. Chem Mater 18:1667–1674
Vold MJ, Funakoshi H, Vold RD (1976) The polymorphism of lithium palmitate. J Phys Chem 80:1753–1761
Lomer TR, Perera K (1974) Anhydrous copper(II) octanoate. Acta Cryst B30:2913–2915
Bird MJ, Lomer TR (1972) The crystal and molecular structure of anhydrous copper butyrate. Acta Cryst B28:242–246
Burrows H, Ellis HA (1982) The thermal behaviour and spectral properties of some long chain copper(II) carboxylates. Thermochim Acta 52:121–129
Berkesi O, Katona T, Dreveni I, Andor JA, Mink J (1995) Temperature-dependent Fourier transform infrared and differential scanning calorimetry studies of zinc carboxylates. Vib Spectrosc 8:167–174
Dreveni I, Berkesi O, Andor JA, Mink J (1996) Influence of the spatial structure of the alkyl chain on the composition of the product of the direct neutralization reaction between aliphatic carboxylic acids and zinc hydroxide. Inorg Chim Acta 249:17–23
Šegedin P, Lah N, Žefran M, Leban I, Golič L (1999) Synthesis and characterization of bis(carboxylato)zinc(II) (C6–C18)—crystal structure of bis(haxanoato)zinc(II) Zn(O2CC5H11)2—form A. Acta Chim Slov 46:173–184
Berkesi O, Dreveni I, Andor JA, Mink J (1992) FT-IR studies on the formation of tetrazinc long straight-chain (even-numbered C6–C18) μ4-oxo-hexa-μ-carboxylates from the corresponding bis(carboxylato)zinc compounds. Inorg Chim Acta 195:169–173
Peultier J, François M, Steinmetz J (1999) Anhydrous polymeric zinc(II) heptanoate. Acta Cryst C55:2064–2065
Lacouture F, Peultier J, François M, Steinmetz J (2000) Anhydrous polymeric zinc(II) octanoate. Acta Cryst C56:556–557
Clegg W, Little IR, Straughan BP (1987) Orthorhombic anhydrous zinc(II) propionate. Acta Cryst C43:456–457
Blair J, Howie RA, Wardell JL (1993) Structure of monoclinic zinc(II) n-butanoate. Acta Cryst C49:219–221
Clegg W, Little IR, Straughan BP (1986) Monoclinic anhydrous zinc(II) acetate. Acta Cryst C42:1701–1703
Ishioka T, Shibata Y, Takahashi M, Kanesaka I, Kitagawa Y, Nakamura KT (1998) Vibrational spectra and structures of zinc carboxylates I. Zinc acetate dehydrate. Spectrochim Acta Part A 54:1827–1836
Ishioka T, Maeda K, Watanabe I, Kawauchi S, Harada M (2000) Infrared and XAFS study on structure and transition behavior of zinc stearate. Spectrochim Acta Part A 56:1731–1737
Mesbah A, Juers C, Francois M, Ricca E, Steinmetz J (2007) Structures of magnesium and zinc long aliphatic chains carboxylates. Z Kristallogr Suppl 26:593–598
Nelson PN, Taylor RA, Ellis HA (2013) The effects of molecular and lattice structures on thermotropic phase behaviour of zinc(II) undecanoate and isomeric zinc(II) undecynoates. J Mol Struct 1034:75–83
Harrison PG, Steel AT (1982) Lead(II) carboxylate structures. J Organomet Chem 239:105
Mesubi MA (1961) J Mol Struct 11:61
Baxter DV, Cayton RH, Chisholm MH, Huffman JC, Putilina EF, Tagg SL, Wesemann JL, Zwanziger JW, Darrington FD (1994) Multiple bonds between metal atoms in ordered assemblies. 2. Quadrupole bonds in the mesomorphic state. J Am Chem Soc 116:4551–4566
Jongen L, Binnemans K, Hinz D, Meyer G (2001) Mesomorphic behaviour of praseodymium(III) alkanoates. Liq Cryst 28(6):819–825
Thalladi VR, Boese R (2000) Why is the melting point of propane the lowest among n-alkanes. New J Chem 24:579–581
Boese R, Weiss H-C, Bläser D (1999) The melting point alternation in the short-chain n-alkanes: single-crystal X-ray analyses of propane at 30 K and of n-butane to n-nonane at 90 K. Angew Chem Int Ed 38:988–992
