
Abstract
Endocrine resistance may develop as a consequence of enhanced growth factor signaling. Fibroblast growth factor 2 (FGF2) consists of a low and several high molecular weight forms (HMW-FGF2). We previously demonstrated that antiprogestin-resistant mammary carcinomas display lower levels of progesterone receptor A isoforms (PRA) than B isoforms (PRB). Our aim was to evaluate the role of FGF2 isoforms in breast cancer progression. We evaluated FGF2 expression, cell proliferation, and pathway activation in models with different PRA/PRB ratios. We performed lentiviral infections of different FGF2 isoforms using the human hormone-responsive T47D-YA cells, engineered to only express PRA, and evaluated tumor growth, metastatic dissemination, and endocrine responsiveness. We assessed FGF2 expression and localization in 81 human breast cancer samples. Antiprogestin-resistant experimental mammary carcinomas with low PRA/PRB ratios and T47D-YB cells, which only express PRB, displayed higher levels of HMW-FGF2 than responsive variants. HMW-FGF2 overexpression in T47D-YA cells induced increased tumor growth, lung metastasis, and antiprogestin resistance compared to control tumors. In human breast carcinomas categorized by their PRA/PRB ratio, we found nuclear FGF2 expression in 55.6% of tumor cells. No differences were found between nuclear FGF2 expression and Ki67 proliferation index, tumor stage, or tumor grade. In low-grade tumor samples, moderate to high nuclear FGF2 levels were associated to carcinomas with low PRA/PRB ratio. In conclusion, we show that HMW-FGF2 isoforms are PRB targets which confer endocrine resistance and are localized in the nuclei of breast cancer samples. Hence, targeting intracellular FGF2 may contribute to overcome tumor progression.
Similar content being viewed by others


Introduction
Two-thirds of breast cancers express estrogen receptor α (ERα) and progesterone receptor (PR) at the time of diagnosis [1] and endocrine therapies that block the ERα pathway are usually the standard treatment, while PR expression is mainly used as a surrogate marker of a functional ERα [2, 3]. Two main PR isoforms have been described, PRA and PRB, which play different roles in mammary gland development [4, 5]. We have previously demonstrated that antiprogestin-responsive mammary carcinomas show higher PRA/PRB ratios than antiprogestin-resistant variants [6].
Despite expressing hormone receptors, most patients develop endocrine resistance with time [7]. Deregulation of growth factor signaling pathways have been proposed as a potential mechanism of hormone resistance [8]. Genomic profiling studies have demonstrated that the fibroblast growth factor receptor (FGFR) pathway is aberrantly regulated in breast cancer [9]. This can occur through different mechanisms, including increased expression of FGFR and ligands [9]. About 22 FGFR ligands have been described, with fibroblast growth factor 2 (FGF2), being one of the prototypical members of this family. A single FGF2 mRNA can give rise to five different protein variants through alternative translation sites, with the higher molecular weight variants (HMW 34, 24, 22.5, and 22 kDa) being co-linear N-terminal extensions of the lower molecular weight variant (LMW 18 kDa). Most attention has been dedicated to the latter, which is mainly secreted by cells to the extracellular matrix, activating cell surface FGFR. On the other hand, the HMW-FGF2 variants rarely leave their producing cells, are accumulated in the nucleus, and can act independently of FGFR [10].
A number of studies have provided comprehensive information on the genomic alterations in FGFR and its paracrine or autocrine activation by LMW-FGF2, while the role of HMW-FGF2 in breast cancer progression remains poorly understood [11].
The aim of this study was to investigate the role of FGF2 isoforms in endocrine resistance and metastatic dissemination. In experimental breast cancer models, we established that hormone-resistant breast carcinomas express higher levels of HMW-FGF2 than hormone-responsive tumors in luminal murine carcinomas and human cell lines. Moreover, we demonstrated that low-grade breast carcinomas with higher levels of PRB than PRA express higher levels of nuclear FGF2 than those with the opposite ratio. These results strongly suggest that targeting intracellular FGF2, in addition to the classical membrane FGFR pathway, may be a promising strategy to overcome tumor progression.
Materials and Methods
Reagents and Antibodies
Mifepristone (MFP), 17β-estradiol (E2), and 4-hydroxytamoxifen (TAM) were purchased from Sigma-Aldrich. Telapristone acetate (TLP) is from Repros Therapeutics (The Woodlands). FGF2 (sc-79), total AKT (sc-8312), ERK (sc-94), pERK (sc-7383), PKCα (sc-208), PR (sc-7208), and ERα (sc-543) were all purchased from Santa Cruz Biotechnology, and phosphorylated Ser473 AKT (4060) was purchased from Cell Signaling Technology. Secondary antibodies for Western blot and immunohistochemistry were obtained from Amersham Pharmacia Biotech and Vector Laboratories, respectively.
Animal Models
Murine tumors or human cell lines were subcutaneously transplanted into the right inguinal flank of 2-month-old virgin female BALB/c mice (IBYME Animal Facility) and NOD/LtSz-scid/IL-2Rgamma null mice (NSG; Jackson Labs), respectively. Animal care and manipulation were in agreement with institutional guidelines.
Murine ERα+ and PR+ breast cancer model: C4-HI/C4-HIR, 59-2-HI/59-2-HIR and C7-2-HI/C7-2-HIR are hormone-independent (HI) variants from the C4, 59, and C7 tumor families, respectively; C4-HI, 59-2-HI, and C7-2-HI are antiprogestin-responsive tumors, whereas C4-HIR, 59-2-HIR, and C7-2-HIR are antiprogestin-resistant variants [12].
