
Abstract
Tamoxifen (TMX) is used as adjuvant therapy for estrogen receptor-positive (ER+) breast cancer cases due to its affinity and inhibitory effects. However, about 30% of cases show drug resistance, resulting in recurrence and metastasis, the leading causes of death. A literature review can help to elucidate the main cellular processes involved in TMX resistance. A scoping review was performed to find clinical studies investigating the association of expression of molecular markers profiles with long-term outcomes in ER+ patients treated with TMX. In silico analysis was performed to assess the interrelationship among the selected markers, evaluating the joint involvement with the biological processes. Forty-five studies were selected according to the inclusion and exclusion criteria. After clustering and gene ontology analysis, 23 molecular markers were significantly associated, forming three clusters of strong correlation with cell cycle regulation, signal transduction of proliferative stimuli, and hormone response involved in morphogenesis and differentiation of mammary gland. Also, it was found that overexpression of markers in selected clusters is a significant indicator of poor overall survival. The proposed review offered a better understanding of independent data from the literature, revealing an integrative network of markers involved in cellular processes that could modulate the response of TMX. Analysis of these mechanisms and their molecular components could improve the effectiveness of TMX.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Breast cancer (BC) is the most prevalent cancer among women in developed and developing countries [1]. Due to its incidence and mortality, it is considered a serious public health problem worldwide [2, 3]. This type of cancer is a complex and heterogeneous disease with several distinct histopathological and molecular subtypes. Among them, about 60 to 70% of all diagnosed BC are positive for hormone receptor expression (HR+), specifically estrogen receptor (ER+) and/or progesterone receptor (PR+).
The stimulation of both receptors by its ligand molecules—estrogen and progesterone—induces mainly transcriptional changes, which trigger biological processes such as cell survival, proliferation, and differentiation. Therefore, in breast cancer, its constant stimulation by sexual hormones could contribute to tumor increasing and disease progression [4, 5], justifying anti-estrogen therapies as an adjuvant treatment for this type of neoplasia [6].
Tamoxifen (TMX) is one of the most used types of adjuvant treatment in ER+ breast cancer. Administered as a prodrug, TMX must be metabolized in the liver to form active metabolites that will perform their estrogen antagonist functions and thus referred to as a selective estrogen receptor modulator (SERM) which blocks the binding of estrogen to the estrogen receptor and consequently inhibit cell proliferation and survival [7,8,9,10,11]. For this reason, TMX is the standard hormone therapy for HR+ breast cancer patients in the premenopausal stage [12,13,14].
Whereas this hormone adjuvant therapy has considerably improved survival in HR molecularly subtyped BC patients in recent decades, the development of pharmacological resistance has been an increasing challenge for oncology, once the lack of responsiveness to treatment is a critical and limiting factor for therapeutic efficacy [15, 16]. Around 30 to 50% of HR+ BC patients, regardless of the expression level of these receptors, presented intrinsic or acquired resistance to TMX [17, 18]. Consequently, the five-year survival rate after resistance occurrence was less than 20% [19, 20].
Several clinical works have been published in the literature bringing information about a new specific marker involved with this frame of resistance, associating its molecular profile with a better or poor outcome in BC patients. Therefore, a better comprehension of the interrelationship among these identified molecular markers associated with TMX resistance becomes increasingly relevant and necessary to strengthen the understanding of what molecular mechanisms and cell processes are changed in tumor cells when they acquire hormone resistance in an attempt to overcome it and improving therapeutic effectiveness.
A major meta-analysis carried out by Wirapati et al. analyzed the gene expression in breast cancer patients and associated it with important biological processes that could be present in all molecular subtypes [21]. Another meta-analysis by Mihály et al. investigated new independent biomarkers that could be related to the tamoxifen response in breast cancer patients [22].
This work presents a systematic review of recent papers in the literature, focusing only on translational studies with a minimum 3-year follow-up. It highlights how individual biomarkers related to TMX resistance impact the survival rate of ER+ breast cancer patients. Then, it employs a clustering technique to find groups of biomarkers whose biological processes show a strong interaction. Based on this clustering analysis, this work evaluates how these biological processes influence the overall survival rate in ER+ breast cancer patients, indicating which processes could be predictive of clinical results in response to treatment with TMX. To the best of the authors’ knowledge, this is the first systematic review that presents such a clustering technique for the conjoint evaluation of multiple biomarkers in the survival rate of ER+ breast cancer patients.
2 Methodology
2.1 Identifying relevant studies
The manuscript selection was performed according to the PRISMA Extension for Scoping Reviews (PRISMA-ScR) methodology: Checklist and Explanation [23]. The literature review was performed to identify clinical studies assessing the correlation between the expression profile of a molecular biomarker and the long-term outcome of HR+ BC patients in response to TMX treatment. The initial inclusion criteria were original manuscripts, published from 2013/01/01 to 2018/06/30, full texts in any language, obtained from EMBASE or PUBMED databases. For manuscript search, three primary keyword descriptors were applied: “breast cancer”, “hormone therapy” and “resistance”, or their variations listed in Supplementary material 1. Case studies, reviews, and comments were not considered.
2.2 Selection of eligible studies
The manuscripts found in the initial search were analyzed in three stages: first, a title reading was performed, selecting only those presenting all primary keyword descriptors or their variations. Duplicates were removed. The second and third stages consisted of abstract and full-text reading, respectively. These stages ensure that the selected manuscripts respected the inclusion and exclusion criteria, according to Table 1. The essential items of the REMARK protocol [24] were considered in this review. Two independent reviewers performed the manuscript search and selection processes, while a third reviewer solved incongruities. Only manuscripts that assessed molecular biomarkers as a prognostic factor and not as a risk factor were considered. Studies only evaluating patient expression databases, in vivo or in vitro experimentation were also excluded.
2.3 Data extraction
After the full-text review stage of the selected manuscripts included in this scoping review, the following information was plotted in Table 2:
-
Population: number of participants and follow-up of only those HR+BC patients treated with Tamoxifen.
-
Marker profile: the marker symbol used by each selected manuscript; the genetic alteration (deletion, mutation, or polymorphisms) when altered; the type of molecule assessed (DNA, RNA, or protein); the molecular status of the analyzed marker in relation to activity, expression, subcellular localization, or post-translational modification, which could alter its functionality.
-
Clinical outcomes and statistical analysis: clinical profile of tested BC patients (survival or recurrence) associated with the alteration of analyzed molecular markers; the statistical analysis of the association between the molecular status of markers and clinical outcome, prioritizing multivariate analysis; the significance level of statistical analysis when available. In order to standardize the collected information, it was plotted molecular profile of markers, which was associated with a poor clinical outcome, except for those presenting polymorphisms or specific subcellular localization.