Thalladi VR, Boese R, Weiss H-C (2000) The melting point alternation in α, ω-alkanedithiols. J Am Chem Soc 122:1186–1190
Polishchuk AP, Timofeeva TV (1993) Metal-containing liquid-crystal phases. Russ Chem Rev 62:291–321
Lawrence ASC (1938) The metal soaps and the gelation of their paraffin solutions. Trans Faraday Soc 34:660
Hattiangdi GS, Vold MJ, Vold RD (1949) Differential thermal analysis of metal soaps. Ind Eng Chem 41:2320
Koga Y, Matsuura R (1961) Mem Fac Sci Kyushu Univ Ser C 4:1
Koga Y, Matsuura R (1965) Chem Abstr 62:13391c
Skoda W (1969) Dilatometric investigation of phase transitions and structures of anhydrous soaps. Kolloid-Z Polym 234:1128
Gallot B, Skoulios A (1966) Structure des savons de rubidium. Mol Cryst 1:263
White NAS, Ellis HA (2009) Thermal behaviour of even chain length lithium n-alkanoates. Mol Cryst Liq Cryst 501:28–42
Gallot B, Skoulios A (1966) Structure des savons alcalins. Kolloid-Z Polym 209:164
Seurin P, Guillon D (1981) Skoulios, Smectogènes Dissymétriques III. Synthèse et Propriétés Mésomorphes des p, n-Alkoxybenzydèneanilines Parasubstituées, A. Mol Cryst Liq Cryst 65:85
Luzzati V (1968) Biological membranes. In: Chapman D (ed) Academic Press, New York, p 71
Skoulios A, Luzzati V (1961) La structure des colloïdes d’association. III. Description des phases mésomorphes des savons de sodium purs, rencontrées au-dessus de 100 °C. Acta Cryst 14:278
Benton DP, Howe PG, Farnard R, Puddington IE (1955) The mesomorphic behaviour of anhydrous soaps: part II. Densities of alkali metal stearates. Can J Chem 33:1798
Vold RD (1941) Anhydrous sodium soaps. Heats of transition and classification of the phases. J Am Chem Soc 63:2915
Vold RD, Vold MJ (1939) Successive phases in the transformation of anhydrous sodium palmitate from crystal to liquid. J Am Chem Soc 61:808
Sokolov NM (1954) Zh Obshch Khim 24:1150
Baum E, Demus D, Sackmann H (1970) Wiss Z Univ Halle 19:37
Ubbelohde AR, Michels HJ, Duruz JJ (1970) Liquid crystals in molten salt systems. Nature 228:50
Ferlani P, Sanesi M, Franzosini PZ (1975) Naturforsch Teil A 30:1447
Storonkin AV, Vasil’kova IV, Vestn SS (1974) Leningr Univ Fiz Khim 10:84
Cingolani A, Spinolo G, Sanesi MZ (1979) Naturforsch Teil A 34:575
Roth J, Meisel T, Seybold K, Halmos Z (1976) Investigation of the thermal behaviour of fatty acid sodium salts. J Therm Anal 10:223
Busico V, Ferraro A, Vacatello M (1985) Thirmotropic smectic liquid crystals of ionic amphiphilic compounds: a general discussion. Mol Cryst Liq Cryst 128:243–261
Duruz JJ, Michels HJ, Ubbelohde AR (1971) Molten fatty acid salts as model ionic liquids. I. Thermodynamic and transport parameters of some organic sodium salts. Proc R Soc Lond A 322:1550
Franzosini P, Westrum EF Jr (1984) Thermophysics of metal alkanoates IV. Heat capacities and thermodynamic properties of potassium 2-methylpropanoate. J Chem Thermodyn 16:127–130
Tse WS, Chiang PY, Lin SJ (1986) A Raman spectral study of the phase transitions in crystalline potassium acetate. Chin J Phys 24(1):63–68
Bolduan OEA, McBain JW, Ross S (1943) Diffraction of X-rays by sodium laurate and sodium palmitate at higher temperatures. J Phys Chem 47:528
Aleixo AI, Oliveira PH, Diogo HP, da Piedade MEM (2005) Enthalpies of formation and lattice enthalpies of alkaline metal acetates. Thermochim Acta 428:131–136
Johnson DA (1982) Some thermodynamic aspects of inorganic chemistry, 2nd edn. Cambridge University Press, Cambridge, pp 29–81
