
Abstract
We determine the chemical changes associated with viscosity reduction when heavy oil is cracked in subcritical water. The viscosity reduction has a temperature threshold for onset of 290 °C—this suggests an enhanced acid cracking regime associated with the maximisation of water dissociation at these conditions aided by the already increased solubility. The mean molecular weight is reduced by nearly 50%. Oxygen and sulphur are reduced by about half of this—either by expelled gas effluent (\({\text{H}}_{2} {\text{S}}\)) or by conversion into mono-aromatic base sulphur-containing structures. The amount of lower branched paraffins is increased.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Classical viscosity breaking of vacuum distillate residue or bitumens requires temperatures well in excess of 400 °C and up to 500 °C. This involves direct heating with coils or in soak drums as the pressure has to stay low to encourage cracking. Another method used to heat heavy feeds is hot water. Low pressure steam aids thermal cracking to make the smallest chemical building blocks such as ethylene and propylene—although this requires even higher temperatures. By contrast, lower temperature steam mobilises heavy hydrocarbons in enhanced oil recovery (EOR) (in situ) as well as post-processing such as bituminous oil (“tar sands”). The viscosity is reduced as long as heat is applied enabling improved mobility. Simultaneously, the steam selectively removes (“strips”) lighter components from the crude, but no cracking takes place as the temperature is well below the 340 °C threshold. Once the heating conditions are removed (i.e. after the heating zone), cooling results in a viscosity increase again.
Permanent reduction in viscosity would require cracking. The application of hot water for doing this is called aquathermolysis. This breaks carbon sulphur bonds in aromatic structures (Kamimura et al. 1998), causing a reduction in sulphur and viscosity (Park and Son 2011). Part of these studies involve the enhanced cracking and dissolving properties of sub- and supercritical water. This technique is particularly prevalent in the green chemistry area (Yang et al. 1998; Ramos et al. 2002; Bicker et al. 2005; Oliveira et al. 2011; Isa et al. 2015). By contrast with steam and supercritical processes, subcritical processes maintain water in the liquid state. (Note that many studies use the term “subcritical” to cover conditions where one of pressure and temperature is supercritical and the other subcritical (Isa et al. 2015; Tumanyan et al. 2015).) In the current study, subcritical water is defined as liquid water in the temperature range between the boiling point and the critical point (374 °C) (Alenezi et al. 2009) with appropriate pressures between 1 and 220 bar to maintain the liquid phase.
A permanent decrease in viscosity is obtained due to two characteristics of subcritical water. Firstly, when water approaches its critical point, it is able to dissolve large organic structures (Miller et al. 1998). The dielectric constant (ϵ) of water decreases from 80 to around 6 as it approaches the critical point (Goto et al. 2004; Alenezi et al. 2009). Secondly, the ionic properties of water peak about 10% below the critical point (Hageman et al. 1996; Peterson et al. 2008). At this point, the dissociation constant (\(K_{w}\)) increases by a factor of 100–1000 compared to its STP value of ca. 10−14. The resulting higher H+/OH- concentration enhances cracking potential (Ryan et al. 1997; Patrick et al. 2001; Akiya and Savage 2002; Golombok and Ineke 2013). By contrast, supercritical water relies solely on its increased thermal cracking potential (Ogunsola and Berkowitz 1995; Kamimura et al. 1998; Deng et al. 2011; Park and Son 2011; Deng et al. 2012; Golombok and Ineke 2013).
SCW has several potential applications, including extracting oil from oil shale lumps (Deng et al. 2011), sulphur removal (Kamimura et al. 1998; Park and Son 2011), tar sands (Park and Son 2011) and the cracking of heavy hydrocarbons into lighter fractions (Duan et al. 2013; Golombok and Ineke 2013), thereby reducing the viscosity. The use of catalysts to enhance near critical effects has recently been extensively reviewed (Tumanyan et al. 2015).