T47D-YA FGF2-infected cell variants were inoculated into the right inguinal flank of NSG mice previously treated with E2 (sc 0.25 mg slow-release pellet; [6]).
Cell Lines
T47D-Y cells, which are PR-, T47D-YA cells, and T47D-YB cells, engineered to only express PRA or PRB, respectively, were cultured as previously described [13]. All cell lines were validated by Genetica DNA Laboratories in 2017.
Primary Cultures and Cell Proliferation Assays
Primary cultures and cell proliferation assays were performed as described [14]. [3H]-thymidine uptake was used to quantify cell proliferation in murine primary cultures: after attachment, cells were starved in 1% charcoal-stripped fetal calf serume (chFCS) for 24 h and then treated for 48 h with FGF2. Treatments were performed in octuplicates.
Total cell counting was used to quantify proliferation in human cells; after attachment, T47D-YA and T47D-YB cells were starved in 1% chFCS for 24 h and then treated for 5 days with the experimental solutions (100 ng/mL FGF2; MFP, TLP, and TAM all at 10−7 M). Treatments were performed in quadruplicates.
Western Blot (WB)
Total cell fractions were processed for WB as described [15, 16] using ERK as a loading control. Cell cultures were starved for 24 h by using culture medium (DMEM/F12 without phenol red, Sigma-Aldrich) without serum and were then incubated with 100 ng/mL FGF2.
Breast cancer samples were pulverized and proteins extracted with NE-PER extraction reagents (Thermo Scientific) [15]; band intensity was quantified using the ImageJ software.
Transient PRB Cell Transfection
T47D-Y cells were transiently transfected with control pSG5 or human PRB expression plasmids [17] using Lipofectamine 2000 (Invitrogen Life Technologies) according to the manufacturer’s protocol. At 16 h post-transfection, the media was changed and 24 h later, the cells were processed for WB.
Immunohistochemistry (IHC)
Formalin-fixed, paraffin-embedded murine and human breast cancer tissue sections were dewaxed in xylene, rehydrated through graded ethanols, and incubated for 30 min at room temperature with 3% H2O2 in distilled water to quench endogenous peroxidase activity. The antigen retrieval was achieved by boiling tissue sections for 50 min in 10 mM sodium citrate buffer (pH 6). The slides were then incubated in 2.5% albumin (Sigma-Aldrich) in PBS for 1 h and allowed to react with the primary antibodies overnight at 4 °C. The sections were washed with PBS and successively incubated for 1 h at room temperature with biotin-conjugated secondary antibodies (Vector Labs), and the avidin/biotin peroxidase complex (Vectastain Elite ABC kit; Vector), and then revealed under microscopic control with liquid 3-3′diaminobenzidine and substrate chromogen system (Dako, Agilent Technologies). The slides were lightly counterstained with 10% hematoxylin (Biopur), dehydrated, cleared, and mounted in DPX (Sigma-Aldrich). Primary and secondary antibodies were used at 1/100 and 1/400 dilutions, respectively.
Immunofluorescence (IF)
T47D-YA FGF2-infected cell variants were grown on coverslips, washed, and fixed in cold 10% formalin for 20 min. The cells were permeabilized with 0.25% methanol-Triton X-100 for 20 min, blocked with 10% FCS, and incubated overnight at 4 °C with the primary anti-FGF2 antibody (1/100) dissolved in 2.5% albumin (Sigma-Aldrich). On the next day, cells were washed and incubated with secondary antibody (1/100 Texas Red-conjugated, Vector). Negative controls lacked incubation with the primary antibody. Stained cells were analyzed using a Nikon Eclipse E800 microscope.
Preparation of Lentiviral Particles and Stable T47D-YA Infection
Lentiviral packaging was performed in HEK-293T cells with polyethylenimine (PEI87K). 5 × 106 HEK-293T cells were seeded in 10% FCS DMEM/F12 in 100 mm2 dishes. The lentiviral vector (12 μg), gag-pol (7.5 μg), and ENV (4 μg) were mixed with 0.15 M NaCl to a final volume of 83 μL. The DNA was added to 750 μL of Optimem (Invitrogen) previously mixed with 35 μL of 25 mM PEI87K. The cells were incubated with the mixture at 37 °C for 5 h. Next, the media was replaced by fresh medium; after 48–72 h, the conditioned media was collected and centrifuged and the supernatant stored at − 70 °C. For infection, exponentially growing T47D-YA cells were mixed for 48 h with lentiviral particles previously diluted 1:2 with fresh medium in the presence of 0.8 μg/mL polybrene (Sigma-Aldrich) in a six-well plate. T47D-YA were stably infected with p6NST50 (empty vector), FGF2-18 kDa, or FGF2-22.5 kDa plasmids [18], monitored for GFP expression and selected with zeocin (Invivogen; 300 μg/mL).
Enzyme-Linked Immunosorbent Assay (ELISA)
FGF2 levels in cell culture supernatants were determined using an ELISA kit (Abcam) following the manufacturer’s instructions. A series of FGF2 concentrations (0–300 pg/mL) were used to plot the standard curve in parallel. Optical densities were determined at 450 nm in a Multiskan MS microplate reader (Thermo Fisher Scientific). All cell culture supernatants were tested in triplicate.
