
Abstract
Background
HSP90 may be a favorable target for investigational therapy in breast cancer. In fact, the HSP90 inhibitor, 17AAG, currently has entered in phase II clinical trials as an anticancer agent in breast and other tumors. Since HSP90 inhibition leads to global depletion of oncogenic proteins involved in multiple pathways we applied global analysis using gene array technology to study new genes and pathways involved in the drug response in breast cancer.
Methods
Gene expression profiling using Whole Human Genome Agilent array technology was applied to a total of six sensitive and two resistant breast cancer cell lines pre-treatment and treated with the 17AAG for 24 and 48 hours.
Results
We have identified a common molecular signature of response to 17AAG composed of 35 genes which include novel pharmacodynamic markers of this drug. In addition, different patterns of HSP90 client transcriptional changes after 17AAG were identified associated to the sensitive cell lines, which could be useful to evaluate drug effectiveness. Finally, we have found differentially expressed pathways associated to resistance to 17AAG. We observed significant activation of NF-κB and MAPK pathways in resistant cells upon treatment, indicating that these pathways could be potentially targeted to overcome resistance.
Conclusions
Our study shows that global mRNA expression analysis is a useful strategy to examine molecular effects of drugs, which allowed us the discovery of new biomarkers of 17AAG activity and provided more insights into the complex mechanism of 17AAG resistance.
Similar content being viewed by others

Background
Considering the complexity of breast cancer, with its multiple genetic abnormalities, targeting a single pathway by inhibiting the activity of one component is unlikely to be effective in a long term. Identification of molecular targets that will modulate multiple components of several signalling pathways would be desired for anticancer treatment. To that end, HSP90 gained lately extreme interest and became an interesting cancer drug target [1]. In breast cancer, preclinical studies have demonstrated sensitivity of HER2+ tumors to HSP90 inhibitor [2–4], lately though it was demonstrated that HSP90 is a very effective target of therapy in triple negative breast cancers [5, 6]. HSP90 is a chaperone for several oncogenic client proteins (ERBB2, B-RAF, CDK4, AKT, mutant p53, among others) involved in transcriptional regulation, signal transduction, and cell cycle control as well as in other crucial steps leading to malignant phenotype [7, 8]. Hsp90 is overexpressed in tumor cells, indicating that these cells are highly dependent on the Hsp90 function [9]. Mutant oncoproteins may depend on the full function of Hsp90 as a conformational buffer to maintain full activity [10–13].
The HSP90 inhibitor, 17-allyloamino-17-demethoxy-geldanamycin (17AAG) a geldanamycin analogue, is currently in phase II clinical trials [14] in a number of cancers [15–19], see http://www.clinicaltrials.gov. At present, most of the drug candidates fail relatively late during the process (phase III) of clinical trials due to lack of efficacy [20, 21]. To save that failure there is a high demand for biomarkers that can adequately and with great specificity, indicate the presence or absence of the desired pharmacological response[22]. Since HSP90 inhibition leads to global depletion of oncogenic proteins involved in multiple signaling pathways, expression signatures have been developed to understand the mechanisms of drug action and to predict the sensitivity to treatment. With the microarray technology, rather than studying effect of the drug on a single gene or protein, we can now look for signatures consisting of multiple genes that are altered in some way, and together define novel set of pharmacodynamic biomarkers of the drug response as well as of resistance[23]. Gene expression and proteomic profiling studies have been done previously in a panel of ovarian and colon cancer cell lines after 17AAG treatment [24, 25] and in pancreatic cancer after treatment with 17AAG partner, IPI 504 [26], however there are no previous studies focused in breast tumors under 17AAG treatment.
Although a basic molecular signature of response to 17AAG has been previously defined [27] with depletion of the levels of client proteins such as c-RAF-1 and cyclin-dependant kinase 4 (CDK4), and upregulation of the inducible isoform of HSP70 (HSP72), the exact mechanism of action of 17AAG has not been clearly defined. To that end a discovery of clinical markers of response and mechanism of resistance to 17AAG are still a matter of time. The need to reveal biomarkers and understand the resistance will help to identify responsive versus non responsive patients to 17AAG. In our current study we performed a global gene expression analysis using Whole Human Genome array technology to understand the molecular mechanism of action of 17AAG in breast cancer. First, we have identified a breast cancer signature of response to 17AAG and suggested biomarkers of 17AAG sensitivity in breast cancer. Secondly, we have studied transcriptional changes in known HSP90 clients. And finally, we have identified gene expression and pathway activity differences in response to 17AAG in sensitive versus resistant cell lines.
All together these results may provide further understanding of the mode of action of 17AAG and suggest potential molecular markers of response and drug resistance in breast cancer.
Methods
Breast Cancer cell lines
Eight breast cancer cell lines were included in this study, MCF-7, MDA-MB-157, Hs578T, HCC1937, MDA-MB-436, UACC3199, MDA-MB-231 and T47D). Breast cancer cell lines, MCF-7, UACC3199, Hs578T, MDA-MB-231 and T47 D were obtained from Cancer Epigenetic Group at Spanish National Cancer Centre, Madrid, Spain. HCC1937 and MDA-MB-157 were kindly provided by Dr. P. Edwards from Department of Pathology, University of Cambridge, Cambridge, UK and MDA-MB-436 by Dr. K.S Massey-Brown from Department of Pharmacology and Toxicology, University of Arizona, Tucson, USA. These tumor cell lines were cultured in RPMI 1640 containing 10% fetal bovine serum (Gibco-BRL, Grand Island, NY, USA) with the exception of MDA-MB-157 being cultured in DMEM/F12 (Gibco-BRL) supplemented with 10% fetal bovine serum and maintained at 37°C in 5% CO2. All media were supplemented with fungizone, and penicillin/streptomycin.
Fresh material from a tumor biopsy corresponding to a breast tumor sample was used to validate results obtained in cell lines. Appropriate ethical committee approval and informed consent was obtained. Tumor was cultured in vitro, and after several passages, tumor cells were treated with 17AAG at the same dose used for the cell lines. These primary tumor cells were grown in F-10 medium (Gibco), supplemented with 10% fetal bovine serum and 2% Ultroser G (PALL Life Sciences).