Since we are seeking to exploit the enhanced cracking and dissolution properties of subcritical water, we prefer conditions below, but close to the critical point. Accordingly, we work in this study within ca. 20% of the critical temperature (in K) (i.e. >250 °C) but below the thermal cracking threshold of 340 °C. The objective of this study is to show that cracking can occur in subcritical water significantly below this thermal cracking threshold and to identify the chemical changes associated with the induced heavy oil viscosity reduction. By understanding the mechanism, one can improve or address conditions for each reservoir specifically, generating a more beneficial and cost-effective recovery process. Section 2 describes our experimental methodology. Next, the main results are presented and discussed to identify the main chemical changes leading to reduced viscosity.
Experimental
Set-up
A mixture of heavy oil and water is placed in a 200-mL Hastelloy C Premex autoclave. An electric heating mantle heats the reactor. A silver gasket ensures sealing at high temperatures. The pressure inside the autoclave is controlled with an Isco pump 500d and a back pressure regulator set to the desired pressure. A magnetically coupled fluid suction stirrer mixes the heavy hydrocarbons and water and later during reaction circulates the mixture. Controllers are provided to set all variables. Bursting discs are added as safety features after the Isco pump and the autoclave. These bursting discs are designed for a rupture pressure of 250 bar at 330 °C. A schematic view of the set-up is shown in Fig. 1.

Schematic of subcritical water processing set-up for cracking heavy hydrocarbons. A Isco 500D syringe pump; B Premex electric heating mantle; C Hastelloy C Premex autoclave; D Premex magnetic stirrer; E back pressure regulator; F Premex lift; G Premex controller; H Isco D-series pump controller; I all control temperature controller; J computer
Feedstock
Traditionally, heavy crude has been defined as any liquid petroleum with a viscosity ranging between 0.01 and 10 Pa.S at 25 °C. The medium heavy crude used in this study originates from Marmul, a site in Oman [API ca. 25° (Gao 2011)], and belongs to the light end of heavy crudes. The specific gravity is 0.9, and the viscosity at room temperature is 570 mPa s (Golombok and Ineke 2013). Table 1 shows the composition of the C30 fraction corresponding to 35% of the total.
Experimental procedure
We loaded equal volumes of both water and crude oil in the reactor and pressurised using the pump. Excess air is expelled by flushing the system with water. The pressure is maintained in the range of 150–200 bar by means of a back pressure regulator. After reaching the desired pressure, the reactor is heated. Sudden pressure eruptions arising from impurities or volatile components vent to external exhaust. The independent operating parameters of the study are thus (1) the water-to-oil ratio; (2) the set temperature T and (3) pressure p sufficient to maintain the liquid phase; and finally (4) the duration of the reaction τ.
After the desired process time, the reactor is cooled and the sample is extracted from the reactor. The content is a two-phase medium containing heavy hydrocarbons and water. The sample is centrifuged for 30 min at 6500 rpm to isolate the hydrocarbons from water. In summary, the independent operating parameters in this study are: the water-to-oil ratio, the set pressure and temperature and the time duration of operation at these conditions.
Analyses
Viscosity measurements were conducted using a Physica MCR 302 Anton Paar rheometer at two temperatures, 25 and 50 °C, respectively. Mean molecular weight analysis was carried out by gel permeation chromatography (GPC), using a Shimadzu Prominence-I LC-2030C 3D apparatus at 25 °C with tetrahydrofuran (THF) as eluent. The molecules are thus separated based on their thermodynamic volumes with the biggest eluted first. The molecular weight was then determined based on polystyrene which was used as a calibration standard.
Elemental analyses were carried out on a Thermo Scientific Flash 2000 combustion analyser with GC and thermal conductivity detector. Simulated distillation (SimDis) was carried out (ASTM D2887), with a maximum temperature for the boiling curve of approximately 720 °C corresponding to C120. Detection was by flame ionisation detector and a sulphur chemiluminescence detector. The relative error on the reported results is <5%, a figure obtained from reproducibility tests, comprising a number of repeated runs (minimum of three runs).