Dasent WE (1982) Inorganic energetics, 2nd edn. Cambridge University Press, Cambridge (chapter 3)
Jenkins HDB, Roobottom HK (2002). In: Lide DR (ed) CRC handbook of chemistry and physics, 83rd edn. CRC Press, Boca Raton, pp 22–36 (section 12)
Boerio-Goates J, de Lopez La Fuente FL, Cheda JAR, Westrum EF Jr (1985) Thermodynamics of thallium alkanoates I. Heat capacity and thermodynamic functions of thallium(I) n-hexanoate. J Chem Thermodyn 17:401–408
Fernandez-Martin F, Lopez de La Fuente FL, Cheda JAR (1984) DSC thermophysics on some lower thallium(I) N-alkanoates. Thermochim Acta 73:109
Ngeyi SP, de Lopez La Fuente FL, Cheda JAR, Fernandez-Martin F, Westrum EF Jr (1985) Thermodynamics of thallium alkanoates II. Heat capacity and thermodynamic functions of thallium(I) n-heptanoate. J Chem Thermodyn 17:409
Ngeyi SP, de Lopez La Fuente FL, Cheda JAR, Fernandez-Martin F, Westrum EF Jr (1987) Thermodynamics of thallium alkanoates III. Heat capacity and thermodynamic functions of thallium(I) n-tetradecanoate from 7 to 450 K. J Chem Thermodyn 19:327
de Lopez La Fuente FL, Westrum EF Jr, Cheda JAR, Fernandez-Martin F (1987) Thermophysics of thallium alkanoates IV. Heat capacity and thermodynamic functions of thallium(I) n-dodecanoate from 7 to 470 K. J Chem Thermodyn 19:1261
Meisel T, Seybold K, Halmos Z, Roth J, Melykuti C (1976) Thermal behaviour or thallium(I) fatty acid salts, I. J Therm Anal 10:419
Lindau J, Diele S, Kruger H, Dorfler HD (1981) Phys Chem 262:775
de Lopez La Fuente FL, Cheda JAR, Fernandez-Martin F, Westrum EF (1988) Thermodynamics of thallium alkanoates V. Heat capacity and thermodynamic functions of thallium(I) n-decanoate from 6 to 480 K. J Chem Thermodyn 20:1137–1148
Peltzl G, Sackmann H (1971) Birefringene of smectic modifications of the homologous thallium soaps. Mol Liq Liq Cryst 15:75
Fernández-García M, García MV, Redondo MI, Cheda JAR, Fernandez-García MA, Westrum EF Jr, Fernandez-Martin F (1997) Molecular association of normal alkanoic acids with their thallium(I) salts: a new homologous series of fatty acid metal soaps. J Lipid Res 38:361
Fernández-García M, Cheda JAR, Westrum EF Jr, Fernández-Martin F (1997) Interactions in the thallium(I) heptanoate and heptanoic acid system: association, aggregation, and phase behavior. J Colloid Interface Sci 185:371
Cheda JAR, Fernandez-García M, Ugarelli P, Ferloni P, Fernández-Martin F (2000) Phase behavior of the n-decanoic acid + thallium(I) n-decanoate system. Langmuir 16:5825
Andreev VM, Burleva LP, Boldyrev VV (1984) Izv Sib Otd Akad Nauk SSSR Ser Khim Nauk 5:3
Andreev VM, Burleva LP, Boldyrev VV, Mikhailov YuI (1983) Izv Sib Otd Akad Nauk SSSR Ser Khim Nauk 4:58
Uvarov NF, Burleva NF, Mizen MB, Whitcomb DR, Zou C (1998) Solid State Ion 107:31
Bokhonov BB, Burleva LP, Whitcomb DR, Usanov YE (2001) Formation of nano-sized silver particles during thermal and photochemical decomposition of silver carboxylates. J Imaging Sci Technol 45:259
Iwig F, Hecht O (1886) Ber 19:238
Judd MD, Plunkett BA, Pope MI (1974) The thermal decomposition of calcium, sodium, silver and copper(II) acetates. J Therm Anal 6:555
Burrows HD, Geraldes CFGC, Pinheiro TJT, Harris RK, Sebald A (1988) 207Pb NMR of lead(II) soaps in solid, liquid-crystalline and liquid phases. Liq Cryst 3:853
Adeosun SO, Kehinde AO, Odesola GA (1979) Electrical conductance, density, viscosity and thermal behaviour of lead(II) and zinc(II) salts of some unsaturated fatty acids. Thermochim Acta 28:133