Drug and treatment protocol
17AAG (Sigma-Aldrich, St.Louis, MO, USA) was prepared as a 1 mM stock in dimethyl sulfoxide (DMSO) and stored at -20°C and freshly dissolved immediately prior to use. Cells were seeded in 10 cm dishes at a moderate density in 20 ml complete medium. At 24 h after plating, cells were treated with 500 nM 17AAG or DMSO (0.1%) as a control. At appropriate intervals 24 H, 48 H upon treatment with the drug, cells were harvested.
RNA extraction, cRNA amplification, labeling and hybridization
Total RNA was extracted with TriReagent (Molecular Research Center, Cincinnati, OH, USA). Purity and integrity of the RNA was assessed with Agilent 2100 BioAnalyzer (Agilent Technologies, Palo Alto, CA, USA). Then, 500 ng of total RNA from samples and Universal Human Reference RNA (Stratagene, La Jolla, CA, USA) were used for amplification and labeling using the Agilent's Low RNA Input Linear Amplification Kit (Agilent Technologies) following the detailed protocol described in the kit manual. Cyanine 5 labeled samples or Cyanine 3 labeled reference cRNAs were purified using QIAGEN's RNeasy mini spin columns and eluted in 30 μl of nuclease-free water. After amplification and labeling, cRNA quantity and cyanine incorporation were determined using a nanodrop ND.1000 UV-VIS-Spectrophotometer version 3.2.1 (Agilent Technologies). For each hybridization,1 μg Cyanine 3 labeled cRNA (reference) and 1 μg of Cyanine 5 labeled cRNA (samples) were mixed, fragmented, and hybridized at 65°C for 17 hours to an Agilent 4 × 44 K Whole Human genome Oligo Microarray containing 45,015 features representing 41,000 unique probes. We have hybridized each sample at each time point (untreated, treated 24 H and 48 H with 17AAG), including some technical replicates. After washing, microarrays were scanned using an Agilent Array scanner (Agilent Technologies). Images were analyzed. Reproducibility and reliability of each single microarray was assessed using Quality Control report data. Data were extracted with Agilent feature extraction software (version 9.5.3) using the GE2-v5_95_Feb07 protocol. Background substraction were carried out using normexp. Lowess and quantiles methods were performed for intra-array and inter-array normalization respectively. Expression ratios were calculated (Cy5 processed signal was divided by Cy3 processed signal) and log2 transformed. Gene patterns containing missing values were discarded. Additionally a filter procedure eliminated genes with uniformly low expression or with low expression variation across the experiments, retaining 20374 genes and transcripts. Microarray dataset is publicly available at GEO database http://www.ncbi.nlm.nih.gov/geo/info/linking.html (GEO accession number, GSE23209).
Differential gene expression analysis
To determine if there were genes differentially expressed between treated and untreated and sensitive versus resistant breast cancer cell lines supervised classification was performed with linear models (limma) implemented in the POMELO II tool, available in Asterias web server http://asterias.bioinfo.cnio.es. The estimated significance level (p value) was corrected for multiple hypotheses testing using Benjamini & Hochberg [28] False Discovery Rate (FDR) adjustment. Those genes with FDR <0.05 were selected as significantly differentially expressed.
Clustering
Average linkage hierarchical clustering (Pearson correlation, uncentered metrics) from Gene Cluster and Treeview http://rana.stanford.edu/software algorithms were used to obtain clustering of the data sets. The level of expression of each gene in each sample, relative to the median level of expression of that gene across all the samples is represented using a red-white-blue color scale. Blue correspond to expression value below median, white, equal to median and red above the median.
Functional Profiling of genes and pathways
We used Gene Set Enrichment Analysis (GSEA) to gain insight into global molecular networks and canonical pathways related to differentially expressed genes associated to resistant phenotype after 17AAG treatment. In our data analysis we have included the Biocarta http://www.biocarta.com as a source of pathway annotation, and whichgenes web-based tool for building 249 gene sets with application in gene set enrichment analysis [29]. The ranking of genes was performed with T test, with an absolute mode for gene list sorting. Gene set permutations were used to assess the statistical significance of the pathways. Those pathways showing FDR <0.05, a well-established cut-off for the identification of biologically relevant gene sets [30], were considered significantly enriched between classes under comparison. The ranking of genes was performed with T test, with an absolute mode for gene list sorting.
Validation of microarray data by quantitative RT-PCR analysis
One μg of total RNA was reverse transcribed using MMLV Reverse Transcriptase (Invitrogen, San Diego, CA, USA) and random primers. The cDNAs were subjected to quantitative real-time PCR (QT-PCR) assay with the use of labeled probes for selected genes (Roche Universal Probe library, Basel, Switzerland) and the TaqMan Universal PCR Mix in an ABI prism 7900 system (Applied Biosystems Inc, Foster City, CA, USA) under manufacturer's recommendations. The PCR amplification was carried out with 10 min at 95°C, followed by 50 cycles of 15 s at 95°C and 1 min at 60°C, using the oligonucleotides shown in Additional file 1: Supplemental Table S1. All experiments were analyzed in triplicate.
Validation of microarray data by western blot analysis
The protein expression changes were examined after 24 H and 48 H of 17AAG treatment. Western blot analysis was performed using standard procedures for whole-cell extracts from cell lines as described previously [31]. A set of antibodies used for immunodetection are listed in the Additional file 1: Supplemental Table S2. The immunoblotting was repeated at least twice in all of the experiments presented in this study.
Validation of microarray data by cell based multi-pathway activity assays
Cignal Reporter Assay was performed according to manufacturer's instructions. Briefly, the transcription factor-responsive reporter, negative control, and positive control constructs were diluted in Opti-MEM (Invitrogen). The diluted nucleic acids were mixed with the diluted SureFECTTM (transfection reagent; SABiosciences) and delivered to 20,000 cells in a 96-well plate format. Culture media were changed 24 H after transfection. Transfection efficiency was estimated by following the expression of GFP (in the positive control wells) using fluorescence microscopy. At 24 H post transfection the cells were treated with the drug. After 48 H of treatment, cells were harvested into cell lysis buffer (Promega). Luciferase activities were determined using the Dual-Luciferase Assay System (Promega) and a multilabel reader (Promega). The Firefly/Renilla activity ratio generated from the transcription factor-responsive reporter transfections was divided by the Firefly/Renilla activity ratio generated from the negative control transfections to obtain the relative luciferase units. At least three independent transfections were carried out in triplicate for each of the conditions tested with each reporter assay. This experimental design was repeated at least once.