Results
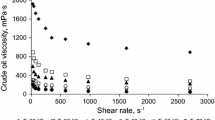
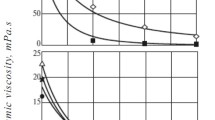
Figure 2a shows that below a temperature threshold of around 280 °C there is no significant viscosity reduction. This shows the effect of subcritical water. We referenced above that traditional (i.e. using purely thermal effects) viscosity breaking requires temperatures above 400 °C: aquathermolysis with steam requires >30 days of “soaking” for a viscosity reduction effect (Clark et al. 1986). We have shown that by carrying out the effect in subcritical liquid water, the threshold temperature is reduced by ca. 120 °C. Moreover, the time for the viscosity reduction is reduced from days to a matter of hours as we have previously shown (Golombok and Ineke 2013). We can combine the effect in a severity given by

Viscosity as function of (a) process temperature (b) severity
with temperature T (°C) and reaction time \(\tau\) (hours). \(a\) is a constant and typically around 0.05 (Golombok et al. 2001). A comparison of our temperature threshold knowledge from Fig. 2a with the severity data (Fig. 2b) shows that most of the viscosity (\(\mu\)) reduction takes place in the first 24 h at process temperatures above 330 °C. We can separate the two effects of lower temperature requirement and much shorter times in this process. The lower temperature requirements derive directly from the fact that we maintain water in the liquid state as opposed to the gaseous state traditionally associated with these studies. The result is the increased solubility of the oil in water. Oil molecules are more easily separated from one another so are more susceptible to cracking rather than any polymerisation effects if they were not separated by water. The shorter times derive from the 1000× higher water dissociation and may be attributed to the simultaneous acid/base catalytic cracking effects.
Figure 3 shows the mean molecular weight (from gel permeation chromatography) as a function of severity. The original crude has a mean molecular weight of 737 g/mol which is reduced by typically ca. 50%. Of course, molecular weight is not in and of itself, directly correlated with viscosity. Branched species typically have higher viscosities because of the restriction on the slippage associated with linear species which enables layers to flow over one another more easily. Nonetheless, as described above, gel permeation chromatography is a form of size exclusion chromatography so it is not surprising that the form in Fig. 3 somewhat matches with that in Fig. 2b despite more scatter in the molecular weight curve compared to the smoother trend for the viscosity as a function of severity.

Mean molecular weight as function of severity
A more detailed breakdown of molecular changes is obtained from a “simulated” distillation (ASTM D2887) analysis. Figure 4a shows the boiling point (TBP) curves for two different samples, the original crude and a typical processed sample. There is a significant difference in distillation curves. Two factors here are E(720)—the fraction recovered at maximum temperature 720 °C, and T(50)—the temperature at which 50% is recovered. T(50) improves from 534 to 463 °C, and E(720) improves from 80 to 92%. This is of course substantially due to the lowered molecular weight. As mentioned above, one needs to take aromaticity and branching into account. Our analysis suggests that the aromaticity has remained constant (Table 1). Figure 4b shows the distribution of carbon number for the original crude feed as well as a typical processed feed. We calculate a light-to-heavy ratio from the mass associated with any carbon number n by:

Lightening of heavy oil by subcritical water processing. (a) Boiling point curves for original and processed sample. (b) Mass distribution as function of carbon number
The original crude has LHR = 0.67 compared to a value of 1 for the processed sample.
Generally, higher branched paraffins are easier to crack (Golombok and Ineke 2013). An analysis of the carbon groups up to C30 is made using GC × GC FID into linear paraffins (P) (mass m l ), methyl paraffins (Me-P), ethyl or dimethyl paraffins (C2-P) and higher branched paraffins (hbr-P) as shown in Fig. 5. We define a branching-to-linear ratio by

Branching distribution of paraffins in a original and b processed Marmul
In the original Marmul, the ratio is 0.75 while when processed this ratio decreases to 0.31. This suggests a conversion of highly branched paraffins into paraffins during the water-induced viscosity reduction, reflecting the dependency of viscosity on carbon chain length and branching.
We have thus determined that there are no changes in aromaticity, but that there is a reduction in molecular weight as well as a reduction in branching. There remains the role of heteroatoms (i.e. sulphur and oxygen) which we now evaluate. Figure 6a shows the oxygen content as function of severity. The most important observation is the decrease in oxygen content over the whole severity range (ca. 65%). This is a constant decrease independent of severity. In Fig. 6b a similar effect is shown for the sulphur content (albeit with greater variability). The average reduction in sulphur is approximately 60%—we shall show below that this is due to cracking of aliphatic sulphur bonds between organic groups. The remaining elements (C, H and N) remain constant (within 4%) over the severity range. Heteroatom removal is supported by similar findings in the literature (Caniaz and Erkey 2014). This suggests that the dominant source of lowered viscosity is reduced molecular weight due to breaking of C–O and C–S as shown in Fig. 6a, b (Kamimura et al. 1998). A part of the sulphur is converted into \({\text{H}}_{2} {\text{S}}\), while a small amount of sulphur-containing aromatics is expelled during pressure excursions above the setting of the backpressure regulator.