Tumor Growth
Tumor growth was measured twice a week using a Vernier caliper. When tumors reached a size of 25–40 mm2, animals were divided in two groups and treated with 6 mg sc MFP or silastic vehicle pellets [19]. At the end of the experiments, the mice were euthanized, and tumors and other tissues were excised, weighed, fixed in 10% buffered formalin, and embedded in paraffin for histological evaluation. Tumor histology and incidence of lung metastasis were evaluated in H&E-stained slides.
Human Tissue Microarrays (hTMA)
Tissue microarrays were constructed and categorized according to the PR isoform ratio (PRA/PRB) as described [15] using specimens from breast cancer patients undergoing breast cancer surgery at the Hospital Magdalena V. de Martínez, General Pacheco, Buenos Aires. The study was approved by the Institutional Review Boards (#CE08/2016). Detailed information of human breast cancer samples is provided in Table 1.
Samples were categorized according to the PR isoform ratio (PRA/PRB) as determined by WB. A nuclear PRA/PRB ratio of 1.2 or greater was considered as high PRA (PRA-H), whereas a PRA/PRB ratio of 0.83 or less was defined as high PRB (PRB-H). Samples with a ratio falling between 1.2 and 0.83 were considered equimolar.
Scoring of Human Slide Sections
Immunohistochemistry staining was reviewed in detail and a blind semiquantitative scoring was performed by a pathologist (MM). The positivity (0–100%) and intensity of the staining was graded as negative (0), weak (1), moderate (2), and strong (3). Cytosolic and nuclear FGF2 staining were expressed as the percentage of stained cells (0–100%) times the intensity (0–3), in a scale ranging from 0 to 300.
Statistical Analysis
Data analysis, unless otherwise indicated, was performed by one-way ANOVA followed by the Tukey test to compare means of multiple samples and Student’s t test to compare the means of two different experimental groups. Tumor growth curves were compared using linear regression analysis.
For clinical data, analysis of continuous variables was performed using one-way ANOVA followed by correlation using linear regression and only variables with p < 0.05 in the univariate analysis were incorporated into the multivariate analysis. Contingency table analysis followed by Fisher’s exact test was performed to compare FGF2 localization in high or low PR+ samples and in PRA-H or PRB-H samples.
p values are indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001. All experiments were performed at least three times, except otherwise informed.
Results
FGF2 Expression in Murine Mammary Carcinomas and Human Cell Lines with Different Hormone Responsiveness
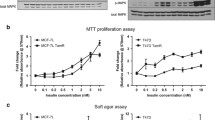
Using the MPA-induced breast cancer model, we have shown that stromal FGF2 induces hormone receptor activation driving tumor growth [16]. These tumors display higher levels of PRA than PRB and regress with antiprogestin therapy. However, after continuous treatment, resistant tumors may arise displaying a low PRA/PRB ratio [12]. To understand the role of FGF2 in tumor progression, we evaluated FGF2 expression in responsive and resistant variants. FGF2 localized mainly in the stromal compartment of responsive tumors, whereas in the resistant variants, the staining was also localized in the tumor parenchyma, being mostly cytosolic with occasional nuclear labeling (Fig. 1a and Supplementary Fig. 1a). WB studies revealed an increased expression of total, LMW-, and HMW-FGF2 in antiprogestin-resistant tumors compared to the responsive variants (Fig. 1b and Supplementary Fig. 1b).

Increased HMW-FGF2 expression and signaling pathway activation in hormone-resistant compared to hormone-responsive tumors. a Immunohistochemistry and b WB of FGF2 in murine C4 tumor samples and human T47D-YA and T47D-YB xenografts. Bar: 40 μm; inset 15 μm. ERK was used as loading control. Immunoreactive bands were quantified and graphed relative to the hormone-responsive variant in each case. c, d Cell proliferation upon FGF2 treatment (1–100 ng/mL) measured by [3H]-thymidine uptake (c) or cell counting (d) in primary cultures of epithelial cells from the C4 family and T47D-YA and T47D-YB cells. e FGFR signaling was evaluated by WB analysis of pERK/ERK, PKCα/ERK, and pAKT/AKT in T47D-YA and T47D-YB whole-cell extracts with or without treatment with exogenous LMW-FGF2 (100 ng/mL; 10 min). Immunoreactive bands were quantified and graphed relative to the T47D-YA cell line or the untreated T47D-YA and T47D-YB cell lines. ERK was used as loading control. f WB analysis of PR and FGF2 in T47D-Y whole-cell extracts transfected with control (pSG5) or PRB plasmids. Immunoreactive bands were quantified and graphed relative to the T47D-Y-pSG5 cell line. ERK was used as loading control. The asterisks in e and f indicate the statistical significance of treated cells (+) vs. untreated cells (−). HMW, high molecular weight; LMW, low molecular weight
Similar results were obtained with human T47D-YA and T47D-YB cells [13], which are antiprogestin responsive or resistant, respectively [6] (Fig. 1a, b).
Effect of LMW-FGF2 on Cell Proliferation and Signaling Pathways
Treatment with exogenous LMW-FGF2 only increased in vitro cell proliferation of responsive variants of murine and human models (Fig. 1c, d). Moreover, LMW-FGF2 induced ERK and AKT phosphorylation in T47D-YA cells while PKCα and AKT pathways are constitutively activated in T47D-YB cells. Further, LMW-FGF2 was able to increase pERK phosphorylation in resistant cells (Fig. 1e).
To confirm that there is a direct link between PRB and FGF2 expression, we transiently transfected a PRB plasmid into T47D-Y cells, which do not express PR, and an increase in HMW-FGF2 expression was observed (Fig. 1f).