Statistical analyses
Analyses of pathway activity differences upon treatment in the resistant and sensitive cell lines were performed using SPSS version 17.0 (SPSS Inc, Chicago, IL). Non-parametric, Mann-Whitney U test was used for our statistical hypothesis testing. P-values reported are two-sided.
Results
Identification of a molecular signature of response to 17AAG
Previous studies have shown feasibility of differential gene expression approach to create expression-based signatures predicting response or resistance in human tumors to therapeutic agents. Here, we investigated the biology underlying the differential gene expression across breast cancer cell lines treated with HSP90 inhibitor, 17AAG.
A total of eight breast cancer cell lines were analyzed. In a previous study, six of them (MCF-7, MDA-MB-157, Hs578T, HCC1937, MDA-MB-436, and UACC3199) were demonstrated to be sensitive to treatment with 17AAG in a dose and time dependent manner with IC50 values between 0.014 ± 0.006 μM and 0.059 ± 0.017 μM at 96 h [31]. In addition, two other cell lines were also analyzed (MDA-MB-231, T47D) and found to be resistant to 17AAG, showing IC50 values of 1.92 ± 0.4 μM and 3.82 ± 0.97 μM, respectively. Then, all cell lines were treated with 500 nM 17AAG for 24 and 48 hours, concentration at which sensitive cells showed cytotoxic and cytostatic effects as well as G2/M arrest and degradation of client proteins [31], and whole genome expression profiling of control and treated cells at this time points were performed.
Unsupervised hierarchical clustering of all samples using more than 20000 genes, as expected, did not separate the treated versus untreated cell lines. Instead, all 17AAG treated cell populations clustered with its control cell line, suggesting that the effect of the drug is very subtle in the whole genome context or that the effect of the drug is largely specific for each cell line (Additional file 1: Supplemental Figure S1). In general, after treatment with 17AAG, there was an increase in the number of genes with expression changes >2-fold over control cells after 48 h than after 24 h. Roughly, the proportion of genes with >2-fold changes at 48 h ranged between 8% and 35% in sensitive cells. Resistant cell lines showed lower number of genes, only 2% (MBA-MD-231) and 2.8% (T47D) of genes showing >2-fold change versus control (Figure 1). Significantly, only a small number of genes were commonly altered in the resistant cell lines after treatment.

Gene expression changes induced by 17AAG. Averaged number of genes with expression changes >2-fold following 17AAG treatment are represented for all breast cancer cell lines, at both time points 24 H and 48 H. Resistant cell lines MDA-MB-231 and T47 D exhibited much lower variation after treatment.
Regardless of the specific changes found in each cell line, we searched for genes commonly associated to sensitivity to 17AAG. Analysis of differentially expressed genes between all treated compared with all untreated 17AAG sensitive breast cancer cell lines was performed. A list of 35 genes with a significant FDR value <0.05 was obtained, which constituted a molecular signature of response to 17AAG in breast cancer cells (Figure 2A). Among the 35 genes, 15 were down-regulated and 20 up-regulated genes following treatment (Table 1). The up-regulated genes following treatment included heat shock family proteins such as HSP90AA1, HSPA8, DNAJB12, HSPA1L, DNAJA1, HSPA4L, consistently with other array studies in different tumor cell types [24, 32, 33]. Among other genes showing increased expression after treatment were ubiquitin-conjugating enzyme E2C (UBE2C), death effector domain containing 2 (DEDD2), zinc finger protein 587 (ZNF587), zinc finger protein 473 (ZNF473), Rac GTPase activating protein 1 (RACGAP1), MHC class I polypeptide-related sequence B (MICB), regulator of G-protein signalling 2 (RGS2), cysteine and histidine-rich domain (CHORD)-containing 1 (CHORDC1), PPAR binding protein. There were also down-regulated genes in response to 17AAG which included e.g. JUNB, CYCLIN D1, NFKBIA, immediate early response 3 (IER3), transmembrane protein 129 (TMEM129) or replication factor C (activator 1) 4 (RFC4).

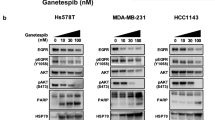
Gene signature of response to 17AAG in breast cancer cell lines. A) Heat-map represents the expression of significant differentially expressed 35 genes included in the molecular signature of response to 17AAG. Untreated samples were compared to treated samples from all sensitive cell lines to obtain common variations. B) Comparison between the changes in expression in the genes included in the signature in sensitive and resistant cells. Fold changes after 17AAG for sensitive cell lines, taken the averaged expression of all of them, compared to fold changes in resistant MDA-MB-231 cells or T47 D.Error bars represent standard deviation. C) Western blot analysis of different HSP70 isoforms before and after 17AAG in resistant MDA-MB-231 and sensitive MCF-7 cell line showed differential HSP70 induction after 17AAG. Lack of induction of the HSPA2 and HSPA1L in resistant cells, and increased expression of HSP72 and HSC70 in both resistant and sensitive cell lines upon treatment is shown. GAPDH was used as loading control.
As expected, most of the 35 genes from the signature of response did not change upon treatment in the resistant cell lines. However, some members of the co-chaperone complex such as, HSP90AA1, HSPA4L, HSPA8, DNAJA1 and CHORDC1 showed induction following treatment, in the resistant MDA-MB-231 cell line, but not in T47 D cells (Figure 2B). Interestingly, HSP90 and HSP70 induction are common markers of response to 17AAG. Indeed, HSP70 is one of the most commonly used pharmacodynamic markers of HSP90 inhibition in clinical trials nowadays. However, we have seen induction of the most common isoforms of HSP70, HSPA8 (HSC70) and HSP72, not only in sensitive cell lines but also in MDA-MB-231 resistant cell line. The induction of HSP90, HSPA8 (HSC70) and HSP72 in MDA-MB-231 cells was confirmed by QT-PCR and western blot (Figure 2C). We also analyzed by western blot other isoforms of HSP70, such as HSPA1L, which was part of the signature of response, and HSPA2 (Figure 2C). These two isoforms, in contrary to HSC70 and HSP72 were found exclusively induced in responsive cell lines upon treatment, suggesting that they could be better markers of sensitivity to 17AAG. Moreover, down or up-regulation of other differentially expressed genes from the molecular signature of response to 17AAG was also validated by QT-PCR (Additional file 1: Supplemental Figure S2).