Attard GS, West YD (1990) Lead(II) alkadiynoates. A novel class of liquid-crystalline precursors to poly(diacetylenes). Liq Cryst 7:487
Baraldi P (1981) Thermal behavior of metal carboxylates—II. Lead formate. Spectrochim Acta 37A:99
Freeman ES, Caroll B (1958) The application of thermoanalytical techniques to reaction kinetics: the thermogravimetric evaluation of the kinetics of the decomposition of calcium oxalate monohydrate. J Phys Chem 62:394
Rasheed A, Bhobe RA (1976) J Indian Chem Soc LIII:442
Anderson DA, Freeman ES (1961) The kinetics of the thermal degradation of polystyrene and polyethylene. J Polym Sci 54:253
Abied H, Guillon D, Skoulios A, Weber P, Giroud-Godquinn AM, Marchon JC (1987) Investigation of the structures of the crystalline and columnar phases of linear chain copper(II) alkanoates. Liq Cryst 2:269
Marchon JC, Maldivi P, Giroud-Godquin AM, Guillon D, Skoulios A, Strommen DP (1990) Columnar liquid crystals derived from long-chain carboxylates of transition metal ions [and discussion]. Philos Trans R Soc Lond Ser A 330:109
Giroud-Godquin AM, Marchon JC, Guillon A, Skoulios A (1984) Discotic mesophases of dirhodium tetracarboxylates. J Phys Lett 45:L681
Takekoshi M, Watanabe N, Tamamaushi B (1975) Copper stearate as a thermotropic liquid crystal. Colloid Polym Sci 256:588
Abied H, Guillon D, Skoulios A, Giroud-Godquin AM, Maldivi P, Marchon JC (1988) Dilatometric investigation of thermotropic linear chain copper(II) alkanoates. Colloid Polym Sci 266:579
Strommen DP, Giroud-Godquin AM, Maldivi P, Marchon JC, Marchon B (1987) Vibrational studies of some dicopper tetracarboxylates which exhibit a thermotropic columnar mesophase. Liq Cryst 2:689
Giroud-Godquin AM, Latour JM, Marchon JC (1985) Magnetic susceptibility as a probe of the solid-discotic phase transition in binuclear copper(II) n-alkanoates. Inorg Chem 24:4452
Abied H, Guillon D, Skoulios A, Dexpert H, Giroud-Godquin AM, Marchon JC (1988) J Phys Fr 49:345
Grant RF (1964) Proton magnetic resonance absorption in copper(II) stearate. Can J Chem 42:951
Ramos Riesco M, Martinez Casado FJ, Lopez-Andres S, Garcia Perez MV, Redondo Yelamos MI, Torres MR, Garrido L, Cheda JAR (2008) Monotropic polymorphism in copper(II) decanoate. Cryst Growth Des 8(7):2547–2554
Busico V, Cernichiaro P, Corradini P, Valcatello M (1983) Polymorphism in anhydrous amphiphilic systems. Long-chain primary n-alkylammonium chlorides. J Phys Chem 87:1631–1635
Busico V, Ferraro A, Valcatello M (1984) J Phys Chem 88:4055–4058
Van My L, Perinet G (1965) Bull Soc Chim Fr 1379
Akanni MS, Ajayi OB, Lambi JN (1986) Pyrolytic decomposition of some even chain length copper (II) carboxylates. J Therm Anal 31:131
Konkoly-Thege I, Ruff I, Adeosun SO, Sime SJ (1978) Properties of molten carboxylates part 6. A quantitative differential thermal analysis study of phase transitions in some zinc and cadmium carboxylates. Thermochim Acta 24:89
Mesubi MA (1982) An infrared study of zinc, cadmium, and lead salts of some fatty acids. J Mol Struct 81:61
Wood JA, Seddon AB (1985) The solubility of zinc dodecanoate in xylene and tetrachloromethane. Colloid Polym Sci 263:600
Dollimore D, Tonge KH (1967) The thermal decomposition of zinc and manganous formats. J Inorg Nucl Chem 29:621
Rasheed A, Bhobe RA (1976) J Indian Chem Soc 53:442