Collectively, these results indicate that the FGF2/FGFR pathway is constitutively activated in resistant tumors and suggest that PRB associates with increased intrinsic FGF2 levels, favoring antiprogestin resistance.
Overexpression of FGF2 Isoforms in T47D-YA Cell Line
To further explore the role of FGF2 isoforms in hormone-resistant breast cancer, we infected T47D-YA cells with viral particles containing the following plasmids: p6NST50 (empty vector), pFGF2-18 kDa (LMW-FGF2) and pFGF2-22.5 kDa (HMW-FGF2). We verified FGF2 overexpression by WB and IF (Fig. 2a, b). T47D-YA-22.5 cells expressed high levels of 22.5 kDa isoform along with low levels of LMW-FGF2 (Fig. 2a). Both FGF2-expressing cells displayed an increased cytosolic and nuclear FGF2-staining (Fig. 2b) and FGF2 was significantly accumulated in the conditioned media of these cell lines (Fig. 2c, d) compared to T47D-YA-p6NST50. WB evaluation of the cell culture supernatant revealed that the main secreted isoform corresponded to the 18-kDa form, although a faint band matching the 22.5-kDa isoform was also detected in the T47D-YA-22.5 cells (Fig. 2d).

Generation of T47D-YA cell lines that overexpress FGF2 isoforms. a, b FGF2 expression and nuclear quantification assessed by WB (a) and IF (b) of T47D-YA-p6NST50, T47D-YA-18, and T47D-YA-22.5 cells growing in culture. Bar 40 μm. Inset 10 μm. c ELISA FGF2 quantification and d WB of secreted FGF2 in cell culture supernatant (CS) of T47D-YA-p6NST50, T47D-YA-18, and T47D-YA-22.5 cells
Effect of Overexpression of FGF2 Isoforms on Cell Proliferation, Tumor Growth, and Endocrine Resistance
Cells with overexpression of the 18- and 22.5-kDa FGF2 isoforms had an increased proliferation compared to T47D-YA-p6NST50 cells (Fig. 3a). In vivo, the final tumor burden of the T47D-YA-22.5 xenografts was higher than those of the other tumor lines (Fig. 3b and Supplementary Fig. 2a). T47D-YA-p6NST50 xenografts are undifferentiated tumors, similar to T47D-YA tumors [6]. All three infected cell lines displayed invasive characteristics in the tumor front and also grew inside the mammary ducts (Supplementary Fig. 2b, c).

Overexpression of 22.5 kDa FGF2 isoform induces T47D-YA cell proliferation, tumor growth, endocrine resistance, and 22.5 metastatic dissemination. a Baseline cell proliferation of T47D-YA-p6NST50, T47D-YA-18, and T47D-YA-22.5 cells was measured by total cell counting. b In vivo growth of T47D-YA-p6NST50, T47D-YA-18, and T47D-YA-22.5 cells (NSG mice, N = 4/group). Tumor size, tumor weight, and metastatic lung foci at the end of the experiment (day 56 after cell injection). The asterisks indicate the statistical significance of experimental groups vs. T47D-YA-p6NST50. c Cell proliferation of T47D-YA-p6NST50, T47D-YA-18, and T47D-YA-22.5 cells was measured by total cell counting. Cells were starved for 24 h in 1% chFCS and then treated during 5 days with 10−7 M of mifepristone (MFP; left), telapristone acetate (TLP; center), or tamoxifen (TAM; right). The asterisks indicate the statistical significance of treated cells (+) vs. untreated cells (−). d In vivo growth of T47D-YA-p6NST50, T47D-YA-18, and T47D-YA-22.5 cells (NSG mice, N = 4/group). The data shown is representative of two independent experiments. Inset, tumor weight at the end of each experiment
T47D-YA-22.5 xenografts display vascular embolia and are highly aggressive: invading arterial walls and adipose and muscle tissues (Supplementary Fig. 2b). All xenografts developed lung metastasis; however, mice bearing T47D-YA-22.5 tumors carried a higher number of metastatic foci compared to mice bearing T47D-YA-p6NST50 and T47D-YA-18 tumors (Fig. 3b). IHC staining confirmed that T47D-YA-22.5 metastatic lung foci also overexpress FGF2 (Supplementary Fig. 2d). Both FGF2-overexpressing xenografts showed positive FGF2 staining, being exclusively nuclear and stronger in T47D-YA-22.5 compared to T47D-YA-18 tumors (Supplementary Fig. 2e).
We then examined whether the overexpression of FGF2 isoforms had an impact on endocrine responsiveness. Whereas control cells are responsive to antiprogestins (MFP and TLP) and antiestrogen (TAM) treatment, FGF2-overexpressing cells are hormone resistant (Fig. 3c). Moreover, the ERα and PR antagonists stimulated the proliferation of the T47D-YA-22.5 cells. In vivo, MFP induced stromal remodeling and inhibited tumor growth only in control tumors (Supplementary Fig. 2c and Fig. 3d). MFP-treated T47D-YA-18 and T47D-YA-22.5 xenografts continued to grow: in the former tumor growth was almost exclusively confined to the mammary ducts and in the latter, xenografts maintained their highly invasive ability (Supplementary Fig. 2c).
FGF2 Expression and Localization in Human Breast Cancer Samples
We analyzed FGF2 expression in a cohort of 81 breast cancer samples (Table 1) classified according to their PR isoform ratio in PRA-H and PRB-H samples [15]. FGF2 nuclear immunostaining in tumor cells was observed in 55.6% (45/81) of tumor samples (Fig. 4a).