Validation of the signature of response in a tumor biopsy
To validate and investigate the significance of the obtained molecular signature of 17AAG response, a breast tumor biopsy from a patient with breast cancer was grown in vitro and treated with the same concentration used for tumor cell lines for 24 h and 48 h. Similarly to what was seen in cell lines [31], 17AAG produced in these primary tumor cells a growth inhibitory effect, with an arrest in G2/M phase of the cell cycle (Figure 3). Then, changes in the expression of a group of seven genes from the signature (HSP90, HSPA8, DNAJA1, CHORDC1, DEDD2, HSPA4L and MICB) were assessed by QT-PCR comparing with untreated cells. Significantly all these genes were confirmed to be 17AAG-responsive genes, also in this primary tumor.

Effect of HSP90 inhibition by 17AAG in a biopsy from a breast cancer patient. A) Cell cycle analysis in untreated cells and cells treated with 17AAG for 24 H and 48 H, exhibiting G2/M arrest at both time points. B) Validation of a group of genes from the signature of response to 17AAG in the primary tumor cells. All these genes showed increased expression after treatment >2fold at 24 H or 48 H, as occurred in the sensitive cell lines.
Transcriptional changes in HSP90 client proteins
The cellular response to 17AAG has a complex nature with effects including protein and transcriptional changes [25]. As the treatment with 17AAG has a global effect in the cell through depletion of client proteins, we were interested in analysing the subsequent changes induced by HSP90 inhibition at transcription level. We characterized the profile of transcriptional changes of the list of 168 well reported Hsp90 client proteins and interactors available on the Picard lab home page http://www.picard.ch/DP/DPhome.html. Since expression of HSP90 client proteins vary according to cell type, we have presented the data of HSP90 interactors' expression as differences in expression in treated cell lines versus their corresponding untreated cells. This analysis revealed that each cell line has an individual pattern of Hsp90 interactors' changes (Figure 4A). As we expected, variations in the expression levels of genes coding for client proteins were much lower in resistant cell lines comparing to the sensitive ones (Figure 4A), suggesting that transcriptional changes of at least some client proteins could be taken into account to measure drug effectiveness.

Transcriptional changes of HSP90 clients. A) Heatmap representing the differences in the mRNA expression found in the list of known HSP90 clients and interactors in treated breast cancer cell lines (increments over untreated cells in red, reductions in blue). Weaker variations in resistant cell lines are shown. B) Significant HSP90 client transcriptional changes in individual cell lines following 17AAG treatment. Genes for which mRNAs exhibited over 2-fold increase or reduction are shown for the different cell lines. Stars indicate genes commonly changing at least in four of the six cell lines.
To identify crucial differentially expressed HSP90 clients in each cell line, expression changes of >2 fold after treatment were obtained. Indeed, the sensitive cell lines showed a much higher number of HSP90 clients that differ following treatment, compared to the resistant cell lines. By selecting HSP90 interactors that change >2 fold at both time points, 24 h and 48 h following 17AAG, Hs578T cells showed 35 genes, MCF-7 showed 9 genes; MDA-MB-157 showed 20 genes; MDA-MB-436 showed 13 genes; HCC1937 showed 17 genes and UACC3199 showed 9 genes (Figure 4B), while the resistant cell lines, T47 D showed only 3 genes and in MDA-MB-231 only 2 genes changed. Moreover, there were only two HSP90 interactors commonly upregulated in all the sensitive cell lines: HSPA1A (HSP72), the inducible isoform of HSP70, and CHORDC1. Interestingly, the resistant cell line MDA-MB-231 showed exactly these two genes HSPA1A and CHORDC1 upregulated at both time points with no other HSP90 interactor significantly changing its expression. Upregulation of HSPA1A was detected also in T47 D resistant cells only after 48 h post treatment.
In addition to HSPA1A and CHORDC1, other client protein transcripts expression such as ASHA1 and CCNB1 were commonly induced, and IRAK1 decreased, with the treatment in at least four of the six cell lines (Figure 4B).
Signaling Pathways related to resistance to 17AAG
It is well established that a major factor in resistance to 17-AAG is expression/activity of DT-diaphorase (NQO1), [34, 35]. The resistant cell line, MDA-MB-231, has very low level of NQO1 [34], and it is likely to be the main cause of 17AAG resistance in this cells. In contrary, the other resistant cell line T47 D has higher levels of NQO1 and may have another, independent mode of resistance to 17AAG. Regardless of the intrinsic mode of resistance in the two cell lines, we investigate genes and pathways differentially modulated in responsive and resistant cells following treatment.
Differential expression profiling between resistant and sensitive cell lines after treatment was performed. As expected, large differences were found, with more than 1500 genes significantly changing (data not shown). Many of these differences could be attributed to the fact that sensitive treated cells represent arrested cells, while resistant cells are proliferating cells. In fact, significant number of these genes was involved in cellular metabolism.
Then, we tried to find whether there were significant signaling pathways that could be differentially expressed between treated sensitive and resistant cells. In order to obtain significant pathways, gene set enrichment analysis (GSEA) was performed. Because of the small number of samples this analysis did not show any pathways associated with 17AAG resistance with a FDR < 0.05. Even though the three top pathways more associated to 17AAG resistance included CDMAC pathway (FDR = 0.157), NF-κB pathway (FDR = 0.205) and ATM pathway (FDR = 0.246) (See Additional file 1: Supplemental Table S3, for a list of the top 10 pathways related to 17AAG resistant cells).
Additionally to the in silico pathway analysis, to look for differentially activated pathways following 17AAG treatment we have applied transcription factor-responsive luciferase reporter assays to resistant (T47 D, MDA-MB-231) and two sensitive cell lines (Hs578T, MCF-7). The latter quantitatively assesses the signal transduction pathway changes by measuring the activities of downstream transcription factors. By this method we evaluated activation of ten different cancer related pathways such as NOTCH, WNT, TGFβ, P53, cell cycle, MYC/MAX, NF-κB, MAPK/ERK, MAPK/JNK and HIF1.
