
Abstract
As water approaches the critical point, the dielectric constant lowers enabling solution of heavy oil components. Simultaneously, enhanced water dissociation enhances cracking to lighter fractions. We evaluated this effect for three heavy oils concentrating on the effect of processing on feed viscosity. Reductions of up to 90 % were observed and these could be achieved in a few hours. Although more applicable to surface processing of oil sands in the short term, ultimately this technique could also be applied to immature oils for in situ processing and recovery. This may already be happening in some steam-assisted gravity drainage scenarios. In situ application would be limited to reservoirs where formation pressure is higher than the pressure required to generate these subcritical effects.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The use of hot water is well established in thermal/steam oil recovery for high viscosity oils (Gray 1994). This can take the form of cyclic steam soaks, steam drive, well bore heating or—for thicker hydrocarbon-bearing regions, steam-assisted gravity drainage (SAGD). The main purpose of the heating is to mobilise the hydrocarbons by heat-induced temporary reduction of the viscosity or by a form of stripping to remove the lighter fractions. The same is true for oil sands, although this is usually carried out as a kind of surface mining operation with intensive treatment in site processing plants. Similar considerations apply to systems for oil shales where mining, crushing, ash-forming mineral removal, but most importantly the liberating heating (or “retorting”) process are applied.
These hot treatments can also pyrolytically crack the long molecular structures. This is particularly the case if water is added in which case better quality oil yields are found. The heating methods—including those for the bituminous oils above—can be separated into four types based on the heating medium: hot combustion gases, hot solids, radiant heating and hot fluids. We are here concerned with the use of hot water—and in particular water at conditions approaching the critical point (Tp = 374 °C, pc = 220 bar). Clay catalysed reactions at high temperatures in water on representative large molecules were reported (Su et al. 2004). Previous work has also identified possibilities for the components of kerogenic oils typified by clusters of two or three aromatic rings connected by linkages (Siskin and Katritzky 1991; Deng et al. 2012). Of course, the possibility of near critical water treatment is also applicable to the heavy ends of matured crude oils to which we referred at the beginning.
The object of the current work is to chemically augment the pyrolysis of heavy components from heavy oil, oil sands and kerogen in oil shales in order to generate crude oil components. For obtaining these effects, we use two effects of near critical water on organic components. These are the ability of water to dissolve organic molecules as the critical point is approached as well as increased cracking potential. This is reviewed in "Background" of this paper where we also describe the optimum behaviour for estimating where acid/base cracking would be optimal. In "Experimental", we describe our experimental apparatus, the hydrocarbons studied and our criteria for ascertaining cracking performance. "Results" describes the results with a conclusion geared towards practical in situ application.
Background
The main effect of heating on hydrocarbons when mixed with very hot water will be pyrolysis as in for example, the processes of steam or thermal cracking. The severity of these reactions—i.e. to what extent the long molecules are broken down—depends on both the temperature and the time duration of exposure to the elevated temperature (Golombok et al. 2001; Nowak and Gunschel 1983). The effectiveness of heat transfer to the hydrocarbon to crack it, is restricted by the miscibility. This is one of the specific interesting properties to be considered, i.e. the miscibility of water and hydrocarbons when approaching the critical point of the former. As the pressure and temperature increase, the spatial ordering between water molecules decreases because of the increase in kinetic energy and the dielectric constant of water fall enabling solution of organic components which it could not do at standard temperature and pressure (Peterson et al. 2008). There are two regimes of interest: the subcritical regime where heavier liquid organics are soluble and the supercritical regime where methane is soluble—we are here interested in the former. For the purpose of pure solubilisation, we would want to be near the critical temperature of 374 °C although the pressure needs to be sufficient to keep the water in the liquid state, i.e. above the vapour pressure. With these parameters in mind, by analogy with oil sands, we would envision an initial application as a surface processing facility in reactors akin to these used on tar sands. The pressures in particular are envisioned to be only 10–20 % below the critical pressure of water and this typically is 2 km of hydrostatic depth. The process would thus not be suitable for in situ application in shallower deposits. Not only the intermolecular polar effects but microscopic intramolecular behaviour of water is altered under near critical conditions. The dipolar nature of water implies a charge separation on the molecules and this itself is associated with the dissociation of water
It is well known that the dissociation constant corresponding to Kw = [H+][OH−] has a value Kw = 10−14 under normal ambient conditions. As temperature increases, KW also increases and can reach values typically 100–1,000 times higher than that associated with water under standard temperature and pressure (Peterson et al. 2008). These augmented values of Kw correspond to increased concentrations of H+ and OH−—effectively the solution is simultaneously more acidic and more basic, or more logically—the dissociated species exist simultaneously in equilibrium. This presents the possibility of initiating acid and base type cracking reactions simultaneously.