Low-grade PRB-H human samples express higher levels of nuclear HMW-FGF2 than low-grade PRA-H samples. a FGF2 immunohistochemistry. Representative images of cytosolic (left) and nuclear (right) FGF2 expression in a subset of luminal breast cancer samples. Bar 50 μm. b Percentage of PRA-H or PRB-H samples with a FGF2 nuclear score > 90. c Western blot analysis and d quantification of FGF2 expression in nuclear cell extracts from human breast cancer samples. Total ERK was used as loading control. FGF2 expression was normalized to a positive control which was run in all gel electrophoresis. nFGF2, nuclear FGF2; HMW, high molecular weight; LMW, low molecular weight
PRB-H samples exhibit lower levels of PR than PRA-H samples [15]. We assessed FGF2 expression and localization and found that samples with low total PR (≤ 25%) are more likely to have a nuclear staining for FGF2 (nFGF2; Fisher, p = 0.08) than tumors with high total PR (≥ 75%). Moreover, we found a trend towards an association between PRB-H carcinomas and a higher score for nuclear FGF2 (nFGF2 > 90; Fisher, p = 0.29; Fig. 4b). In fact, when analyzing only low-grade tumor samples (1 and 2), higher nuclear FGF2 levels were significantly related to PRB-H carcinomas (nFGF2 > 90; Fisher, p = 0.02; Fig. 4b). We found no significant association between nuclear FGF2 expression and Ki67 proliferation index, tumor stage, or tumor grade.
We also determined whether nuclear FGF2 staining in PRB-H samples was associated with expression of HMW-FGF2 isoforms. As observed in Fig. 4c, d, increased levels of LMW- and HMW-FGF2 isoforms were confirmed in the PRB-H samples. Our results suggest that nuclear FGF2 is associated with low-grade PRB-H samples and thus, targeting FGF2 in addition to the classical pathway through FGFR, may be a promising strategy to overcome endocrine resistance.
Discussion
The major contribution of this study is that we found that intrinsic expression of FGF2, and particularly HMW-FGF2, is associated with endocrine-resistant breast cancer, suggesting that high intracellular FGF2 levels may be indicative of breast cancer progression.
Few studies show that HMW-FGF2 contributes to chemoresistance [11, 20, 21] and radioresistance in experimental models [22,23,24]. Our results represent the first experimental evidence which demonstrates that increased expression of FGF2 isoforms is associated with hormone resistance in breast cancer. However, hormone receptor activation through growth factor signaling has been proposed as a mechanism underlying endocrine resistance and several studies have suggested that FGFR inhibition may reverse endocrine resistance [25,26,27,28,29]. Moreover, Shee et al. have recently demonstrated that LMW-FGF2, secreted by the tumor microenvironment, mediates antiestrogen resistance in ER+ breast cancer cells [30]. We have focused in antiprogestin treatment under the hypothesis that PR targeting should be added to the standard antihormone armamentarium, specifically in patients with PRA-H tumors. The hypothesis is supported by our results, showing that high levels of HMW-FGF2 confer resistance to both antiprogestins and tamoxifen.
Clinical data regarding FGF2 expression in breast cancer samples is scarce and controversial [31, 32]. Although high extracellular FGF2 levels have been determined in the urine [33], nipple fluid [34], and milk of breastfeeding patients with breast cancer compared to healthy individuals [35], these studies do not distinguish between LMW and HMW-FGF2, and neither have they investigated a possible association between FGF2 expression and endocrine resistance. As previously mentioned, HMW-FGF2 isoforms are mainly localized in the cell nucleus, while LMW-FGF2 is predominantly secreted outside of the cell [10, 36]. In line with our findings, Li et al. recently showed that, in a subset of patients with triple negative breast cancer, neoadjuvant chemotherapy treatment induced enrichment in nuclear FGF2-positive cells [11]. Moreover, we found that most breast cancer samples with high HMW-FGF2 levels also have high LMW-FGF2, although increased expression of LMW-FGF2 does not always correlate with increased HMW-FGF2 levels suggesting that nuclear FGF2 expression correlates with high FGF2 levels in breast cancer samples.
As opposed to the human samples and to the T47D-YA cells engineered to overexpress FGF2, the increase in FGF2 IHC-staining in the tumor cells of T47D-YB and resistant murine models was mainly cytosolic. The reason for this difference remains to be investigated; however, it may be possible that with increased time exposures, nuclear staining might have been detected, as HMW-FGF2 isoforms were observed by WB using purified nuclear extracts (not shown).
The prognostic value of PR isoforms is still controversial. Bamberger et al. [37] and Hopp et al. [38] suggested that high total PRA levels correlate with worse patient prognosis. Conversely, Pathiraja et al. have associated PRA silencing with poor outcome [39] and, previous studies from our lab, which focused on nuclear expression of PR to determine the PRA/PRB ratio, showed that patients with PRB-H tumors may have a worse prognosis than PRA-H tumors. PRB-H carcinomas had increased tumor size, Ki67 and HER-2 expression, higher histological grades (less differentiation), and lower PR levels compared to PRA-H tumors [15]. Herein, we show that low-grade PRB-H tumors also express high levels of nuclear FGF2, which is in line with their worse prognosis. Moreover, future work with a larger cohort will determine if total FGF2 expression levels and sub-cellular localization in luminal breast cancer is a useful predictive marker of tumor progression and recurrence. The fact that overexpression of 22.5 kDa FGF2 isoform induced a more aggressive phenotype than LMW-FGF2 in the human T47D-YA cell line, with increased nuclear FGF2 staining, reinforces the association of nuclear FGF2 localization with poor prognosis in human carcinomas.