Odilora CA (1989) On the thermal degradation of poly(vinyl chloride). Synergistic effect of mixtures of lead and zinc carboxylates. Acta Polym 40:541
Vold RD, Hattiangdi GS (1949) Characterization of Heavy Metal Soaps by X-Ray Diffraction. Ind Eng Chem 41:2311
Ellis HA (1986) Thermal Behaviour of Mercury(II) Carboxylates. Mol Cryst Liq Cryst 138:321
Adeosun SO (1978) A differential thermal analysis study of phase transitions in some mercury(II) carboxylates. J Therm Anal 14:235
Akanni MS, Borrows HD, Begun PB (1984) Product analysis, reaction mechanism and kinetics of the thermal decomposition of some even chain-length mercury(II) carboxylates. Thermochim Acta 81:45
Spegt PA, Skoulios AE (1963) La structure des colloïdes d’association. VIII. Description de la structure des savons de cadmium à température élevée. Acta Crystallogr 16:301
Prasad S, Ranganathchar T, Krishna V (1985) J Indian Chem Soc 35:267
Mirnaya TA, Sudovtsova LS, Yaremchuk GG, Tolochko AS, Lisetskii LN (2004) Russ J Inorg Chem 49:1440
Yakerson VI (1967) Izv Akad Nauk SSSR Otd Khim Nauk 1003
Wilcox DE, Bromley LA (1963) Computer estimation of heat and free energy of formation for simple inorganic compounds. Ind Eng Chem 55:32
Methrotra KN, Sharma N (1996) Studies on molar volume and rheology of terbium soaps in a benzene-DMF mixture. Monatsh Chem 127:257
Upadhyaya SK, Sharma PSJ (1993) Indian Chem Soc 70:735
Binnemans K, Jongen L, Görller-Walrand C, D’Olieslanger W, Hinz D, Meyer G (2000) Lanthanide(III) dodecanoates: structure, thermal behaviour, and ion-size effects on the mesomorphism. Eur J Inorg Chem 2000(7):1429–1436
Li H, Bu W, Qi W, Wu L (2005) Self-assembled multibilayers of europium alkanoates: structure, photophysics, and mesomorphic behavior. J Phys Chem B 109:21669–21676
Nagle JF (1980) Theory of the main lipid bilayer phase transition. Annu Rev Phys Chem 2:157
Binnemans K, Heinrich B, Guillon D, Bruce DW (1999) On the mesomorphism of lanthanum(III) alkanoates. Liq Cryst 26(11):1717–1721
Giroud-Godquin AM (1998) My 20 years of research in the chemistry of metal containing liquid crystals. Coord Chem Rev 178–180:1485–1499
Cukiernik FD, Ibn-Elhaj M, Chaia ZD, Marchon J-D, Giroud-Godquin A-M, Guillon D, Skoulios A, Maldavi P (1998) Mixed-valent diruthenium (II, III) long-chain carboxylates. 1. Molecular design of columnar liquid-crystalline order. Chem Mater 10:83–91
Chisholm MH, Wilson PJ, Woodward PM (2002) Intermolecular recognition and crystal packing in molybdenum and tungsten coordination polymers as deduced from powder X-ray diffraction data. Chem Commun 566–567. doi:10.1039/B200362G
Montmitonnet P, Monasse B, Haudin JM, Delamore F (1985) Calorimetric and microscopic study of barium stearate. Mater Lett 3:98
Spegt P, Skoulios A (1970) Acad C R Sci Paris 251:2199
Spegt P, Skoulios A (1962) Acad C R Sci Paris 254:4316
Spegt PA, Skoulios AE (1964) La structure des colloïdes d’association. X. Description de la structure des savons de calcium à température ordinaire et à température élevée. Acta Crystallogr 17:198
Spegt P, Skoulios A (1965) J Chim Phys 62:377
Spegt P, Skoulios A (1965) J Chim Phys 62:418
Spegt PA, Skoulios AE (1966) Structure des savons de strontium en fonction de la température. Acta Crystallogr 21:892
Winsor PA (1975) Liquid crystals and plastic crystals, vol 1, chapter 5. Ellis Harwood, Chichester
Reichmuth G, Dubler E (1985) Synthetic and biogenic calcium fatty acid salts: stoichiometry and phase transition behavior. Thermochim Acta 89:485