2.4 In silico analysis
2.4.1 Design of networks
After a full-length analysis of manuscripts and data extraction, the official symbol of all selected molecular markers was confirmed in the HUGO Gene Nomenclature Committee (HGNC) website for in silico analysis (Supplementary Table 1). Those presenting statically significant associations (p < 0.05) with some clinical outcomes in HR+ BC patients were assessed together using the ©STRING CONSORTIUM 2019 (version 11.0) web tool. The minimum interaction score adopted in this analysis is 0.700 (high confidence) for each connection (edges), including all active interaction sources used by the tool, excluding text mining. The resulted network was presented with an interaction score of each connection between two markers for further modular analysis of networks. Posteriorly, the unconnected markers were separately analyzed. Only those markers whose molecular profile of expression/activity direct influence the clinical outcomes of HR+ BC patients were considered, excluding micro RNAs. For polymorphisms, only the affected biomarker was included for network design, not considering the polymorphic variant itself.
2.4.2 Modular analysis of network
From the STRING resulting network, it was used ClusterONE plugin [25] of Cytoscape software. The cluster establishment was assessed considering the following criteria: at least three proteins compounding a cluster, density and interaction quality values above 0.5, and significant p-value (p < 0.05). Molecular markers that were not included in any cluster were called unclustered markers.
2.4.3 Gene ontology analysis
Once the clusters within a molecular marker network were identified, it was performed the gene ontology (GO) analysis using the Biological Network Gene Ontology (BiNGO) plugin (version 3.0.3) in Cytoscape software to assess the possible biological processes that could be involved with the specific set of markers grouped in each cluster. Statistically significant clusters were assessed, considering only their members or added to their direct neighbor markers (networked markers directly linked with clustered markers). Statistical significance was measured using a hypergeometric test with the multiple testing correction of Benjamini & Hochberg False Discovery Rate (FDR). Only biological processes with a p-value < 0.05 were considered significant. Based on biological knowledge about BC resistance, the five biological processes often involved with this event were selected to be illustrated in the results and further discussed.
2.4.4 The predictive power of different sets of TMX-resistance markers
In order to assess the predictive power of each set of markers among those significantly associated with any clinical outcome in TMX-treated HR+ BC patients, the Kaplan Meier-plotter web software [26] was used. It was assessed a general analysis for all selected markers, those connected and unconnected markers from STRING analysis, and those clusters obtained from ClusterOne plugin (with or without their direct neighbor markers). The data extracted from the original papers showing high expression/activity of molecular markers associated with poor outcome were directly related in the KM-plotter analysis settings, inverting those presenting low or absence expression/activity associated with poor prognosis status (Supplementary Table 1). Equal weights were attributed to all tested markers. The Kaplan–Meier graphs represent overall survival (OS) with a follow-up threshold of 240 months, splitting patients by median, using all probe sets per gene, without any restriction of breast cancer subtypes or selected cohorts studies as dataset source.
3 Results
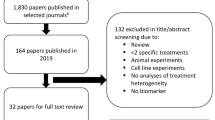
It was identified 2.487 articles in EMBASE and PUBMED databases from the keyword descriptors and their variations. The flow diagram of literature analysis, manuscript selection, and in silico analysis is illustrated in Fig. 1.

PRISMA diagram of selection of studies and in silico analysis. The upper section corresponds to the step-by-step manuscript selection process according to the established inclusion and exclusion criteria. The Lower section includes the sequence of in silico analysis performed with all selected biomarkers to search a better and integrated understanding of the influence of each protein in TMX resistance
The application of the eligibility criteria resulted in the exclusion of 2.443 papers that were not adequate to one or more selection factors or did not provide the needed information. The main excluding factors were: (1) in vitro experimentation; (2) in vivo assays; (3) clinical studies assessing less than 35 patients (4) the absence of prognostic factor assessing in clinical studies; (5) the absence of hormone therapy with TMX; (6) manuscripts using of transcription or genomic sequencing data deposited in public databases; (7) works testing other types of hormone therapy, differently of TMX; (8) and review manuscripts. Thereby, 45 manuscripts were selected from the eligibility analysis, analyzing 60 different molecular targets, as shown in Table 2.
The total number of analyzed patients in each selected study has varied between 37 [29] and 1082 [62]. The follow-up of most studies was 5 years or more, and 6 other studies presented a follow-up of 3 years [36, 45, 58, 59, 61, 68] or less [57]. The main reported alterations in assessed molecular targets were changes in their expression level or activity. However, some structural modifications at the DNA, RNA, or protein level were also found, such as polymorphisms, post-translational modifications (phosphorylation), and mutations (deletion or splicing variant).
Fifteen of the 60 selected markers were excluded from the in silico analysis (Fig. 1). Thereby, only 45 of 60 selected markers were eligible for in silico analysis. STRING analysis resulted in a functional association among 23 of 45 tested markers, forming two networks with 5 and 18 nodes, respectively (Fig. 2a).

In silico analysis. A 3 clusters were formed with strongly linked molecular markers, cluster 1(red), cluster 2 (green), and cluster 3 (blue). The markers in gray were not included in the clusters, remaining just interconnected proteins among clusters inside the network. B unconnected markers. A and B still reveal the functional status of each molecular marker. 12 markers (dashed borderline in the molecular markers) decrease in functional activity associated to poor outcomes in BC patients, the other 33 markers presented an elevated functional activity associated to worst outcomes (full borderline in the molecular markers), accordingly information of their original manuscripts. C Gene ontology analysis for biological processes: CLUSTER 1 presented significant involvement with molecular events related to cell proliferation, CLUSTER 2 with the modulation of proliferative mechanisms, and CLUSTER 3 with the mammary gland development and its hormone stimulation. D Genetic ontology analysis of clusters 2 and 3, taking into account not only its constituent markers but with its direct neighbors
Furthermore, to find densely associated subgroups of molecular markers inside the obtained networks, ClusterONE analysis revealed the presence of 3 clusters with strongly linked molecular markers (red, green, and blue markers). Five proteins (AURKA, MK167, AURKB, FOXM1, RRM2) constituted the CLUSTER 1 (red markers) that presented a density value = 1,000 and interaction quality = 1000, displaying a high interactive power among these markers, represented by a p-value = 0.002. The CLUSTER 2 (green markers) was configured with 7 proteins (STAT3, PTPN2, FYN, SRC, PAK2, FRS2, EGFR), presenting a density value = 0.571 and quality = 0.600 and a p-value = 0.022. The CLUSTER 3 (blue markers) was established with 5 proteins (ESR1, GATA3, PGR, NCOR2, EBAG9), presenting density = 0.600, interaction quality = 0.545 and p-value = 0.055, considered a borderline, accordingly the established criteria.
Not all networked molecular markers were included in the clusters, remaining just interconnected proteins among clusters inside the network (gray markers). This subgroup was called unclustered markers and was constituted by six proteins (SOX2, POU5F1, BCL2, IGF1R, TGFBR2, YAP1). Among these, SOX2 and POU5F1 were directly linked only with CLUSTER 2, and TGFBR2 and YAP1 only with CLUSTER 3. On the other hand, BCL2 and IGF1R seem to establish a link between CLUSTER 2 and 3, acting as an intermediate node between them. The other remaining 22 markers presented no interaction among them (Fig. 2b).