We have previously demonstrated that exogenous LMW-FGF2 added to cultures of mammary carcinomas induced the ligand-independent activation of PR which interacted with FGFR2 and STAT5 at the progesterone-responsive sites (PRE) within the MYC promoter [16, 40]. On the other hand, using microarray technology, we showed that progestins increased FGF2 expression in MPA-induced mouse mammary carcinomas [41]. In the normal endometrium, progesterone also modulates FGF2 expression which undergoes cyclic variations [42].
Our results suggest that in breast cancer, it is PRB that exclusively triggers HMW-FGF2 expression, since T47D-YB cells and PRB-transfected T47D-Y cells display high levels of FGF2 compared to T47D-YA cells and T47D-Y-pSG5, respectively. This may be due to a direct effect of PRB acting on the FGF2 promoter which contains a PRE hemisite (-1154 to -1150 positions).
Overall, our studies indicate that high intracellular FGF2 confers an aggressive behavior to breast carcinomas. The development of new therapies that target intracellular components of the FGF2 pathway in addition to blocking membrane FGFR may prove more effective and thus can help to overcome endocrine resistance and reduce breast cancer progression.
References
Santen RJ, Manni A, Harvey H, Redmond C (1990) Endocrine treatment of breast cancer in women. Endocr Rev 11(2):221–265
Horwitz KB, McGuire WL (1975) Predicting response to endocrine therapy in human breast cancer: a hypothesis. Science 189(4204):726–727
Nicolini A, Ferrari P, Duffy MJ (2017) Prognostic and predictive biomarkers in breast cancer: past, present and future. Semin Cancer Biol. https://doi.org/10.1016/j.semcancer.2017.08.010
Kariagina A, Aupperlee MD, Haslam SZ (2008) Progesterone receptor isoform functions in normal breast development and breast cancer. Crit Rev Eukaryot Gene Expr 18(1):11–33
Diep CH, Daniel A, Mauro L, Knutson T, Lange C (2015) Progesterone action in breast, uterine, and ovarian cancers. J Mol Endocrinol 54(2):R31–R53
Wargon V, Riggio M, Giulianelli S, Sequeira GR, Rojas P, May M, Polo ML, Gorostiaga MA, Jacobsen B, Molinolo A, Novaro V, Lanari C (2015) Progestin and antiprogestin responsiveness in breast cancer is driven by the PRA/PRB ratio via AIB1 or SMRT recruitment to the CCND1 and MYC promoters. Int J Cancer 136(11):2680–2692
Normanno N, Di Maio M, De Maio E, De Luca A, de Matteis A, Giordano A, Perrone F (2005) Mechanisms of endocrine resistance and novel therapeutic strategies in breast cancer. Endocr Relat Cancer 12(4):721–747
Abdel-Hafiz HA, Horwitz KB (2015) Role of epigenetic modifications in luminal breast cancer. Epigenomics 7(5):847–62
Andre F, Cortes J (2015) Rationale for targeting fibroblast growth factor receptor signaling in breast cancer. Breast Cancer Res Treat 150(1):1–8
Yu PJ, Ferrari G, Galloway AC, Mignatti P, Pintucci G (2007) Basic fibroblast growth factor (FGF-2): the high molecular weight forms come of age. J Cell Biochem 100(5):1100–1108
Li S, Payne S, Wang F, Claus P, Su Z, Groth J, Geradts J, de Ridder G, Alvarez R, Marcom PK, Pizzo SV, Bachelder RE (2015) Nuclear basic fibroblast growth factor regulates triple-negative breast cancer chemo-resistance. Breast Cancer Res 17:91
Wargon V, Helguero LA, Bolado J, Rojas P, Novaro V, Molinolo A, Lanari C (2009) Reversal of antiprogestin resistance and progesterone receptor isoform ratio in acquired resistant mammary carcinomas. Breast Cancer Res Treat 116(3):449–460
Jacobsen BM, Richer JK, Schittone SA, Horwitz KB (2002) New human breast Cancer cells to study progesterone receptor isoform ratio effects and ligand-independent gene regulation. J Biol Chem 277(31):27793–27800
Lamb C, Simian M, Molinolo A, Pazos P, Lanari C (1999) Regulation of cell growth of a progestin-dependent murine mammary carcinoma in vitro: progesterone receptor involvement in serum or growth factor-induced cell proliferation. J Steroid Biochem Mol Biol 70(4–6):133–142
Rojas PA, May M, Sequeira GR, Elia A, Alvarez M, Martinez P, Gonzalez P, Hewitt S, He X, Perou CM et al (2017) Progesterone receptor isoform ratio: a breast cancer prognostic and predictive factor for antiprogestin responsiveness. J Natl Cancer Inst 109(7). https://doi.org/10.1093/jnci/djw317
Giulianelli S, Cerliani JP, Lamb CA, Fabris VT, Bottino MC, Gorostiaga MA, Novaro V, Góngora A, Baldi A, Molinolo A, Lanari C (2008) Carcinoma-associated fibroblasts activate progesterone receptors and induce hormone independent mammary tumor growth: a role for the FGF-2/FGFR-2 axis. Int J Cancer 123(11):2518–2531