Using subcritical water to mobilise heavy oils requires a balance to be found between (1) dissolving the heavy oil component within this novel medium (determined by dielectric constant ε) and (2) cracking it simultaneously by using the enhanced [H+] and [OH−], which are present. Although the highest temperature gives the best values of ε for ensuring high oil solubility in water, the best values for acid/base cracking would be somewhat below these temperatures at around 290 °C. At 290 °C, the dielectric constant of water already matches that of acetone. This is sufficiently low to dissolve a substantial component of the heavy oil (HO) while at the same time high enough to stop dissolved salts precipitating out (Kumar and Gupta 2009). If there is simultaneously removal of hydrocarbon components heavy oil (HO) via a cracking mechanism to a light oil (LO) then we can consider the reaction to be
The subscripts refer, respectively, to the hydrocarbon components present as pure water-insoluble liquid as found at ambient conditions (l), or dissolved in water under subcritical conditions (aq). [H+] and [OH−] are the concentrations of hydrogen and hydroxide ions arising from dissociation of water as in Eq. 1. In general, heavy organics (containing polyaromatics) will be more soluble than light cracked components. Thus, the light components will form separate phases from water more easily. As the more soluble heavy oils are cracked, then more heavy oil dissolves. Thus, it is not necessary to operate at p, T levels where the dielectric constants are at the low level of pentane solubility. Operation at a substantially subcritical “acetone” level is sufficient. This is important for maximising water dissociation.
It could be that many of the “mobilisation” effects with steam injection observed to date have made unwitting use of these effects but in a non-optimal fashion. For example, Imperial Oil reports a steam injection scenario, whereby after initial back production of mainly water, the bitumen production is characterised by relatively constant water oil ratio (Batycky et al. 1997). They reported injection at 100 bar corresponding to around 310 °C. Thus, subcritical dissolution may explain the creation of “bound” water, which becomes emulsion during production. At any rate, the aim here is to explore (1) how near-critical enhanced miscibility increases the cracking potential for heavy hydrocarbons (2) can acid and base catalysis from the greater availability of H+ and OH− at near critical conditions affect the process?
The cracking process we are aiming at thus has a similar object to the thermal cracking process used on the remnants of crude oil left over after vacuum and atmospheric distillation—so-called short and long residues. The process aim there is also to reduce the viscosities of the heavy components—hence the name viscosity or vis-breaking. (This avoids having to cut the residues with more valuable gas oil to reduce viscosity.) The only purpose of water in the vis-breaking process is to limit excessive coke formation by increasing turbulence to reduce the temperature at hot surfaces in the reactor. The regime of operation is far below that associated with the increased solubility in which we are interested.