Figure 2a and 2b reveal the functional status of each molecular marker that is associated with a worsening outcome in patients, according to the information of their original manuscripts. From 45 tested markers, 12 of them presented a decrease in functional activity associated with poor outcomes in BC patients (dashed borderline in the molecular markers), which may be due to an inactivating polymorphism, a gene deletion, a gene/protein expression decrease, or still an activity reduction. Further, the other 33 markers presented an elevated functional activity associated with the worst outcomes due to an activating polymorphism, elevated gene/protein expression, or increasing functional activity.
In the gene ontology analysis (Fig. 2c), CLUSTER 1 presented significant involvement with molecular events related to cell proliferation, strongly suggesting that this subset of molecular markers could directly influence the mitotic mechanism regulation. The analysis of CLUSTER 2 presented significant implications in biological processes directly connected with signaling pathways involved with the modulation of proliferative mechanisms. Lastly, CLUSTER 3 showed a significant relationship with processes related to the development of the mammary gland and its hormone stimulation, suggesting a possible involvement with mechanisms of differentiation and signaling pathways of sexual hormone response in the breast tissue.
CLUSTER 2, together with its direct neighbors (BCL2, SOX, POU5F1, GATA3, YAP1. ESR1, PGR, IGF1R) (Fig. 2d), has shown significant implication with biological processes as “gland morphogenesis,” “enzyme-linked receptor pathway,” “transmembrane RTK pathway,” “cell differentiation” and “protein autophosphorylation.” The same approach was followed for CLUSTER 3 and its direct neighbors (BCL2, STAT3, SRC, IGF1R), resulting in significant involvement with cellular mechanisms such as “gland morphogenesis,” “response to estrogen stimulus,” “anti-apoptosis,” “negative regulation of DNA-dependent transcription” and “cell proliferation.” It was found that some cell processes had changed between CLUSTER 2 and 3 when direct neighbors were added in analysis, highlighting their proximity inside the network and the possible involvement of both clusters in similar or the same cellular processes.
In order to complement our data, a more in-depth analysis was carried out to test if these clusters would have a predictive power of overall survival in BC patients when treated with TMX (Fig. 3).

Predictive analysis of overall survival for each set of markers in BC patients using the Kaplan–Meier plotter tool. A Analysis of all 45 selected markers. B Only the networked markers (23 markers). C Unconnected markers (22 markers). D Analysis of clusters 1, 2, and 3. E Cluster 2 and 3 with their direct neighbors. Significant p-values (< 0.05) are highlighted in bold
For this analysis, Kaplan Meier-plotter web software [26] was used, assessing the overall survival (OS) with a follow-up of 240 months. A first analysis of all 45 selected markers shown hazard ratio (HR) 1.51 (95% CI 1.1–2.08) and a p-value = 0.0095 (Fig. 3a). A significant predictive power was found when only those networked markers were analyzed, with an HR 1.44 (95% CI 1.05–1.97), p-value = 0.023 (Fig. 3b). As expected, the joint analysis of non-connected markers was not significant in predicting overall survival (Fig. 3c).
Furthermore, it was assessed if the specific molecular clusters would present predictive power for overall survival. Figure 3d shows Kaplan–Meier for each analyzed cluster, and, among them, only CLUSTER 1 has presented significant predictive capability, displaying HR = 2.0 (95% CI 1.6–2.49), p-value < 0.00001. Conversely, knowing the proximity and possible functional overlap between CLUSTER 2 and 3, it was assessed the predictive power of specific clusters together with their direct neighbors (Fig. 3e). Only CLUSTER 3 with its direct neighbors displayed significant results, with HR 1.4 (95% CI 1.02–1.91), p-value = 0.037.
4 Discussion
This review found evidence in the literature about possible molecular targets capable of individually predicting TMX resistance in patients with BC, demonstrating a strong association between their molecular status and specific clinical outcome, such as tumor survival and/or recurrence. Therefore, an in silico analysis was performed to aid in data analysis. An interrelation between some molecular targets was found, forming three distinct clusters, showing significant involvement in biological processes. Our method, based on a dense analysis from literature datasets, showed that there might be groups of cellular processes that could be more involved with resistance to TMX in HR+BC patients. It is worth highlighting that the same weight was assigned for all analyzed markers in the KM-plot analysis since this information is not commonly evaluated in the study models of the original data sources. Therefore, it might represent a limitation in the present analysis since each marker can have different influence degrees within a signaling pathway to control a specific biological process.
The markers MKI67, AURKA, AURKB, FOXM1 in CLUSTER 1 presented significant involvement with molecular events related to cell proliferation (Fig. 2c), as observed by their original manuscripts [32, 35, 39, 44, 64]. However, this cellular process is highly complex, involving several components that could regulate it positively or negatively, and it has been widely studied as it is crucial for resistant subpopulation rising, not only to TMX but also to other drugs [72,73,74,75].
Analysis of overall survival of CLUSTER 1 showed a significant association with a worse outcome of the TMX-treated BC patients (Fig. 3d) when strongly expressed or activated. Bergamaschi et al. showed that the analysis of a gene signature related to mitosis control, orchestrated by marker 14-3-3ζ, was able to control the resistance profile to endocrine therapy in BC, influencing the expression of markers involved with CLUSTER 1, such as FOXM1 and AURKB [76]. Perhaps, if the Thistle JE. et al., who analyzed the involvement of 14-3-3ζ with a worse outcome in TMX+ treated ER+ patients, had presented statistical correlation values, this marker would be highly related to CLUSTER1 and could result in an increase of predictive power for this set of genes for TMX therapy response [62].
The uncontrolled cell cycle is considered a hallmark of cancer [77], as shown in the meta-analysis that evaluated three key biological processes for the development of breast cancer (cell proliferation, ER, and HER2 signaling). This analysis highlighted cell proliferation as an important prognostic role. Further, the results showed that the subtypes ER+/HER2- with low proliferation showed a higher DRFS, whereas the ER−/HER2− and HER2+ showed a higher level of cell proliferation and worse outcomes. However, there was no analysis of the markers with regard to the different hormonal treatments [21].
Besides, there are studies providing information about possible combined effects among these markers associated with a worse prognosis in patients with other BC subtypes [78, 79]. However, no studies simultaneously evaluate the expression profile of five markers composing CLUSTER 1 in tumor cells, which is a promising molecular target to be better explored in future research about this predictive power in response to TMX treatment.
No significant correlation was found with the clinical outcomes for CLUSTERS 2 and 3 (considering only the constituent markers) (Fig. 3d). However, individually, each molecular marker has presented an association with worse outcomes in their original studies. This controversial effect of joint analysis demonstrates the importance of simultaneously analyzing as many markers as possible and jointly correlating with clinical outcomes. The more extensive and more complete the molecular profile of tumor cells is, the closer to reality this analysis will be, increasing the representativeness of the experimental data.