Kastner P, Bocquel MT, Turcotte B, Garnier JM, Horwitz KB, Chambon P, Gronemeyer H (1990) Transient expression of human and chicken progesterone receptors does not support alternative translational initiation from a single mRNA as the mechanism generating two receptor isoforms. J Biol Chem 265(21):12163–12167
Valtink M, Knels L, Stanke N, Engelmann K, Funk RH, Lindemann D (2012) Overexpression of human HMW FGF-2 but not LMW FGF-2 reduces the cytotoxic effect of lentiviral gene transfer in human corneal endothelial cells. Invest Ophthalmol Vis Sci 53(6):3207–3214
Sahores A, Luque GM, Wargon V, May M, Molinolo A, Becu-Villalobos D, Lanari C, Lamb CA (2013) Novel, low cost, highly effective, handmade steroid pellets for experimental studies. PLoSONE 8(5):e64049
Dini G, Funghini S, Witort E, Magnelli L, Fanti E, Rifkin DB, Del Rosso M (2002) Overexpression of the 18 kDa and 22/24 kDa FGF-2 isoforms results in differential drug resistance and amplification potential. J Cell Physiol 193(1):64–72
Gruber G, Schwarzmeier JD, Shehata M, Hilgarth M, Berger R (1999) Basic fibroblast growth factor is expressed by CD19/CD11c-positive cells in hairy cell leukemia. Blood 94(3):1077–1085
Cohen-Jonathan E, Toulas C, Monteil S, Couderc B, Maret A, Bard JJ, Prats H, Daly-Schveitzer N, Favre G (1997) Radioresistance induced by the high molecular forms of the basic fibroblast growth factor is associated with an increased G2 delay and a hyperphosphorylation of p34CDC2 in HeLa cells. Cancer Res 57(7):1364–1370
Delrieu I, Arnaud E, Ferjoux G, Bayard F, Faye JC (1998) Overexpression of the FGF-2 24-kDa isoform up-regulates IL-6 transcription in NIH-3T3 cells. FEBS Lett 436(1):17–22
Ader I, Muller C, Bonnet J, Favre G, Cohen-Jonathan E, Salles B, Toulas C (2002) The radioprotective effect of the 24 kDa FGF-2 isoform in HeLa cells is related to an increased expression and activity of the DNA dependent protein kinase (DNA-PK) catalytic subunit. Oncogene 21(42):6471–6479
Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, Natrajan R, Marchio C, Iorns E, Mackay A, Gillett C, Grigoriadis A, Tutt A, Reis-Filho JS, Ashworth A (2010) FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res 70(5):2085–2094
Piasecka D, Kitowska K, Czaplinska D, Mieczkowski K, Mieszkowska M, Turczyk L, Skladanowski AC, Zaczek AJ, Biernat W, Kordek R, Romanska HM, Sadej R (2016) Fibroblast growth factor signalling induces loss of progesterone receptor in breast cancer cells. Oncotarget 7(52):86011–86025
Andre F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, Turner N, Rugo H, Smith JW, Deudon S, Shi M, Zhang Y, Kay A, Graus Porta D, Yovine A, Baselga J (2013) Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res 19(13):3693–3702
Musolino A, Campone M, Neven P, Denduluri N, Barrios CH, Cortes J, Blackwell K, Soliman H, Kahan Z, Bonnefoi H, Squires M, Zhang Y, Deudon S, Shi MM, André F (2017) Phase II, randomized, placebo-controlled study of dovitinib in combination with fulvestrant in postmenopausal patients with HR(+), HER2(-) breast cancer that had progressed during or after prior endocrine therapy. Breast Cancer Res 19(1):18
Formisano L, Stauffer KM, Young CD, Bhola NE, Guerrero-Zotano AL, Jansen VM, Estrada MM, Hutchinson KE, Giltnane JM, Schwarz LJ, Lu Y, Balko JM, Deas O, Cairo S, Judde JG, Mayer IA, Sanders M, Dugger TC, Bianco R, Stricker T, Arteaga CL (2017) Association of FGFR1 with ERalpha maintains ligand-independent ER transcription and mediates resistance to estrogen deprivation in ER(+) breast cancer. Clin Cancer Res 23(20):6138–6150
Shee K, Yang W, Hinds JW, Hampsch RA, Varn FS, Traphagen NA, Patel K, Cheng C, Jenkins NP, Kettenbach AN, Demidenko E, Owens P, Faber AC, Golub TR, Straussman R, Miller TW (2018) Therapeutically targeting tumor microenvironment-mediated drug resistance in estrogen receptor-positive breast cancer. J Exp Med 215(3):895–910
Yiangou C, Gomm JJ, Coope RC, Law M, Luqmani YA, Shousha S, Coombes RC, Johnston CL (1997) Fibroblast growth factor 2 in breast cancer: occurrence and prognostic significance. Br J Cancer 75(1):28–33
Faridi A, Rudlowski C, Biesterfeld S, Schuh S, Rath W, Schroder W (2002) Long-term follow-up and prognostic significance of angiogenic basic fibroblast growth factor (bFGF) expression in patients with breast cancer. Pathol Res Pract 198(1):1–5
Nguyen M, Watanabe H, Budson AE, Richie JP, Hayes DF, Folkman J (1994) Elevated levels of an angiogenic peptide, basic fibroblast growth factor, in the urine of patients with a wide spectrum of cancers. J Natl Cancer Inst 86(5):356–361
Sartippour MR, Zhang L, Lu M, Wang HJ, Brooks MN (2005) Nipple fluid basic fibroblast growth factor in patients with breast cancer. Cancer Epidemiol Biomark Prev 14(12):2995–2998