Peterson et al. (2008) have given an extensive review of biofuel production in near critical water. In the early 80s, Shell looked at subcritical fluid injection because of its enhanced solubility possibilities. Although it was recognised that for the pressure effects, considerable depth would be required which ruled out in situ upgrading for the Canadian oil sands, the possibilities for subcritical enhanced cracking were not recognised (Offeringa et al. 1981). The work of Kumar and Gupta (2009) clearly reflects the more recent upsurge of interest for biomass processing in this medium. There have been studies oriented towards heavy oils: Sinag (2004) processed an oil shale using both sub and supercritical water—although he was aiming more at a combined solvent and distillation process rather than cracking. He found that conversion was optimised at precisely the critical temperature. Ogunsola and Berkowitz (1995) describe processing oil shales with “sub” critical water, however, this appears to refer only to pressures being subcritical—temperatures were all in excess of 400 °C so that there was no liquid/liquid mixing possible nor any beneficial potential from the generation of extra hydrogen or hydroxide ions identified above.
Kamimura et al. (1998) makes the connection with SAGD but then proceeds to do a study in the supercritical regime (>400 °C). In some studies the observed improvements are purely related to supercritical temperatures being above 374 °C and thus in excess of the cracking threshold of 340 °C—the effective limit for distillation for example. This problem is explicitly admitted in a much later paper from the same group (Kishita et al. 2009) using what appears to be the same equipment. Here the authors are again concentrating on the supercritical regime but show that this along with alkali inhibits cracking. The most important observation was that viscosity increases with reaction pressure. This is a clear indication that the authors are observing pure traditional thermal cracking because pressure inhibits pyrolysis. Finally there have been quite recent attempts to extract oil both from bitumen and oil shales (Park and Son 2011; Deng et al. 2011). These concentrated on the chemical composition of the products rather than the improved mobility and process itself but nonetheless demonstrate the extension of previous work to kerogenic compounds.
Experimental
Apparatus
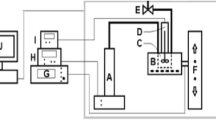
Experiments were carried out in a 200 ml. Hastelloy C Premex high pressure/high temperature autoclave. Figure 1 shows a schematic of the set-up. Typically 20 ml of the heavy oil sample is placed in the autoclave along with 180 ml demineralised water. Because of the relatively extreme conditions (pressures up to ca. 200 bar, and temperatures up to ca. 350 °C), a metal sealing rather than o-rings is used between the lid and autoclave. After closing the top the system is pressurised to 50 bar. To avoid a (too) high pressure, a back pressure regulator (which is used during heating) is installed for pressure regulation. Positive excursions in pressure are controlled by the constant pressure regulator valve. Such sudden increases can arise from rapid boiling of more volatile oil components or from the formation of lighter cracked components. An All Control temperature system heats and regulates the temperature of the autoclave. A 500 ml Isco pump ensures that sufficient pressure maintains the liquid state as the temperature increases (Fig. 1) and to keep it that way during an experimental run. Both Isco pump and autoclave have bursting discs set at 230 bar. A Premex suction magnetically driven stirrer is used to ensure mixing during heating of the two phases.

Schematic of setup for testing of effects of subcritical water on different types of heavy oil component. A Isco pump; B electric heating mantle, C Premex autoclave; D magnetic stirrer; E pressure control
Analyses
The basic property which steam heating aims at is reduction of the viscosity at higher temperatures. In our case the properties of the feed and products were determined by two tests. Viscosity was measured on an Anton Paar Physica MCR 301 Couette type bob and cup viscometer. Most viscosities were measured at 25 °C in a Couette cell. With some of the Marmul samples, on some occasions the annulus between the bob and cup was not properly filled. In that case measurements were then also carried out at 50 °C. We show below that the relative changes in viscosity between feed and product are, to first order, temperature independent.
The other main test carried out was the standard ASTM D2887 simulated distillation curve. Thermogravimetric analysis and differential scanning calorimetry (DSC) were used to more accurately identify changes to components in certain boiling ranges as a result of the reactions in near critical water. This was carried out on a Mettler instrument.
Feeds
The traditional definition of heavy oil is with a viscosity range 10–10,000 mPa s. Oils above this range are normally classed as bitumen (Strausz 1989). For demonstration purposes, the crudes were selected for being in the light range of “heavy oil”. As was the case for the SARA analyses reported in Table 1, the oil samples used were “dead”, i.e. gas-free. Table 2 shows the parameters associated with two heating properties: the distillation curve (ASTM D2887—see below) and viscosity measurement. The viscosities range from 20 to 760 mPa s. The effect on these are the main way of assessing successful processing to lighter components. Table 3 shows the associated reservoir properties.