Heteroatom content as a function of severity a oxygen b sulphur
Finally, the organic sulphur compounds were investigated using GC × GC and sulphur chemiluminescence detection. Figure 7 shows a significant increase in benzothiophene (BT) and dibenzothiophene (DiBT) compounds arising from the break down of heavier components. During the subcritical water treatment, the high molecular weight aromatic sulphur polymers are cracked, which may be the source of these one- and two-ring species in the product. BT and DiBT appear to no longer be susceptible to further cracking. This means the principal cracking mechanism involves C–S bond cleavage in addition to the aforementioned breaking of aliphatic C–C bonds. This confirms similar findings by Kamimura et al. (1998).

Mass distribution of sulphur as function of organic sulphur types. (Th thiophene, DS disulphides, Ar-S aryl sulphides, BT benzothiophene, etc.)
Conclusion
-
1.
A reduction of up to 80% in viscosity is obtained during a relatively short dissolution/cracking process in subcritical water.
-
2.
The chemical source behind the reduction in viscosity is the breakage of aliphatic C–C, C–O and C–S bonds. The aromatic monomer compounds increase in concentration in tandem with an increase in light-to-heavy ratio.
-
3.
The mass fraction boiling below 300 °C increases by 8%. This corresponds to molecules cracked to lower molecular weights.
-
4.
The decrease in sulphur content is due to breakage of C–S bonds linking aromatic components—mainly benzothiophenes and dibenzothiophenes.
Abbreviations
- C2-P:
-
Ethyl/dimethyl paraffins
- \(K_{W}\) :
-
Dissociation constant
- \(M_{W}\) :
-
Molecular weight
- \(\epsilon\) :
-
Dielectric constant
- μ :
-
Viscosity
- DiBT:
-
(di) benzothiophene
- EOR:
-
Enhanced oil recovery
- FID:
-
Flame ionisation detector
- hbrP:
-
Higher branched paraffins
- MeP:
-
Methyl paraffins
- P:
-
Paraffins
- SCD:
-
Sulphur chemiluminescence detector
- SCW:
-
Subcritical water
- SimDis:
-
Simulated distillation
- STP:
-
Standard temperature and pressure
- TBP:
-
True boiling point
References
Akiya N, Savage PE (2002) Roles of water for chemical reactions in high-temperature water. Chem Rev 102:2725–2750
Alenezi R, Leeke GA, Santos RCD, Khan AR (2009) Hydrolysis kinetics of sunflower oil under subcritical water conditions. Chem Eng Res Des 87:867–873. doi:10.1016/j.cherd.2008.12.009
Bicker M, Endres S, Ott L, Vogel H (2005) Catalytical conversion of carbohydrates in subcritical water: a new chemical process for lactic acid production. J Mol Catal A Chem 239:151–157. doi:10.1016/j.molcata.2005.06.017
Caniaz RO, Erkey C (2014) Process intensification for heavy oil upgrading using supercritical water. Chem Eng Res Des 92:1845–1863. doi:10.1016/j.cherd.2014.06.007
Clark PD, Clarke RA, Hyne JB (1986) Aquathermolysis: the chemical reaction of steam with heavy oils—emphasizing the critical role of sulphur compounds. ASRL Q Bull XXIII(1,2):1–20
Deng S, Wang Z, Gu Q et al (2011) Extracting hydrocarbons from Huadian oil shale by sub-critical water. Fuel Process Technol 92:1062–1067. doi:10.1016/j.fuproc.2011.01.001
Deng S, Wang Z, Gao Y et al (2012) Sub-critical water extraction of bitumen from Huadian oil shale lumps. J Anal Appl Pyrolysis 98:151–158. doi:10.1016/j.jaap.2012.07.011
Duan P, Xu Y, Bai X (2013) Upgrading of crude duckweed bio-oil in subcritical water. Energy Fuels 27:4729–4738. doi:10.1021/ef4009168