Reinforcing this concept, when only constituting markers of CLUSTER 3 were analyzed, no association was found with some clinical outcome. However, when other close markers were included in this analysis, a significant association with overall survival was observed (Fig. 3e). The combination of CLUSTER 3 and its direct neighbors have shown involvement with cellular processes linked to mammary gland development and differentiation, in addition to sexual hormone response and gene transcription regulation (Fig. 2d). The latter has been associated with Decitabine (DAC) chemoresistance, also in BC [80]. However, some of these cellular processes are quite generic and could be related to several molecular markers. Therefore, more evidence about this subset of markers is required to increase the number of molecular components integrating this cluster, restrict the involvement with generic cellular processes, and improve its predictive power concerning clinical outcomes.
The investigation of gene signatures relating a specific cell profile with specific clinical outcomes has gained traction, not only for BC but also for other tumor types. Toshimitsu et al. revealed a specific molecular profile of cisplatin resistance in esophageal cancer based on in vitro studies using drug-resistant cell lineage [81]. Other works have discovered gene signatures related to BC resistance for the most varied treatments, such as neoadjuvant therapy [82], inhibitory molecules [83], and hormone therapy [84], among others. These studies make use of multiple strategies, such as transcriptome sequencing and microarray, to simultaneously investigate a range of markers in the same tumor sample and compare it with the resistant condition.
The study used a gene expression assay to quantify the likelihood of recurrence in patients with positive breast cancer, negative nodule, and ER+ treated with TMX. The study managed to identify 16 genes that were related to cancer recurrence that belonged to either of the following biological processes: proliferation (such as Ki-67 and cyclin B1), estrogen receptor (including genes such as ER and PR), HER2 (including HER2 and GRB7), and cell invasion (including genes like Stromelysin 3 and Cathepsin L2) [85]. A follow-up study was developed to investigate the ESR1 marker, which has been reported as a strong predictor of resistance to TMX in ER-positive patients with low levels of ESR1 [86].
Mihály et al. performed a meta-analysis looking for studies that evaluated expression-based markers to provide independent information regarding TMX treatment. Out of 68 biomarkers with probable connection to the TMX resistance, three (PGR, MAPT, and SLC7A5) were the most promising observed in patients treated with TMX. However, only independent markers were evaluated without making a joint analysis to investigate their main biological processes. According to our review, we can also observe that many markers remain under study [22].
In conclusion, this systematic review is the first of its kind to adopt a clustering technique to evaluate the independent data on the literature regarding TMX resistance in ER+ breast cancer patients. This analysis revealed three networks of biomarkers involved in biological processes: cell cycle, signal transduction of proliferative stimuli, and hormone response involved in morphogenesis and differentiation of mammary gland. As expected, CLUSTER 1 corroborated that the cell proliferation pathway is strongly linked to the prediction of TMX response. Further, CLUSTER 2 and 3 indicate that there are other resistance pathways that must be thoroughly investigated as currently available data are not sufficient to reach a statistically significant conclusion. When the direct cluster neighbors are considered, CLUSTER 3 showed a strong link between TMX resistance and the development of the mammary gland and its hormone stimulation. Thus, our data found are hypothesis generators, which suggest promising mechanisms and biomarkers for predicting resistance to TMX and contributing to future studies and personalized medicine.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- TMX:
-
Tamoxifen
- ER+:
-
Estrogen of receptor-positive
- BC:
-
Breast cancer
- HR+:
-
Hormone receptor expression
- PR+:
-
Progesterone receptor
- SERM:
-
Selective estrogen receptor modulator
- REMARK:
-
Reporting recommendations for tumor marker prognostic
- GO:
-
Gene ontology
- BiNGO:
-
Biological Network Gene Ontology
- FDR:
-
False discovery rate
- OS:
-
Overall survival
- AURKA:
-
Aurora kinase A
- MK167:
-
Marker of proliferation Ki-67
- AURKB:
-
Aurora kinase B
- FOXM1:
-
Forkhead box M1
- RRM2:
-
Ribonucleotide reductase regulatory subunit M2
- STAT3:
-
Signal transducer and activator of transcription 3
- PTPN2:
-
Protein tyrosine phosphatase non-receptor type 2
- FYN:
-
Protein-tyrosine kinase oncogene family
- SRC:
-
Proto-oncogene, non-receptor tyrosine kinase
- PAK2:
-
Serine/threonine protein kinase
- FRS2:
-
Fibroblast growth factor receptor substrate 2
- EGFR:
-
Epidermal growth factor receptor
- ESR 1:
-
Estrogen receptor 1
- GATA3:
-
Transcription factor
- PGR:
-
Progesterone receptor
- NCOR2:
-
Nuclear receptor corepressor 2
- EBAG9:
-
Estrogen receptor binding site associated antigen 9
- SOX2:
-
SRY-box transcription factor 2
- POU5F1:
-
POU class 5 homeobox 1
- BCL2:
-
B-cell lymphoma 2
- IGF1R:
-
Insulin like growth factor 1 receptor
- TGFBR2:
-
Transforming growth factor beta receptor 2
- YAP1:
-
Yes-associated protein 1
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN, 2012. Int J Cancer. 2015;136:E359-386. https://doi.org/10.1002/ijc.29210.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. https://doi.org/10.3322/caac.20107.
Yersal O, Barutca S. Biological subtypes of breast cancer: prognostic and therapeutic implications. World J Clin Oncol. 2014;5:412–24. https://doi.org/10.5306/wjco.v5.i3.412.
Mueller SO, Korach KS. Estrogen receptors and endocrine diseases: lessons from estrogen receptor knockout mice. Curr Opin Pharmacol. 2001;1:613–9. https://doi.org/10.1016/s1471-4892(01)00105-9.
Lei JT, Gou X, Seker S, Ellis MJ. ESR1 alterations and metastasis in estrogen receptor positive breast cancer. J Cancer Metastasis Treat. 2019. https://doi.org/10.20517/2394-4722.2019.12.
Morotti M, Bridges E, Valli A, Choudhry H, Sheldon H, Wigfield S, Gray N, Zois CE, Grimm F, Jones D, Teoh EJ, Cheng WC, Lord S, Anastasiou D, Haider S, McIntyre A, Goberdhan DCI, Buffa F, Harris AL. Hypoxia-induced switch in SNAT2/SLC38A2 regulation generates endocrine resistance in breast cancer. Proc Natl Acad Sci USA. 2019;116:12452–61. https://doi.org/10.1073/pnas.1818521116.
Osborne CK, Zhao H, Fuqua SA. Selective estrogen receptor modulators: structure, function, and clinical use. J Clin Oncol. 2000;18:3172–86. https://doi.org/10.1200/JCO.2000.18.17.3172.
Goldstein SR, Siddhanti S, Ciaccia AV, Plouffe L. A pharmacological review of selective oestrogen receptor modulators. Hum Reprod Update. 2000;6:212–24. https://doi.org/10.1093/humupd/6.3.212.