Note that these oils do not count as particularly heavy ones and are selected for demonstration purposes only since they are in the light range of “heavy oil”. The techniques under research reported here are thus applied to oil from already producing reservoirs as an illustration and are not intended for deployment in either surface facility or by subsurface injection.
Procedure
A typical experiment was carried out by loading a total sample volume of ca. 200 ml liquid in the reactor. For base run pyrolysis tests this would be pure hydrocarbon. For the effect of water, either equal volumes of oil and water were used (i.e. 100 + 100 ml) or excess water, i.e. 20 ml of oil and 180 ml of water. Runs of varying duration were carried out. Sudden pressure fluctuations arising from flashing or cracking are controlled by the constant pressure valve which release pressure if it exceeds the set-pressure. After cooling the reactor the oil/water mixture was centrifuged for 30 min at 30,000 rpm in order to extract the oil phase.
Results
Viscosity
The primary interest for subcritical processing described here is to reduce the viscosity. For surface processing of oil sands, this improves transportability in pipeline flow. In a more distant projected application in the subsurface (either for bituminous or for kerogenic oils where the rock permeability permits), viscosity reduction improves mobility for subsequent oil displacement. Existing thermal methods such as SAGD and “huff and puff” already use thermal reduction of viscosity. In these cases, the viscosity improvement only applies as long as heating is maintained. Cooling viscosity goes up again as there is no change in the composition of the oil. In contrast, the method under discussion here also induces a change of the composition towards cracked lighter fractions with a permanent lowered viscosity even when heating is no longer applied.
In both cases, the processing time for conversion needs to be short to keep firing costs to a minimum. For our experiment this translates into the time needed for a permanent (i.e. not just temperature induced) viscosity reduction. Figure 2 compares the effect of identical runs for 20 and 88 h of processing on the Marmul oil. The temperature was 330 °C and pressure 150 bar with a 9:1 water oil ratio. Most of the improvement is obtained in 20 h. Further subcritical processing beyond this time does not produce much improvement. Similar results were obtained for the Bonga feed with similar oil/water ratios. For example, 3 h of processing at 350 °C and 200 bar, produced the same 40 % viscosity reduction as 40 h at 338 °C and 155 bar. The values cannot be directly compared for different oils because the effects are influenced by the proximity of operation conditions to the critical values—see below for a quantification of this. In addition, there are different base oil starting viscosities although we present a formalism below for normalising these effects enabling direct comparison.

Behaviour of viscosity as a function of run time for Marmul feed processed at 150 bar in water at 330 °C
The next step is to compare operation without and with water. We thus measure the permanent pyrolytic vis-breaking effect of temperature with no water present and compare that to the effect of operating with water near the critical regime. The base case (pyrolysis with no water) is shown in Fig. 3, where Marmul feed is simply heated to high temperatures. From Fig. 3, we see that the viscosity was reduced by water-free pyrolysis to 150 mPa s at 50 °C compared to the feed processed in subcritical water which had a viscosity 104 mPa s at 25 °C, i.e. a lower viscosity at a lower measurement temperature showing increased mobility. Therefore, the water can be presumed to have played a role in the viscosity reduction. We non-dimensionalise these effects below to show the temperature independence of induced viscosity changes—see Fig. 5 below. We note that this is not classical “steam cracking” where water is present as vapour, as in this case the pressure is sufficiently high to maintain water in its liquid state.