Interestingly, the results revealed that NF-κB and MAPK/JNK pathway were found significantly activated in both resistant compared to the sensitive cell lines (Figure 5). Although below statistical significance, GSEA revealed NF-κB as one of the most relevant pathways associated to 17AAG resistance. The increased activity of the NF-κB, may indicate that this pathway plays an important role in cell survival upon treatment. Additionally, MAPK pathways could have also some role in the resistance to 17AAG, as MAPK/JNK was significantly activated in both resistant T47 D and MDA-MB-231 cell lines, and the MAPK/ERK was induced in T47 D. Cell cycle pathway was significantly activated only in MDA-MB-231 resistant cell line, what may reflect the higher proliferation status of these cells. Sensitive MCF-7 cells showed a critical reduction in the activity of all of the pathways, which correlate with the stop in proliferation and death induced after the treatment (Figure 5). The other sensitive cell line, Hs578T also showed inhibition of most of the pathways except NOTCH, TGF and Hypoxia pathway.

Differential signaling pathway activation in resistant and sensitive breast cancer cells upon treatment with 17AAG. Transfections were carried out in the sensitive (MCF-7 and Hs578T) and resistant (MDA MB 231 and T47D), cells with the reporter constructs for the typical cancer biology pathways: NOTCH, WNT, TGFβ, P53, cell cycle, MYC/MAX, NF-κB, MAPK/ERK, MAPK/JNK and HIF, followed by 17AAG or DMSO treatment. Dual-luciferase assay was performed, and promoter activity values are expressed as arbitrary units using a Renilla reporter for internal normalization. The experiment performed itself in triplicates was repeated at least twice of each cell line studied. The error bars represent standard deviation. Pathways significantly (p-value < 0.05) repressed or activated are marked with a star.
Discussion
In an attempt to identify detailed molecular mechanisms of drug response and resistance to treatment with 17AAG we performed gene expression profiling of eight breast cancer cell lines treated with the drug. Although there are many studies analyzing the molecular effect of HSP90 inhibition, global expression changes after 17AAG in breast cancer have not been analyzed in-depth. In this study we examined the expression changes in the sensitive (MCF-7, MDA-MB-157, Hs578T, UACC3199, HCC1937, MDA-MB-436) and two resistant (MDA-MB-231, T47D) breast cancer cell lines to the HSP90 inhibitor, 17AAG.
Gene expression profiling after 17AAG, showed different number of 17AAG responsive genes associated to the different cell lines. As HSP90 inhibition results in degradation of client proteins, it is possible that intrinsic differences in the abundance of HSP90 clients in the cells cause subsequent transcriptional changes in a cell line dependent manner. Although it was clear that HSP90 inhibition produced cell line dependent changes we could not associate them to the fact that these cell lines belong to different molecular breast cancer subtypes. Since one of the resistant cell lines, MDA-MB-231, is reported to be basal B and the other, T47 D, as luminal, and the rest of the sensitive cell lines being all basal B except for MCF7 (luminal) and HCC 1937 (basal A) [36] it is likely that sensitivity to 17AAG is not connected to known breast cancer subtypes.
However, there were a group of genes commonly regulated in all sensitive cell lines upon treatment. We have identified a breast cancer associated molecular signature of response to 17AAG, consisting of 35 17AAG-responsive genes. This gene signature of 17AAG response included, similar as previously reported in other studies [24–26], members of the chaperon complex, HSP90 itself and HSP70 (HSPA8). These changes were identified in a study done in an ovarian cancer cell line as likely on-target effects of the drug, which are induced as a direct consequence of HSP90 inhibition [25]. Importantly, HSP70 isoforms have been used as pharmacodynamic end point in clinical trials [27]. In addition to these chaperons, we also identified up-regulation of other members of heat shock response family such as HSPA4L, HSPA1L, HSP40 (DNAJA1 and DNAJB12) and in CHORDC1, another HSP90 binding protein. Interestingly, we found transcriptional induction of HSP90, HSP70 (HSPA8) and HSPA4L in one of the resistant cell lines analyzed, MDA-MB-231. Moreover, HSC70/HSPA8 and HSP72 induction was confirmed by western blot in these cells. The up-regulation after treatment of HSP90 and HSP70 (both HSC70 and HSP72) might suggest that HSP90 is inhibited at 500 nM 17AAG in both sensitive and MDA-MB-231 resistant cell line. Yet, the resistance in the MDA-MB-231cell lines might be caused by very low levels of NQO1, as reported previously [37]. HSC70 and HSP72 also have an antiapoptotic role [38], so their induction in MDA-MB-231 might also contribute to the 17AAG resistance in these cells. The other resistant T47 D cells seem not to show the induction of Hsp70 isoforms following treatment what could suggest lack of HSP90 target inhibition by 17AAG. Interestingly, T47 D cells have some expression of NQO1 and probably an alternative mechanism of resistance.
The analysis of a number of HSP70 isoforms by immunoblotting revealed, in addition to induction of HSC70 and HSP72, that some other HSP70 isoforms were also induced by 17AAG. Up-regulation of HSPA1L and HSPA2 were found in sensitive cells following exposure to 17AAG. However, they showed lack of induction in resistant MDA-MB-231 cells. We suggest that HSPA1L and HSPA2 could represent potential biomarkers to follow up the effectiveness of 17AAG in breast cancer, although the mechanism underlying this effect is still unclear.
Other genes from the signature also exhibiting increased expression in response to 17AAG were Rac GTP-ase activating protein (RACGAP1), ubiquitin conjugating enzyme E2C (UBE2C), zinc fingers proteins (ZNF473, ZNF587) and MHC class I antigen (MICB). These data are in line with previous work done on 17AAG treated ovarian cancer cell lines [25]. Additionally, in a recent microarray study of novel HSP90 inhibitor (IPI-504) in pancreatic cancer, Song and colleagues [26] identified similar class of up-regulated genes following treatment along with GTPase activating proteins, zinc finger proteins, heat shock proteins and ribosomal proteins.
There were also genes with decreased expression following HSP90 inhibition by 17AAG. Some of them clearly represent cell cycle regulators (CCND1, PLK3) and important proliferation signaling pathways mediators (JUNB, NFKBIA). The decreased expression of them might be a consequence of cell cycle arrest produced after 17AAG.