Ao A, Morrison BJ, Wang H, López JA, Reynolds BA, Lu J. Response of estrogen receptor-positive breast cancer tumorspheres to antiestrogen treatments. PLoS ONE. 2011;6: e18810. https://doi.org/10.1371/journal.pone.0018810.
Antunes MV, Linden R, Santos TV, Wallemacq P, Haufroid V, Classen JF, Andreolla H, Costa N, Fontanive TO, Rosa DD. Endoxifen levels and its association with CYP2D6 genotype and phenotype: evaluation of a southern Brazilian population under tamoxifen pharmacotherapy. Ther Drug Monit. 2012;34:422–31. https://doi.org/10.1097/FTD.0b013e318260b46e.
An KC. Selective estrogen receptor modulators. Asian Spine J. 2016;10:787–91. https://doi.org/10.4184/asj.2016.10.4.787.
Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631–43. https://doi.org/10.1038/nrc2713.
Del Re M, Michelucci A, Simi P, Danesi R. Pharmacogenetics of anti-estrogen treatment of breast cancer. Cancer Treat Rev. 2012;38:442–50. https://doi.org/10.1016/j.ctrv.2011.08.003.
Droog M, Beelen K, Linn S, Zwart W. Tamoxifen resistance: from bench to bedside. Eur J Pharmacol. 2013;717:47–57. https://doi.org/10.1016/j.ejphar.2012.11.071.
Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–47. https://doi.org/10.1146/annurev-med-070909-182917.
Yu KD, Wu J, Shen ZZ, Shao ZM. Hazard of breast cancer-specific mortality among women with estrogen receptor-positive breast cancer after five years from diagnosis: implication for extended endocrine therapy. J Clin Endocrinol Metab. 2012;97:E2201-2209. https://doi.org/10.1210/jc.2012-2423.
Cho SK, Pedram A, Levin ER, Kwon YJ. Acid-degradable core-shell nanoparticles for reversed tamoxifen-resistance in breast cancer by silencing manganese superoxide dismutase (MnSOD). Biomaterials. 2013;34(38):10228–37. https://doi.org/10.1016/j.biomaterials.2013.09.003.
Abdel-Hafiz HA. Epigenetic mechanisms of tamoxifen resistance in luminal breast cancer. Diseases. 2017. https://doi.org/10.3390/diseases5030016.
Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J, Chang JC. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–9. https://doi.org/10.1093/jnci/djn123.
Sotgia F, Fiorillo M, Lisanti MP. Mitochondrial markers predict recurrence, metastasis and tamoxifen-resistance in breast cancer patients: early detection of treatment failure with companion diagnostics. Oncotarget. 2017;8:68730–45. https://doi.org/10.18632/oncotarget.19612.
Wirapati P, Sotiriou C, Kunkel S, Farmer P, Pradervand S, Haibe-Kains B, Desmedt C, Ignatiadis M, Sengstag T, Schutz F, Goldstein DR, Piccart M, Delorenzi M. Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res. 2008;10:R65. https://doi.org/10.1186/bcr2124.
Mihaly Z, Kormos M, Lanczky A, Dank M, Budczies J, Szasz MA, Gyorffy B. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res Treat. 2013;140:219–32. https://doi.org/10.1007/s10549-013-2622-y.
Tricco AC, Lillie E, Zarin W, O’Brien KK, Colquhoun H, Levac D, Moher D, Peters MDJ, Horsley T, Weeks L, Hempel S, Akl EA, Chang C, McGowan J, Stewart L, Hartling L, Aldcroft A, Wilson MG, Garritty C, Lewin S, Godfrey CM, Macdonald MT, Langlois EV, Soares-Weiser K, Moriarty J, Clifford T, Tunçalp O, Straus SE. PRISMA extension for scoping reviews (PRISMA-ScR): checklist and explanation. Ann Intern Med. 2018;169:467–73. https://doi.org/10.7326/m18-0850.
McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM. Statistics Subcommittee of NCIEWGoCD REporting recommendations for tumor MARKer prognostic studies (REMARK). Breast Cancer Res Treat. 2006;100:229–35. https://doi.org/10.1007/s10549-006-9242-8.
Nepusz T, Yu H, Paccanaro A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat Methods. 2012;9:471–2. https://doi.org/10.1038/nmeth.1938.
Györffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123:725–31. https://doi.org/10.1007/s10549-009-0674-9.
Browne B, Hochgrafe F, Wu J, Millar E, Barraclough J, Stone A, McCloy R, Lee C, Roberts C, Ali N, Boulghourjian A, Schmich F, Linding R, Farrow L, Gee J, Nicholson R, O’Toole S, Sutherland R, Musgrove E, Butt A, Daly R. Global characterization of signalling networks associated with tamoxifen resistance in breast cancer. FEBS J. 2013;280:5237–57. https://doi.org/10.1111/febs.12441.
Margolin S, Lindh JD, Thorén L, Xie H, Koukel L, Dahl ML, Eliasson E. CYP2D6 and adjuvant tamoxifen: possible differences of outcome in pre- and post-menopausal patients. Pharmacogenomics. 2013;14:613–22. https://doi.org/10.2217/pgs.13.47.
Zhang L, Gong C, Lau SL, Yang N, Wong OG, Cheung AN, Tsang JW, Chan KY, Khoo US. SpliceArray profiling of breast cancer reveals a novel variant of NCOR2/SMRT that is associated with tamoxifen resistance and control of ERα transcriptional activity. Cancer Res. 2013;73:246–55. https://doi.org/10.1158/0008-5472.Can-12-2241.
Ijichi N, Shigekawa T, Ikeda K, Miyazaki T, Horie-Inoue K, Shimizu C, Saji S, Aogi K, Tsuda H, Osaki A, Saeki T, Inoue S. Association of positive EBAG9 immunoreactivity with unfavorable prognosis in breast cancer patients treated with tamoxifen. Clin Breast Cancer. 2013;13:465–70. https://doi.org/10.1016/j.clbc.2013.08.015.
Piva M, Domenici G, Iriondo O, Rábano M, Simões BM, Comaills V, Barredo I, López-Ruiz JA, Zabalza I, Kypta R, Vivanco M. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med. 2014;6:66–79. https://doi.org/10.1002/emmm.201303411.
Elzawahry HM, Saber MM, Mokhtar NM, Zeeneldin AA, Ismail YM, Alieldin NH. Role of Ki67 in predicting resistance to adjuvant tamoxifen in postmenopausal breast cancer patients. J Egypt Natl Canc Inst. 2013;25:181–91. https://doi.org/10.1016/j.jnci.2013.02.001.
Hrstka R, Brychtova V, Fabian P, Vojtesek B, Svoboda M. AGR2 predicts tamoxifen resistance in postmenopausal breast cancer patients. Dis Markers. 2013;35:207–12. https://doi.org/10.1155/2013/761537.
Chen M, Cui YK, Huang WH, Man K, Zhang GJ. Phosphorylation of Y.M. estrogen receptor α at serine 118 is correlated with breast cancer resistance to tamoxifen. Oncol Lett. 2013;6:118–24. https://doi.org/10.3892/ol.2013.1324.