Viscosity of feed and pyrolysed (no water) Marmul feed at two temperatures; p = 150 bar; run time = 24 h
Experiments on the other crudes also show that mere pyrolysis with no water present did not achieve the viscosity reduction associated with cracking in water. In general, the largest benefits were associated with the most viscous oil feeds. The relevance of the approach to the subcritical condition is shown in two sets of measurements on another oil—a Brunei crude. Figure 4a shows the effect of increasing pressure at a temperature of 315 °C and Fig. 4b shows the effect of increasing the temperature at constant pressure of 150 bar. The critical pressure and temperature levels are marked on the corresponding figures. At constant temperature (Fig. 4a), there is almost no viscosity decrease until at least 150 bar, i.e. 70 bar from the critical pressure of 220 bar. Similarly, at constant pressure (Fig. 4b), there is no viscosity decrease until at least 310 °C, i.e. 66 °C from the critical temperature of 376 °C. Thus, at constant temperature, a viscosity decrease of 32 % occurs within 30 % of the critical pressure whereas at constant pressure a viscosity decrease of 52 % occurs within 10 % of the critical temperature. This is reflected in the gradients of the lines showing that temperature is the more important parameter.

Subcritical water processing of a Brunei crude of viscosity 52 mPa s showing the effect of approaching critical point a changing pressure at constant temperature T = 315 °C, t = 19 h with critical pressure pc = 220 bar b changing temperature at constant pressure p = 150 bar, t = 20 h with critical temperature Tc = 376 °C
In the above analysis, we have used relative viscosity changes: this is necessary to compare the effect of processing conditions on feeds of different viscosity. At temperature T, we define this relative change in viscosity of product μ compared to the feed viscosity μ0 by
We show (in the "Appendix") that for the feeds considered in this study, then increasing the temperature from 25 to 50 °C very roughly halves the viscosity. This results in the δ μ factor being roughly independent of the reference measurement temperature (see "Appendix"), providing sample feed and product are both measured at the same reference temperature for any particular comparison. This means that in the case of very heavy feeds where the viscosity measurements need to be carried out at higher temperatures (50 °C), then the relative reduction in viscosity can to a first approximation be compared with the viscosity changes in other feeds whose viscosity was measured at lower temperatures (25 °C).
The same notion of relative value with respect to a reference (in this case critical) point, can also be applied to the independent control variables, i.e. temperature T and pressure p. The relative deviation from the critical points for each of these two variables then become measures for the proximity to the critical temperature Tc and critical pressure pc, respectively:
Figure 5 shows these relative improvements of up to 90 % (for Marmul) in viscosities for the three oils studied as a function of these proximity parameters for temperature (a) and pressure (b). Earlier results had led us to expect that the proximity to the critical temperature (Eq. 4a) would be the more important parameter for determining the increase in the mobility as indicated by the relative size in the decrease in viscosity (Eq. 3). However, Fig. 5a shows that for a number of very similar values of δ T , then the values of relative decrease in viscosity δ μ are spread across the whole possible range of values. It appears rather that the proximity to the critical pressure (δ p ) as shown in Fig. 5b is more significant. This is in line with the observation in the introduction that sufficient pressure would be required to maintain the water in its liquid state, i.e. subcritical operation. In all cases, the set-point of the Isco pump was higher than the saturated vapour pressure for the temperature of operation, ensuring liquid state operation.

Another observation is that the best results for similar values of relative set point are obtained with the heaviest oils, i.e. the Marmul oil. This suggests a higher intensity of cracking to which a heavier feed would be more susceptible—we have shown above that pure pyrolysis does not reduce viscosity as much as cracking in subcritical water for the same processing time. Thus, viscosity lowering can be achieved in shorter times. Oil maturation in shales might be accelerated with subcritical water processing rather than long term heating (“retorting”). In addition to the mean molecular weight, there will be also other compositional elements (linear and branched composition, for example) which we now consider.
Other parameters
A variety of other tests were performed to assess the cracking performance. TBP distributions showed no significant change in boiling curve for the lighter feeds such as Brunei or Bonga—only for the heaviest feed (Marmul) were there observable changes in the boiling point curve (Fig. 6 which shows two typical contrasting results for processing with and without water). The defining factor of the Marmul feed (compared to the other feeds used in the tests described here) is that not all of a sample is recovered at the maximum temperature (600 °C) associated with the highest temperature, i.e. for the feed, the fraction evaporated at 600 °C [E(600)] is 62 %. Pyrolysis actually increases this value to just over 70 % whereas the subcritical water processing actually decreases the amount of low boiling fractions to around 40 %.