The fact that the expression changes seen in the primary tumor sample after treatment with 17AAG resembled the changes in cell lines, suggests that this set of genes would constitute a robust signature of response in breast cancer. Further studies in additional tumor biopsies are required to better establish the value of the biomarkers identified in this study.
Since effects of 17AAG are driven by HSP90 client proteins degradation, we were interested in studying whether protein depletion also results in transcriptional changes of known client proteins following treatment. Changes in the mRNA levels of a number of client proteins were evident in cell lines responsive to 17AAG, while resistant cell lines demonstrated insignificant variations in transcriptional levels of HSP90 interactors. This observation suggests that the use of transcriptional changes of HSP90 client proteins may facilitate the selection of potentially responsive patients to 17AAG therapy. It is known that client proteins are variable in different types of tumor [10]. It is reasonable then, to find cell line specific transcriptional changes profiles in the 17AAG sensitive cell lines. This finding could be of interest in order to define, in further studies, key client proteins for specific tumor subtypes, with potential clinical significance. In addition, consistently up or down-regulated HSP90 client transcripts following treatment were identified shared by some of the cell lines analyzed (AHSA1, CCNB1, IRAK1), that could represent important HSP90 clients in breast cancer. It is clear that biological processes are regulated not only at transcriptional level, but also protein levels or posttranscriptional modifications of proteins are important when analyzing the effects of HSP90 inhibitors. However, mRNA changes could be helpful in order to evaluate the effect of the drug in clinical samples.
Mechanisms of resistance to 17AAG remain largely unknown. We analyze global expression changes after 17AAG occurring in resistant cells, to define genes or pathways commonly involved in insensitivity to this drug. The identification of pathways in relation to 17AAG resistance would be important to develop in future candidate treatments to be used in combination with 17AAG to induce growth inhibitory effects in the insensitive cell lines. Functional studies with the pathway-focused reporter assays shown significant up-regulation of NF-κB pathway in resistant cells after exposure to 17AAG. It is interesting that GSEA also showed NF-κB pathway as potentially involved in resistance to 17AAG. In addition, there are two other resistance associated pathways in the top-ten list that were related to inflammation/immune response (NTHI_PATHWAY and INFLAM_PATHWAY) (Additional file 1: Supplemental Table S3). Thus it is possible that resistant breast cancer cell lines make use of the inflammation/immune response machinery to evade cell cycle arrest or apoptosis after 17AAG treatment. NF-κB activation was already observed after treatment with cancer chemotherapeutic agents such as gemcitabine [39–41], thereby inducing resistance to apoptosis which results in poor clinical outcome. NF-κB is a ubiquitously expressed transcription factor that is involved in a wide spectrum of cellular functions including cell cycle control, stress adaptation, inflammation and control of apoptosis [42]. Activation of NF-κB has been implicated in the development of a number of human malignancies, and it appears to be important for the survival of cancer cells, as well as the conferring of more aggressive tumor phenotype and resistance to drug therapies [43–46]. Moreover, high basal levels of NF-kB have been related with resistance to gemcitabine in pancreatic carcinoma cell lines [41, 45]. The finding of NF-kB pathway activation after 17AAG treatment significantly associated to resistant cell lines suggests that simultaneous use of anti-tumor agents that block NF-κB activity together with HSP90 inhibitors may have a greater therapeutic value. MAPK/JNK, and also MAPK/ERK pathway were activated in the resistance cell lines, suggesting these pathways may be important for survival of cells after 17AAG treatment.
In summary, in this study we have established a 35 gene-based molecular signature of response to 17AAG in breast cancer, which revealed novel pharmacodynamic markers of drug response. Secondly, we have defined transcriptional changes in known HSP90 client proteins, which may be useful to monitor drug efficacy and finally, we have identified signaling pathways differentially activated following 17AAG in resistant cell lines.
Conclusions
This study confirms gene expression profiling as a useful tool in further understand molecular response to drugs and in the discovery of novel pharmacodynamic markers. By analyzing gene expression changes induced by 17AAG in breast cancer cells we have identified a novel breast cancer associated molecular signature of response to 17AAG, consisting of 35 17AAG-responsive genes which might be potential biomarkers of sensitivity to evaluate clinical response. Analysis of mRNA changes of known HSP90 clients demonstrated cell line specific patterns associated with responsive cells, so this approach could be useful for the follow up of treatment response and could facilitate the discovery of key clients. In addition, signaling pathways associated with resistance to 17AAG were found, such as induction in DNA-binding activity of NF-κB in resistant cells after treatment. The identification of pathways associated to resistance would enable the design of combined therapies to overcome 17AAG resistance.
Abbreviations
- 17AAG :
-
17-allyloamino-17-demethoxy-geldanamycin
- DMSO :
-
Dimethyl sulfoxide
- FDR :
-
False Discovery Rate
- GSEA :
-
Gene Set Enrichment Analysis
- QT-PCR :
-
Quantitative real-time Polymerase Chain Reaction.
References
Eccles SA, Massey A, Raynaud FI, Sharp SY, Box G, Valenti M, Patterson L, de Haven Brandon A, Gowan S, Boxall F, et al: NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008, 68 (8): 2850-2860. 10.1158/0008-5472.CAN-07-5256.
Chiosis G, Timaul MN, Lucas B, Munster PN, Zheng FF, Sepp-Lorenzino L, Rosen N: A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem Biol. 2001, 8 (3): 289-299. 10.1016/S1074-5521(01)00015-1.
Neckers L: Heat shock protein 90 inhibition by 17-allylamino-17- demethoxygeldanamycin: a novel therapeutic approach for treating hormone-refractory prostate cancer. Clin Cancer Res. 2002, 8 (5): 962-966.
Modi S, Stopeck AT, Gordon MS, Mendelson D, Solit DB, Bagatell R, Ma W, Wheler J, Rosen N, Norton L, et al: Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: a phase I dose-escalation study. J Clin Oncol. 2007, 25 (34): 5410-5417. 10.1200/JCO.2007.11.7960.
Caldas-Lopes E, Cerchietti L, Ahn JH, Clement CC, Robles AI, Rodina A, Moulick K, Taldone T, Gozman A, Guo Y, et al: Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc Natl Acad Sci USA. 2009, 106 (20): 8368-8373. 10.1073/pnas.0903392106.