Thrane S, Pedersen AM, Thomsen MB, Kirkegaard T, Rasmussen BB, Duun-Henriksen AK, Lænkholm AV, Bak M, Lykkesfeldt AE. A kinase inhibitor screen identifies Mcl-1 and Aurora kinase A as novel treatment targets in antiestrogen-resistant breast cancer cells. Oncogene. 2014;34:4199–210. https://doi.org/10.1038/onc.2014.351.
Reijm EA, Timmermans AM, Look MP, Meijer-van Gelder ME, Stobbe CK, van Deurzen CH, Martens JW, Sleijfer S, Foekens JA, Berns PM, Jansen MP. High protein expression of EZH2 is related to unfavorable outcome to tamoxifen in metastatic breast cancer. Ann Oncol. 2014;25:2185–90. https://doi.org/10.1093/annonc/mdu391.
Huang R, Faratian D, Sims A, Wilson D, Thomas J, Harrison D, Langdon S. Increased STAT1 signaling in endocrine-resistant breast cancer. PLoS ONE. 2014;9: e94226. https://doi.org/10.1371/journal.pone.0094226.
Wong PP, Yeoh CC, Ahmad AS, Chelala C, Gillett C, Speirs V, Jones JL, Hurst HC. Identification of MAGEA antigens as causal players in the development of tamoxifen-resistant breast cancer. Oncogene. 2014;33:4579–88. https://doi.org/10.1038/onc.2014.45.
Putluri N, Maity S, Kommagani R, Kommangani R, Creighton CJ, Putluri V, Chen F, Nanda S, Bhowmik SK, Terunuma A, Dorsey T, Nardone A, Fu X, Shaw C, Sarkar TR, Schiff R, Lydon JP, O’Malley BW, Ambs S, Das GM, Michailidis G, Sreekumar A. Pathway-centric integrative analysis identifies RRM2 as a prognostic marker in breast cancer associated with poor survival and tamoxifen resistance. Neoplasia. 2014;16:390–402. https://doi.org/10.1016/j.neo.2014.05.007.
Lehn S, Tobin NP, Sims AH, Stål O, Jirström K, Axelson H, Landberg G. Decreased expression of Yes-associated protein is associated with outcome in the luminal A breast cancer subgroup and with an impaired tamoxifen response. BMC Cancer. 2014;14:119. https://doi.org/10.1186/1471-2407-14-119.
Wei C, Cao Y, Yang X, Zheng Z, Guan K, Wang Q, Tai Y, Zhang Y, Ma S, Ge X, Xu C, Li J, Yan H, Ling Y, Song T, Zhu L, Zhang B, Xu Q, Hu C, Bian XW, He X, Zhong H. Elevated expression of TANK-binding kinase 1 enhances tamoxifen resistance in breast cancer. Proc Natl Acad Sci USA. 2014;111:E601-610. https://doi.org/10.1073/pnas.1316255111.
Winder T, Giamas G, Wilson PM, Zhang W, Yang D, Bohanes P, Ning Y, Gerger A, Stebbing J, Lenz HJ. Insulin-like growth factor receptor polymorphism defines clinical outcome in estrogen receptor-positive breast cancer patients treated with tamoxifen. Pharmacogenomics J. 2014;14:28–34. https://doi.org/10.1038/tpj.2013.8.
Redmond AM, Byrne C, Bane FT, Brown GD, Tibbitts P, O’Brien K, Hill AD, Carroll JS, Young LS. Genomic interaction between ER and HMGB2 identifies DDX18 as a novel driver of endocrine resistance in breast cancer cells. Oncogene. 2015;34:3871–80. https://doi.org/10.1038/onc.2014.323.
Bergamaschi A, Madak-Erdogan Z, Kim YJ, Choi YL, Lu H, Katzenellenbogen BS. The forkhead transcription factor FOXM1 promotes endocrine resistance and invasiveness in estrogen receptor-positive breast cancer by expansion of stem-like cancer cells. Breast Cancer Res. 2014;16:436. https://doi.org/10.1186/s13058-014-0436-4.
Nagelkerke A, Sieuwerts AM, Bussink J, Sweep FC, Look MP, Foekens JA, Martens JW, Span PN. LAMP3 is involved in tamoxifen resistance in breast cancer cells through the modulation of autophagy. Endocr Relat Cancer. 2014;21:101–12. https://doi.org/10.1530/erc-13-0183.
Karlsson E, Veenstra C, Emin S, Dutta C, Pérez-Tenorio G, Nordenskjöld B, FornandeR T, Stål O. Loss of protein tyrosine phosphatase, non-receptor type 2 is associated with activation of AKT and tamoxifen resistance in breast cancer. Breast Cancer Res Treat. 2015;153:31–40. https://doi.org/10.1007/s10549-015-3516-y.
Elias D, Vever H, Lænkholm AV, Gjerstorff MF, Yde CW, Lykkesfeldt AE, Ditzel HJ. Correction: gene expression profiling identifies FYN as an important molecule in tamoxifen resistance and a predictor of early recurrence in patients treated with endocrine therapy. Oncogene. 2018;37:5585–6. https://doi.org/10.1038/s41388-018-0495-6.
Hato Y, Kondo N, Yoshimoto N, Endo Y, Asano T, Dong Y, Nishimoto M, Takahashi S, Fujii Y, Nakanishi R, Toyama T. Prognostic impact of a single-nucleotide polymorphism near the CTSO gene in hormone receptor-positive breast cancer patients. Int J Clin Oncol. 2015;21:539–47. https://doi.org/10.1007/s10147-015-0913-5.
Honma N, Horii R, Ito Y, Saji S, Younes M, Iwase T, Akiyama F. Differences in clinical importance of Bcl-2 in breast cancer according to hormone receptors status or adjuvant endocrine therapy. BMC Cancer. 2015. https://doi.org/10.1186/s12885-015-1686-y.
Busch S, Sims AH, Stål O, Fernö M, Landberg G. Loss of TGFβ receptor type 2 expression impairs estrogen response and confers tamoxifen resistance. Cancer Res. 2015;75:1457–69. https://doi.org/10.1158/0008-5472.Can-14-1583.
Larsen SL, Laenkholm AV, Duun-Henriksen AK, Bak M, Lykkesfeldt AE, Kirkegaard T. SRC drives growth of antiestrogen resistant breast cancer cell lines and is a marker for reduced benefit of tamoxifen treatment. PLoS ONE. 2015;10: e0118346. https://doi.org/10.1371/journal.pone.0118346.
Argalacsova S, Slanar O, Vitek P, Tesarova P, Bakhouche H, Drazdakova M, Bartosova O, Zima T, Pertuzelka L. Contribution of ABCB1 and CYP2D6 genotypes to the outcome of tamoxifen adjuvant treatment in premenopausal women with breast cancer. Physiol Res. 2015;64:S539–47. https://doi.org/10.33549/physiolres.933234.