True boiling point curves showing difference between Marmul feed processed for 24 h at 150 bar and 330 °C either with water (“subcritical water”) or with no water (“pyrolysis”)
Figure 7 shows a plot of δ μ as a function of E(600) for a range of runs either simply pyrolysed (p) or with subcritical water (scw) of which Fig. 6 presented just two contrasting examples. The most advantageous viscosity change is seen to be associated with decreases in E(600) compared to the feed whereas one would expect a lightening of the product to give higher values of E(600). In general, lower boiling points are associated with lower viscosity feeds, although one needs to take into account factors such as aromaticity and branching. The correlation thus appears counter-intuitive. However, thermogravimetric analyses do indicate some transfer from high boiling fractions (>600 °C) to the accessible top range of TBP measurement (400–600 °C). This is confirmed by DSC where feed peak power rates shifts from 500 °C for the feed to 550 °C for cracked product. The TBP change is thus associated with mass transfer from the (for the TBP analyses) inaccessible high boiling fraction to lower boiling fractions during subcritical water processing.

Relative change in viscosity produced as a function of the fraction recovered at 600 °C [E(600)] for Marmul feed: “scw” refers to processing with subcritical water; “p” refers to pure pyrolysis (no water)
Generally, a large branched paraffinic portion of a molecule is easier to crack. The viscosity is dependent not only on molecular size, but also on the interactions between aromatic components or other compositional elements (linear vs. branched, etc.). Nonetheless to a first approximation, this may explain why the heaviest oil feed (Marmul) showed the best relative viscosity improvement. These compositional elements suggest that other heavy oil sources such as immature oils might be of interest for subcritical processing.
Conclusion
-
1.
Findings Relatively short duration cracking in subcritical water lowers the viscosity more than purely thermal pyrolysis. The heaviest oil (Marmul) yielded a permanent viscosity reduction of 90 % when conditions were within 10 % of the critical point. This suggests that it is solubilisation of the heavy oil which drives the advantaged performance compared to pyrolysis. There was a reduction in the heavy ends tail (i.e. >600 °C) of at least a few %.
-
2.
Application Various heavy oil components of interest to the petroleum industry become accessible if they are produced exploiting more than merely the heating properties of hot water. The sources include oil sands, kerogenic deposits (“oil shales”) and indeed normal oil reservoirs with high viscosity oils. The enablers are the properties of near critical water: enhanced solubility and higher cracking potential due to higher water dissociation near the critical point. The optimum points of operation are not the same for the solubilisation and cracking processes. Whereas many oil shales schemes require long term heat soaking to stimulate and simulate the maturation associated with traditional catagenesis, exploitation of the enhanced properties associated with near critical water indicate that judicious p, T operation would achieve this with shorter times and lower energy inputs. This may enable increased efficiency of operation and yields from many more reservoirs than are currently considered viable.
-
3.
Future work The experiments reported here used demineralised water. In reality brines would be applied and the critical point parameters change accordingly. Optimisation of both the p, T regime for solution in water (from dielectric constant) and acid/base cracking (from conductivity) could be determined from impedance spectroscopy. [The effect of other ions (as in brines) could be eliminated by frequency filtering.]