Bao R, Lai CJ, Qu H, Wang D, Yin L, Zifcak B, Atoyan R, Wang J, Samson M, Forrester J, et al: CUDC-305, a novel synthetic HSP90 inhibitor with unique pharmacologic properties for cancer therapy. Clin Cancer Res. 2009, 15 (12): 4046-4057. 10.1158/1078-0432.CCR-09-0152.
Maloney A, Workman P: HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert Opin Biol Ther. 2002, 2 (1): 3-24. 10.1517/14712598.2.1.3.
Neckers L: Heat shock protein 90: the cancer chaperone. J Biosci. 2007, 32 (3): 517-530. 10.1007/s12038-007-0051-y.
Yano M, Naito Z, Tanaka S, Asano G: Expression and roles of heat shock proteins in human breast cancer. Jpn J Cancer Res. 1996, 87 (9): 908-915.
Grbovic OM, Basso AD, Sawai A, Ye Q, Friedlander P, Solit D, Rosen N: V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc Natl Acad Sci USA. 2006, 103 (1): 57-62. 10.1073/pnas.0609973103.
Shimamura T, Lowell AM, Engelman JA, Shapiro GI: Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res. 2005, 65 (14): 6401-6408. 10.1158/0008-5472.CAN-05-0933.
da Rocha Dias S, Friedlos F, Light Y, Springer C, Workman P, Marais R: Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2005, 65 (23): 10686-10691. 10.1158/0008-5472.CAN-05-2632.
Blagosklonny MV: Hsp-90-associated oncoproteins: multiple targets of geldanamycin and its analogs. Leukemia. 2002, 16 (4): 455-462. 10.1038/sj.leu.2402415.
Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ: A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003, 425 (6956): 407-410. 10.1038/nature01913.
Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Kung AL, Davies FE, Morgan G, Akiyama M, Shringarpure R, Munshi NC, et al: Antimyeloma activity of heat shock protein-90 inhibition. Blood. 2006, 107 (3): 1092-1100. 10.1182/blood-2005-03-1158.
Senju M, Sueoka N, Sato A, Iwanaga K, Sakao Y, Tomimitsu S, Tominaga M, Irie K, Hayashi S, Sueoka E: Hsp90 inhibitors cause G2/M arrest associated with the reduction of Cdc25C and Cdc2 in lung cancer cell lines. J Cancer Res Clin Oncol. 2006, 132 (3): 150-158. 10.1007/s00432-005-0047-7.
Schwock J, Pham NA, Cao MP, Hedley DW: Efficacy of Hsp90 inhibition for induction of apoptosis and inhibition of growth in cervical carcinoma cells in vitro and in vivo. Cancer Chemother Pharmacol. 2008, 61 (4): 669-681. 10.1007/s00280-007-0522-8.
Williams CR, Tabios R, Linehan WM, Neckers L: Intratumor injection of the Hsp90 inhibitor 17AAG decreases tumor growth and induces apoptosis in a prostate cancer xenograft model. J Urol. 2007, 178 (4 Pt 1): 1528-1532. 10.1016/j.juro.2007.05.120.
Solit DB, Osman I, Polsky D, Panageas KS, Daud A, Goydos JS, Teitcher J, Wolchok JD, Germino FJ, Krown SE, et al: Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin Cancer Res. 2008, 14 (24): 8302-8307. 10.1158/1078-0432.CCR-08-1002.
Lewin DA, Weiner MP: Molecular biomarkers in drug development. Drug Discov Today. 2004, 9 (22): 976-983. 10.1016/S1359-6446(04)03272-6.
Rolan P, Atkinson AJ, Lesko LJ: Use of biomarkers from drug discovery through clinical practice: report of the Ninth European Federation of Pharmaceutical Sciences Conference on Optimizing Drug Development. Clin Pharmacol Ther. 2003, 73 (4): 284-291. 10.1016/S0009-9236(02)17625-9.
Dancey JE, Dobbin KK, Groshen S, Jessup JM, Hruszkewycz AH, Koehler M, Parchment R, Ratain MJ, Shankar LK, Stadler WM, et al: Guidelines for the development and incorporation of biomarker studies in early clinical trials of novel agents. Clin Cancer Res. 16 (6): 1745-1755. 10.1158/1078-0432.CCR-09-2167.
Kulasingam V, Diamandis EP: Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nat Clin Pract Oncol. 2008, 5 (10): 588-599. 10.1038/ncponc1187.
Clarke PA, Hostein I, Banerji U, Stefano FD, Maloney A, Walton M, Judson I, Workman P: Gene expression profiling of human colon cancer cells following inhibition of signal transduction by 17-allylamino-17-demethoxygeldanamycin, an inhibitor of the hsp90 molecular chaperone. Oncogene. 2000, 19 (36): 4125-4133. 10.1038/sj.onc.1203753.
Maloney A, Clarke PA, Naaby-Hansen S, Stein R, Koopman JO, Akpan A, Yang A, Zvelebil M, Cramer R, Stimson L, et al: Gene and protein expression profiling of human ovarian cancer cells treated with the heat shock protein 90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2007, 67 (7): 3239-3253. 10.1158/0008-5472.CAN-06-2968.
Song D, Chaerkady R, Tan AC, Garcia-Garcia E, Nalli A, Suarez-Gauthier A, Lopez-Rios F, Zhang XF, Solomon A, Tong J, et al: Antitumor activity and molecular effects of the novel heat shock protein 90 inhibitor, IPI-504, in pancreatic cancer. Mol Cancer Ther. 2008, 7 (10): 3275-3284. 10.1158/1535-7163.MCT-08-0508.
Banerji U, O'Donnell A, Scurr M, Pacey S, Stapleton S, Asad Y, Simmons L, Maloney A, Raynaud F, Campbell M, et al: Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol. 2005, 23 (18): 4152-4161. 10.1200/JCO.2005.00.612.
Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I: Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001, 125 (1-2): 279-284. 10.1016/S0166-4328(01)00297-2.
Glez-Pena D, Gomez-Lopez G, Pisano DG, Fdez-Riverola F: WhichGenes: a web-based tool for gathering, building, storing and exporting gene sets with application in gene set enrichment analysis. Nucleic Acids Res. 2009, W329-334. 10.1093/nar/gkp263. 37 Web Server
Hu Z, Fan C, Livasy C, He X, Oh DS, Ewend MG, Carey LA, Subramanian S, West R, Ikpatt F, et al: A compact VEGF signature associated with distant metastases and poor outcomes. BMC Med. 2009, 7: 9-10.1186/1741-7015-7-9.