Bentin Toaldo C, Alexi X, Beelen K, Kok M, Hauptmann M, Jansen M, Berns E, Neefjes J, Linn S, Michalides R, Zwart W. Protein Kinase A-induced tamoxifen resistance is mediated by anchoring protein AKAP13. BMC Cancer. 2015. https://doi.org/10.1186/s12885-015-1591-4.
Zhong X, Xie G, Zhang Z, Wang Z, Wang Y, Qiu Y, Li L, Bu H, Li J, Zheng H. MiR-4653–3p and its target gene FRS2 are prognostic biomarkers for hormone receptor positive breast cancer patients receiving tamoxifen as adjuvant endocrine therapy. Oncotarget. 2016;7:61166–82. https://doi.org/10.18632/oncotarget.11278.
Ahern TP, Cronin-Fenton DP, Lash TL, Sørensen HT, Ording AG, Hamilton-Dutoit SJ, Hellberg Y. Pak1, adjuvant tamoxifen therapy, and breast cancer recurrence risk in a Danish population-based study. Acta Oncol. 2016;55:734–41. https://doi.org/10.3109/0284186x.2016.1150606.
Babyshkina N, Vtorushin S, Zavyalova M, Patalyak S, Dronova T, Litviakov N, Slonimskaya E, Kzhyshkowska J, Cherdyntseva N, Choynzonov E. The distribution pattern of ERα expression, ESR1 genetic variation and expression of growth factor receptors: association with breast cancer prognosis in Russian patients treated with adjuvant tamoxifen. Clin Exp Med. 2016;17:383–93. https://doi.org/10.1007/s10238-016-0428-z.
Liu J, Prager-van der Smissen WJ, Look MP, Sieuwerts AM, Smid M, Meijer-van ME, Gelder ME, FoekenS JA, Hollestelle A, Martens JW. GATA3 mRNA expression, but not mutation, associates with longer progression-free survival in ER-positive breast cancer patients treated with first-line tamoxifen for recurrent disease. Cancer Lett. 2016;376:104–9. https://doi.org/10.1016/j.canlet.2016.03.038.
De Marchi T, Timmermans AM, Smid M, Look MP, Stingl C, Opdam M, Linn SC, Sweep FC, Span PN, Kliffen M, van Deurzen CH, Luider TM, Foekens JA, Martens JW, Umar A. Annexin-A1 and caldesmon are associated with resistance to tamoxifen in estrogen receptor positive recurrent breast cancer. Oncotarget. 2016;7:3098–110. https://doi.org/10.18632/oncotarget.6521.
De Marchi T, Liu NQ, Stingl C, Timmermans MA, Smid M, Look MP, Tjoa M, Braakman RB, Opdam M, Linn SC, Sweep FC, Span PN, Kliffen M, Luider TM, Foekens JA, Martens JW, Umar A. 4-protein signature predicting tamoxifen treatment outcome in recurrent breast cancer. Mol Oncol. 2016;10:24–39. https://doi.org/10.1016/j.molonc.2015.07.004.
Sensorn I, Sukasem C, Sirachainan E, Chamnanphon M, Pasomsub E, Trachu N, Supavilai P, Pinthong D, Wongwaisayawan S. ABCB1 and ABCC2 and the risk of distant metastasis in Thai breast cancer patients treated with tamoxifen. Onco Targets Ther. 2016;9:2121–9. https://doi.org/10.2147/ott.S100905.
van der Willik KD, Timmermans MM, van Deurzen CH, Look MP, Reijm EA, van Zundert WJ, Foekens R, Trapman-Jansen AM, den Bakker MA, Westenend PJ, Martens JW, Berns EM, Jansen MP. SIAH2 protein expression in breast cancer is inversely related with ER status and outcome to tamoxifen therapy. Am J Cancer Res. 2016;6:270–84.
Thistle JE, Hellberg Y, Mortensen K, Hamilton-Dutoit S, Kjærsgaard A, Cronin-Fenton D, Sørensen HT, Lash TL. The effect of 14–3-3ζ expression on tamoxifen resistance and breast cancer recurrence: a Danish population-based study. Breast Cancer Res Treat. 2017;165:633–43. https://doi.org/10.1007/s10549-017-4289-2.
Gwak JM, Kim M, Kim HJ, Jang MH, Park SY. Expression of embryonal stem cell transcription factors in breast cancer: Oct4 as an indicator for poor clinical outcome and tamoxifen resistance. Oncotarget. 2017;8:36305–18. https://doi.org/10.18632/oncotarget.16750.
Abudureyimu K, Guo Y, Maimaiti Y, Liu Z, Tan J, Liu C, Nie X, Huang B, Zhou J, Huang T. Aurora kinase B is an exceptional prognostic biomarker for invasive ductal carcinoma and may be a new target for endocrine therapy-resistant breast cancer. Int J Clin Exp Med. 2017;10:3337–44.
Snell CE, Gough M, Middleton K, Hsieh M, Furnas L, Seidl B, Gibbons K, Pyke C, Shannon C, Woodward N, Armes JE. Absent progesterone receptor expression in the lymph node metastases of ER-positive, HER2-negative breast cancer is associated with relapse on tamoxifen. J Clin Pathol. 2017;70:954–60. https://doi.org/10.1136/jclinpath-2016-204304.
Baldacchino S, Wastall LM, Saliba C, Hughes TA, Scerri C, Berwick A, Speirs V, Hanby AM, Grech G. CIP2A expression predicts recurrences of tamoxifen-treated breast cancer. Tumour Biol. 2017;39:1010428317722064. https://doi.org/10.1177/1010428317722064.
Han SH, Kim HJ, Gwak JM, Kim M, Chung YR, Park SY. MicroRNA-222 expression as a predictive marker for tumor progression in hormone receptor-positive breast cancer. J Breast Cancer. 2017;20:35–44. https://doi.org/10.4048/jbc.2017.20.1.35.
Zhang Y, Wester L, He J, Geiger T, Moerkens M, Siddappa R, Helmijr JA, Timmermans MM, Look MP, van Deurzen CHM, Martens JWM, Pont C, de Graauw M, Danen EHJ, Berns EMJJ, Meerman JHN, Jansen MPHM, van de Water B. IGF1R signaling drives antiestrogen resistance through PAK2/PIX activation in luminal breast cancer. Oncogene. 2018;37:1869–84. https://doi.org/10.1038/s41388-017-0027-9.
Moon YW, An HJ, Koo JS, Kim GM, Han H, Park S, Kim SI, Park HS, Kim S, Kim SK, Lee SA, Hwang S, Son GW, Sohn J. CD44/CD24 and aldehyde dehydrogenase 1 in estrogen receptor-positive early breast cancer treated with tamoxifen: CD24 positivity is a poor prognosticator. Oncotarget. 2018;9:2622–30. https://doi.org/10.18632/oncotarget.23519.