Montmitonnet P, Delamore F (1982) Determination of the plastic and viscoelastic properties of a metallic soap by a hot hardness measurement. J Mater Sci 17:121
Shiba S (1961) Differential thermal analysis of aluminium soaps. Bull Chem Soc Jpn 34:804
Adeosun SO (1979) Electrical conductance and thermal behaviour of some manganese(II) carboxylates. Can J Chem 57:151
Ghosh SK, Pathak GK, Chaudhuri AK (1978) Indian J Chem 16A:670
Giroud-Godquin AM, Marchon JC, Guillon D, Skoulios A (1986) Discotic mesophases of dirhodium tetracarboxylates. J Phys Chem 90:5502
Poizat O, Strommen DP, Maldivi P, Giroud-Godquin AM, Marchon JC (1990) Raman spectroscopy: a convenient probe of solid-liquid crystal-phase transitions in binuclear rhodium(II) n-alkanoates. Inorg Chem 29:4851
Maldivi P, Giroud-Godquin AM, Marchon JC, Guillon D, Skoulios A (1989) Diruthenium(II, II) tetra-μ-alkylcarboxylates: magnetic susceptibility studies of their electronic configuration and thermotropic liquid crystalline mesophase. Chem Phys Lett 157:552
Hagino K (1964) Kogyo Kagaku Zasshi 67:597
Adeosun SO, Ellis HA (1979) Binary phase diagrams of some bivalent metal carboxylate systems. Thermochim Acta 28:313–321
Jongen L, Hinz D, Meyer G, Binnemans K (2001) Induced mesophases in binary mixtures of lanthanide(III) dodecanoates. Chem Mater 13:2243–2246
Gray GW (1962) Molecular structure and the properties of liquid crystals. Academic Press, New York, p 314
Martı´nez Casado FJ, Ramos Riesco M, da Silva I, Labrador A, Redondo MI, Garcı´a Pe´rez MV, Lo´pez-Andre´s S, Rodrı´guez Cheda JA (2010) Thermal and structural study of the crystal phases and mesophases in the lithium and thallium(I) propanoates and pentanoates binary systems: formation of mixed salts and stabilization of the ionic liquid crystal phase. J Phys Chem B 114:10075–10085
Mirnaya TA, Polishchuk AP, Bereznitski YV, Ferloni P (1996) Phase diagram of the binary system of barium and sodium n-butanoates. J Chem Eng Data 41(6):1337
Ellis HA (1988) Thermal behaviour of the system: lead(II) decanoate/lead(II) octadecanoate. Thermochim Acta 130:281
Burrows HD, Geraldes CFGC, Miguel MGM, Pinheiro TJT, Cruz Pinto JJC (1992) The phase behaviour of some mixtures of lead(II) carboxylates with phosphatidylcholines. Thermochim Acta 206:203
Adeosun SO, Sime WJ, Sime SJ (1977) Properties of molten carboxylates part 5. A quantitative differential thermal analysis study of mesophase formation in the systems lead(II)dodecanoate/lead(II) oxide and lead(II)dodecanoate/hendecane. Thermochim Acta 19:275
Adeosun SO (1978) Thermal behaviour of the system lead(II) dodecanoate/lead acetate. Thermochim Acta 25:333
Adeosun SO, Akanni MS (1978) Differential thermal analysis study of the system lead(II) dodecanoate/dodecanoic acid. Thermochim Acta 27:133
O’Regan B, Grfitzeli M (1991) A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 353:737
Wang P, Zakeeruddin SM, Moser J-E, Grfitzeli M (2003) A new ionic liquid electrolyte enhances the conversion efficiency of dye-sensitized solar cells. J Phys Chem B 107:13280
Conflict of interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Nelson, P.N., Taylor, R.A. Theories and experimental investigations of the structural and thermotropic mesomorphic phase behaviors of metal carboxylates. Appl Petrochem Res 4, 253–285 (2014). https://doi.org/10.1007/s13203-014-0044-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-014-0044-3