References
Batycky JP, Leaute RP, Dawe BA (1997) A mechanistic model of cyclic steam stimulation, SPE 37550
Deng S, Wang Z, Gu Q, Meng F, Li J, Wang H (2011) Extracting hydrocarbons from huadian oil shale by sub-critical water. Fuel Process Technol 92(5):1062–1067
Deng S, Wang Z, Gao Y, Gu Q, Cui X, Wang H (2012) Subcritical water extraction of bitumen from Huadian oil shale lumps. J Anal Appl Pyrol 98:151
Golombok M, van der Bijl J, Kornegoor M (2001) Severity parameters for steam cracking. Ind Eng Chem Res 470:40
Gray MR (1994) Upgrading petroleum residues and heavy oils. Marcel Dekker, New York
Kamimura H, Takahashi S, Kishita A, Moriya T, Hong CX, Enomoto H (1998) Upgrading of bitumen with supercritical water for a system combined with SAGD. ACS Div Fuel Chem 43(3):741
Kishita A, Watanabe N, Perez JV (2009) Observation of the heavy crude oil dissolution behavior under supercritical condition of water, IPTC 13891 (Int. Pet. Tech. Conf.)
Kumar S, Gupta RB (2009) Biocrude production from switchgrass using subcritical water. Energy Fuels 23(10):5151
Mehrotra AK (1995) A simple equation for predicting the viscosity of crude oil fractions. Trans I Chem 73A:87
Nowak S, Gunschel H (1983) Pyrolysis of petroleum liquids: napthas to crudes. In: Albright LF, Crynes BL, Corcoran WH (eds) Pyrolysis: theory and industrial practice. Academic Press, New York
Offeringa J, Barthel R, Weijdema J (1981) The interplay between research and field operations in the development of thermal recovery methods. In: Fayers FJ (ed) Proceedings of the 3rd Europ. Symp. Enhanced oil recovery, Dev. Pet. Sci, vol 13. Elsevier, Amsterdam, p 527
Ogunsola OM, Berkowitz N (1995) Extraction of oil shales with sub- and near-critical water. Fuel Process Technol 45(2):95–107
Park JH, Son SH (2011) Extraction of bitumen with sub- and supercritical water. Korean J Chem Eng 28(2):455–460
Peterson AA, Vogel F, Lachance RP, Froeling M, Antal MJ Jr, Tester JW (2008) Themochemical biofuel production in hydrothermal media: a review of sub and supercritical water technologies. Energy Environ Sci 1(1):32
Sinag A (2004) Sub and super-critical water extraction of Goynuk oil shale. Energy Sources 26:885
Siskin M, Katritzky A (1991) Reactivity of organic compounds in hot water, geochemical and technological implications. Science 254:231
Strausz OP (1989) Bitumen and heavy oil chemistry. In: Hepler LG, Hsi C (eds) AOSTRA technical handbook on oil sands, bitumens and heavy oils. Alberta Oil Sands Technology and Research Authority, Edmonton, AB
Su X, Zhao Y, Bi J (2004) Investigation on degradation of polyethylene to oils in supercritical water. Fuel Proc Tec 85(8):1249
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
Temperature independence of relative viscosity shift
As described in the experimental section above, viscosity measurements were carried out at 25 and 50 °C. We need a reference temperature-free measure of the viscosity decrease. We use the relative change in viscosity of product compared to the feed defined by
The validity of this approximation can be checked against various models of hydrocarbon mixture viscosity temperature response such as the Eyring model (Mehrotra 1995). This gives a typical relationship in the form of
where A and B are constants associated with a particular crude oil and T is the temperature (K).
We now consider a heavy oil feed (f) and the subcritical cracked product (p). This gives the following pair relationships
Equation A1 now becomes
An examination of Table 1 shows that to a first order of approximation μf(50) ≈ 0.5μf(25). Substituting in Eq. A3a then gives the relationship between the two coefficients Bf ≈ −353Af and we may assume that this will also apply to the cracked product stream as well Bp ≈ −353Ap. Inserting into Eq. A4 then enables the temperature dependence to be cancelled out and we end up with
so that to a first order of approximation the δ μ values may be considered to be roughly comparable if based on 25 or 50 °C as in the experimental results reported above.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Golombok, M., Ineke, E. Oil mobilisation by subcritical water processing. J Petrol Explor Prod Technol 3, 255–263 (2013). https://doi.org/10.1007/s13202-013-0066-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13202-013-0066-x