Choi HJ, Joo HS, Won HY, Min KW, Kim HY, Son T, Oh YH, Lee JY, Kong G. Role of RBP2-induced ER and IGF1R-ErbB signaling in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2018. https://doi.org/10.1093/jnci/djx207.
Bekele RT, Venkatraman G, Liu RZ, Tang X, Mi S, Benesch MG, Mackey JR, Godbout R, Curtis JM, McMullen TP, Brindley DN. Oxidative stress contributes to the tamoxifen-induced killing of breast cancer cells: implications for tamoxifen therapy and resistance. Sci Rep. 2016;6:21164. https://doi.org/10.1038/srep21164.
Haricharan S, Punturi N, Singh P, Holloway KR, Anurag M, Schmelz J, Schmidt C, Lei JT, Suman V, Hunt K, Olson JA, Hoog J, Li S, Huang S, Edwards DP, Kavuri SM, Bainbridge MN, Ma CX, Ellis MJ. Loss of MutL disrupts CHK2-dependent cell-cycle control through CDK4/6 to promote intrinsic endocrine therapy resistance in primary breast cancer. Cancer Discov. 2017;7:1168–83. https://doi.org/10.1158/2159-8290.Cd-16-1179.
Luo L, Gao W, Wang J, Wang D, Peng X, Jia Z, Jiang Y, Li G, Tang D, Wang Y. Study on the mechanism of cell cycle checkpoint kinase 2 (CHEK2) gene dysfunction in chemotherapeutic drug resistance of triple negative breast cancer cells. Med Sci Monit. 2018;24:3176–83. https://doi.org/10.12659/msm.907256.
Dökümcü K, Farahani RM. Evolution of resistance in cancer: a cell cycle perspective. Front Oncol. 2019. https://doi.org/10.3389/fonc.2019.00376.
Tormo E, Ballester S, Adam-Artigues A, Burgués O, Alonso E, Bermejo B, Menéndez S, Zazo S, Madoz-Gúrpide J, Rovira A, Albanell J, Rojo F, Lluch A, Eroles P. The miRNA-449 family mediates doxorubicin resistance in triple-negative breast cancer by regulating cell cycle factors. Sci Rep. 2019;9:5316. https://doi.org/10.1038/s41598-019-41472-y.
Bergamaschi A, Christensen BL, Katzenellenbogen BS. Reversal of endocrine resistance in breast cancer: interrelationships among 14-3-3ζ, FOXM1, and a gene signature associated with mitosis. Breast Cancer Res. 2011. https://doi.org/10.1186/bcr2913.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. https://doi.org/10.1016/j.cell.2011.02.013.
Ahn SG, Lee HM, Lee HW, Lee SA, Lee SR, Leem SH, Jeong J, Chu IS. Prognostic discrimination using a 70-gene signature among patients with estrogen receptor-positive breast cancer and an intermediate 21-gene recurrence score. In J Mol Sci. 2013;14:23685–99. https://doi.org/10.3390/ijms141223685.
Liao Y, Li J, Fan Y, Xu B. Polymorphisms in AURKA an AURKB are associated with the survival of triple-negative breast cancer patients treated with taxane-based adjuvant chemotherapy. Cancer Manag Res. 2018;10:3801–8. https://doi.org/10.2147/CMAR.S174735.
He DX, Xia YD, Gu XT, Jin J, Ma X. A transcription/translation-based gene signature predicts resistance to chemotherapy in breast cancer. J Pharm Biomed Anal. 2015;102:500–8. https://doi.org/10.1016/j.jpba.2014.10.018.
Toshimitsu H, Hashimoto K, Tangoku A, Iizuka N, Yamamoto K, Kawauchi S, Oga A, Furuya T, Oka M, Sasaki K. Molecular signature linked to acquired resistance to cisplatin in esophageal cancer cells. Cancer Lett. 2004;211:69–78. https://doi.org/10.1016/j.canlet.2004.01.038.
Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Becette V, André S, Piccart M, Campone M, Brain E, Macgrogan G, Petit T, Jassem J, Bibeau F, Blot E, Bogaerts J, Aguet M, Bergh J, Iggo R, Delorenzi M. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med. 2009;15:68–74. https://doi.org/10.1038/nm.1908.
Zajac M, Gomez G, Benitez J, Martínez-Delgado B. Molecular signature of response and potential pathways related to resistance to the HSP90 inhibitor, 17AAG, in breast cancer. BMC Med Genomics. 2010;3:44. https://doi.org/10.1186/1755-8794-3-44.
Miller TW, Balko JM, Ghazoui Z, Dunbier A, Anderson H, Dowsett M, González-Angulo AM, Mills GB, Miller WR, Wu H, Shyr Y, Arteaga CL. A gene expression signature from human breast cancer cells with acquired hormone independence identifies MYC as a mediator of antiestrogen resistance. Clin Cancer Res. 2011;17:2024–34. https://doi.org/10.1158/1078-0432.CCR-10-2567.
Paik S, Shak S, Tang G, Kim C, Bake J, Cronin M, Baehner FL, Walker MG, Watson D, Park T, Hiller W, Fisher ER, Wickerham DL, Bryant J, Wolmark N. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–26. https://doi.org/10.1056/NEJMoa041588.
Kim C, Tang G, Pogue-Geile KL, Costantino JP, Baehner FL, Baker J, Cronin MT, Watson D, Shak S, Bohn OL, Fumagalli D, Taniyama Y, Lee A, Reilly ML, Vogel VG, McCaskill-Stevens W, Ford LG, Geyer CE, Wickerham DL, Wolmark N, Paik S. Estrogen receptor (ESR1) mRNA expression and benefit from tamoxifen in the treatment and prevention of estrogen receptor-positive breast cancer. J Clin Oncol. 2011;29:4160–7. https://doi.org/10.1200/JCO.2010.32.9615.
Acknowledgements
This research was supported by Universidade Federal de Ciências da Saúde de Porto Alegre (UFCSPA), the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-Brazil (CAPES) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). The authors gratefully acknowledge the support of the Translational Research Group in Clinical Oncology and students of Scientific Initiation Camila De Paula Macedo and Mariana Debastiani during research process.
Funding
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-Brasil (CAPES)-Finance Code 001.
Author information
Authors and Affiliations
Contributions
Conceptualization: MDB, GTS, AVS, CGB; Data curation: MDB, GTS, AVS; Formal analysis: AOS, FB, IBMC, CGB; Funding acquisition: JEV, RJVA, CGB; Investigation: MDB, GTS, FB, IBMC; Methodology and Project administration; MDB, GTS, AVS, RJVA, AOS, JEV, CGB. Final review: All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent to publication.
Competing interests
The authors declare that they have no potential conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dal Berto, M., dos Santos, G.T., dos Santos, A.V. et al. Molecular markers associated with the outcome of tamoxifen treatment in estrogen receptor-positive breast cancer patients: scoping review and in silico analysis. Discov Onc 12, 37 (2021). https://doi.org/10.1007/s12672-021-00432-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-021-00432-7