Gao CH (2011) Scientific research and field applications of polymer flooding in heavy oil recovery. J Pet Explor Prod Technol 1:65–70. doi:10.1007/s13202-011-0014-6
Golombok M, Ineke E (2013) Oil mobilisation by subcritical water processing. J Pet Explor Prod Technol 3:255–263. doi:10.1007/s13202-013-0066-x
Golombok M, van der Bijl J, Kornegoor M (2001) Severity parameters for steam cracking. Ind Eng Chem Res 40:470–472. doi:10.1021/ie990436r
Goto M, Obuchi R, Hirose T et al (2004) Hydrothermal conversion of municipal organic waste into resources. Bioresour Technol 93:279–284. doi:10.1016/j.biortech.2003.11.017
Hageman KJ, Mazeas L, Grabanski CB et al (1996) Coupled subcritical water extraction with solid-phase microextraction for determining semivolatile organics in environmental solids. Anal Chem 68:3892–3898. doi:10.1021/ac960751c
Isa KM, Snape CE, Uguna C et al (2015) Pyrolysis oil upgrading in high conversions using sub- and supercritical water above 400°C. J Anal Appl Pyrolysis 119:180–188. doi:10.1016/j.jaap.2016.03.004
Kamimura H, Takahashi S, Kishita A, ChengXie H, Enomoto H (1998) Upgrading of bitumen with supercritical water for a system combined with sagd. Abstr Pap Am Chem Soc 216:U879
Miller DJ, Hawthorne SB, Gizir AM, Clifford AA (1998) Solubility of polycyclic aromatic hydrocarbons in subcritical water from 298 to 498 K. J Chem Eng Data 43:1043–1047. doi:10.1021/je980094g
Ogunsola OM, Berkowitz N (1995) Extraction of oil shales with sub- and near-critical water. Fuel Process Technol 45:95–107. doi:10.1016/0378-3820(95)00036-7
Oliveira ELG, Silvestre AJD, Silva CM (2011) Review of kinetic models for supercritical fluid extraction. Chem Eng Res Des 89:1104–1117. doi:10.1016/j.cherd.2010.10.025
Park JH, Son SH (2011) Extraction of bitumen with sub- and supercritical water. Korean J Chem Eng 28:455–460. doi:10.1007/s11814-010-0358-5
Patrick HR, Patrick HR, Griffith K et al (2001) Near-critical water: a benign medium for catalytic reactions. Ind Eng Chem Res 40:6063–6067. doi:10.1021/ie010178+
Peterson RA, Vogel F, Lachance RP et al (2008) Thermochemical biofuel production in hydrothermal media: a review of sub- and supercritical water technologies. Energy Environ Sci 1:32. doi:10.1039/b810100k
Ramos L, Kristenson EM, Brinkman UT (2002) Current use of pressurised liquid extraction and subcritical water extraction in environmental analysis. J Chromatogr A 975:3–29. doi:10.1016/S0021-9673(02)01336-5
Ryan ET, Xiang T, Johnston KP, Fox MA (1997) Absorption and fluorescence studies of acridine in subcritical and supercritical water. J Phys Chem A 101:1827–1835. doi:10.1021/jp962676f
Tumanyan BP, Petrukhina NN, Kayukova GP et al (2015) Aquathermolysis of crude oils and natural bitumen: chemistry, catalysts and prospects for industrial implementation. Russ Chem Rev 84:1145–1175. doi:10.1070/RCR4500
Yang Y, Belghazi M, Lagadec A et al (1998) Elution of organic solutes from different polarity sorbents using subcritical water. J Chromatogr A 810:149–159. doi:10.1016/S0021-9673(98)00222-2
Acknowledgements
Funding was provided by Shell (Grant No. PT 13670).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Cuijpers, M.C.M., Boot, M.D. & Golombok, M. Enhanced viscosity reduction in heavy oils by subcritical water. J Petrol Explor Prod Technol 8, 291–298 (2018). https://doi.org/10.1007/s13202-017-0370-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13202-017-0370-y