Zajac M, Moneo MV, Carnero A, Benitez J, Martinez-Delgado B: Mitotic catastrophe cell death induced by heat shock protein 90 inhibitor in BRCA1-deficient breast cancer cell lines. Mol Cancer Ther. 2008, 7 (8): 2358-2366. 10.1158/1535-7163.MCT-08-0327.
Amundson SA, Bittner M, Chen Y, Trent J, Meltzer P, Fornace AJ: Fluorescent cDNA microarray hybridization reveals complexity and heterogeneity of cellular genotoxic stress responses. Oncogene. 1999, 18 (24): 3666-3672. 10.1038/sj.onc.1202676.
Fambrough D, McClure K, Kazlauskas A, Lander ES: Diverse signaling pathways activated by growth factor receptors induce broadly overlapping, rather than independent, sets of genes. Cell. 1999, 97 (6): 727-741. 10.1016/S0092-8674(00)80785-0.
Kelland LR, Sharp SY, Rogers PM, Myers TG, Workman P: DT-Diaphorase expression and tumor cell sensitivity to 17-allylamino, 17-demethoxygeldanamycin, an inhibitor of heat shock protein 90. J Natl Cancer Inst. 1999, 91 (22): 1940-1949. 10.1093/jnci/91.22.1940.
Gaspar N, Sharp SY, Pacey S, Jones C, Walton M, Vassal G, Eccles S, Pearson A, Workman P: Acquired resistance to 17-allylamino-17-demethoxygeldanamycin (17-AAG, tanespimycin) in glioblastoma cells. Cancer Res. 2009, 69 (5): 1966-1975. 10.1158/0008-5472.CAN-08-3131.
Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al: A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006, 10 (6): 515-527. 10.1016/j.ccr.2006.10.008.
Han Y, Shen H, Carr BI, Wipf P, Lazo JS, Pan SS: NAD(P)H:quinone oxidoreductase-1-dependent and -independent cytotoxicity of potent quinone Cdc25 phosphatase inhibitors. J Pharmacol Exp Ther. 2004, 309 (1): 64-70. 10.1124/jpet.103.059477.
Powers MV, Clarke PA, Workman P: Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell. 2008, 14 (3): 250-262. 10.1016/j.ccr.2008.08.002.
Steiner T, Junker U, Henzgen B, Nuske K, Durum SK, Schubert J: Interferon-alpha suppresses the antiapoptotic effect of NF-kB and sensitizes renal cell carcinoma cells in vitro to chemotherapeutic drugs. Eur Urol. 2001, 39 (4): 478-483. 10.1159/000052489.
Lind DS, Hochwald SN, Malaty J, Rekkas S, Hebig P, Mishra G, Moldawer LL, Copeland EM, Mackay S: Nuclear factor-kappa B is upregulated in colorectal cancer. Surgery. 2001, 130 (2): 363-369. 10.1067/msy.2001.116672.
Hernandez-Vargas H, Rodriguez-Pinilla SM, Julian-Tendero M, Sanchez-Rovira P, Cuevas C, Anton A, Rios MJ, Palacios J, Moreno-Bueno G: Gene expression profiling of breast cancer cells in response to gemcitabine: NF-kappaB pathway activation as a potential mechanism of resistance. Breast Cancer Res Treat. 2007, 102 (2): 157-172. 10.1007/s10549-006-9322-9.
Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB, Iglehart JD: NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci USA. 2004, 101 (27): 10137-10142. 10.1073/pnas.0403621101.
Dong G, Chen Z, Kato T, Van Waes C: The host environment promotes the constitutive activation of nuclear factor-kappaB and proinflammatory cytokine expression during metastatic tumor progression of murine squamous cell carcinoma. Cancer Res. 1999, 59 (14): 3495-3504.
Arlt A, Grobe O, Sieke A, Kruse ML, Folsch UR, Schmidt WE, Schafer H: Expression of the NF-kappa B target gene IEX-1 (p22/PRG1) does not prevent cell death but instead triggers apoptosis in Hela cells. Oncogene. 2001, 20 (1): 69-76. 10.1038/sj.onc.1204061.
Arlt A, Gehrz A, Muerkoster S, Vorndamm J, Kruse ML, Folsch UR, Schafer H: Role of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene. 2003, 22 (21): 3243-3251. 10.1038/sj.onc.1206390.
Biswas DK, Martin KJ, McAlister C, Cruz AP, Graner E, Dai SC, Pardee AB: Apoptosis caused by chemotherapeutic inhibition of nuclear factor-kappaB activation. Cancer Res. 2003, 63 (2): 290-295.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1755-8794/3/44/prepub
Acknowledgements
This work was supported by grants from Spanish Fondo de Investigaciones Sanitarias (FIS-PI081298) and Grant from Ministerio de Education y Ciencia (MEC), SAF06-06140.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MZ carried out hybridizations, participated in the analysis of results and have been involved in drafting the manuscript. GG gave support for the analysis and interpretation of microarray data and performed the statistical analysis. JB participated in the design of the study and analysis of results. BMD conception and design of the study, supervision of experiments and helped to draft the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12920_2010_179_MOESM1_ESM.DOC
Additional file 1: Supplemental tables and figures. Supplemental Table S1: Oligonucleotides used for quantitative RT-PCR to validate expression of the selected from microarray study. Supplemental Table S2: List of the antibodies used for western blot. Supplemental Table S3: Top-ten pathways more associated to resistance to 17AAG. Supplemental Figure S1: Unsupervised clustering showed that samples mostly grouped by cell line. Some samples have technical replicates (marked with a star). Supplemental Figure S2: QT-PCR measurements of selected genes from the signature, confirmed the down regulation of five genes from the molecular signature (CYCLIN D1, JUNB, TMEM129, PLK3, NFKBIA) and up-regulation of UBE2C after treatment. (DOC 548 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zajac, M., Gomez, G., Benitez, J. et al. Molecular signature of response and potential pathways related to resistance to the HSP90 inhibitor, 17AAG, in breast cancer. BMC Med Genomics 3, 44 (2010). https://doi.org/10.1186/1755-8794-3-44
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8794-3-44