Arcaro KF, Browne EP, Qin W, Zhang K, Anderton DL, Sauter ER (2012) Differential expression of cancer-related proteins in paired breast milk samples from women with breast cancer. J Hum Lact 28(4):543–546
Sorensen V, Nilsen T, Wiedlocha A (2006) Functional diversity of FGF-2 isoforms by intracellular sorting. Bioessays 28(5):504–514
Bamberger AM, Milde-Langosch K, Schulte HM, Loning T (2000) Progesterone receptor isoforms, PR-B and PR-A, in breast cancer: correlations with clinicopathologic tumor parameters and expression of AP-1 factors. Horm Res 54(1):32–37
Hopp TA, Weiss HL, Hilsenbeck SG, Cui Y, Allred DC, Horwitz KB, Fuqua SA (2004) Breast cancer patients with progesterone receptor PR-A-rich tumors have poorer disease-free survival rates. Clin Cancer Res 10(8):2751–2760
Pathiraja TN, Shetty PB, Jelinek J, He R, Hartmaier R, Margossian AL, Hilsenbeck SG, Issa JPJ, Oesterreich S (2011) Progesterone receptor isoform-specific promoter methylation: association of PRA promoter methylation with worse outcome in breast cancer patients. Clin Cancer Res 17(12):4177–4186
Cerliani JP, Guillardoy T, Giulianelli S, Vaque JP, Gutkind JS, Vanzulli SI, Martins R, Zeitlin E, Lamb CA, Lanari C (2011) Interaction between FGFR-2, STAT5, and progesterone receptors in breast cancer. Cancer Res 71(10):3720–3731
Giulianelli S, Herschkowitz JI, Patel V, Lamb CA, Gutkind JS, Molinolo A, Perou CM, Lanari C (2011) MPA-induced gene expression and stromal and parenchymal gene expression profiles in luminal murine mammary carcinomas with different hormonal requirements. Breast Cancer Res Treat 129(1):49–67
Okumu LA, Forde N, Mamo S, McGettigan P, Mehta JP, Roche JF, Lonergan P (2014) Temporal regulation of fibroblast growth factors and their receptors in the endometrium and conceptus during the pre-implantation period of pregnancy in cattle. Reproduction 147(6):825–834
Acknowledgements
The authors are very grateful to Dr. A. Baldi (IBYME, Buenos Aires) for the FGF2 ligand, to Repros Therapeutics (The Woodlands) for the telapristone acetate (Proellex; TLP), to Dr. D. Lindemann (University of Technology, Dresden) for the FGF2 expression plasmids, to Dr. K. Horwitz (University of Colorado, Denver) for sharing the T47D-Y, T47D-YA, and T47D-YB cells, and to Annabelle Nelson who assisted in the proofreading of this manuscript.
Funding
This work was supported by Instituto Nacional del Cáncer (2016–2017), Agencia Nacional de Promoción de Ciencia y Tecnología (ANAPCYT; PICT 2015/1022, PID 2012/84), CONICET (PIP 603, 2013-15), Fundación Roemmers, Fundación René Barón, and Fundación Fiorini. Fundación Sales supported the study until March 2014. AS, VF, MM, and GRS are CONICET fellows and PR, CL, and CAL are members of the Research Career; AE and MMA are fellows of Instituto Nacional de Cáncer (INC). AM is supported at 25% effort from the Cancer Center Support Grants as Director of Biorepository and Tissue Technology Shared Resource [3 P30 CA023100 (Lippman, Scott), 05/01/2014–04/30/2019].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study was approved by the Institutional Review Boards (#CE08/2016 and CE29/2015).
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Adrián Rubstein is an independent cosultant
Electronic supplementary material
Supplementary Fig. 1
Increased HMW-FGF2 expression in hormone resistant murine mammary carcinomas compared to hormone responsive tumors. a Immunohistochemical evaluation of FGF2 in tumor samples from C7, and 59 families. Bar: 60 μm. b WB analysis and quantification of whole-cell murine tumor extracts from C7 family, and immunoblotted for FGF2. ERK was used as loading control. Immunoreactive bands were quantified and graphed relative to the hormone responsive variant in each case. HMW, high molecular weight; LMW, low molecular weight (PNG 1406 kb)
Supplementary Fig. 2
T47D-YA-22.5 xenografts display increased tumor growth and metastasis. FGF2-overexpressing tumors and metastasis express high FGF2 levels. a In vivo growth curve of T47D-YA-p6NST50, -18, and -22.5 cells (NSG mice, N = 4/group). b H&E staining of T47D-YA-22.5 xenografts displaying tumor embolism (i), and invasion of adjacent fat (ii), muscle (iii) and nervous tissue (iv). Arrowheads indicate tumor epithelium and arrows show tumor embolism (i), adipocytes (ii), muscle fibers (iii) and arterial wall (iv). Bar: 100 μm. c H&E staining of T47D-YA-p6NST50, -18 and -22.5 xenografts growing in NSG mice treated with control or 6 mg MFP pellets. Bar: 160 μm. d Immunohistochemical analysis of FGF2 expression in a lung metastatic foci from a mice carrying a T47D-YA-22.5 xenograft. Bar: 25 μm. e FGF2 immunohistochemistry in T47D-YA-p6NST50, -18 and -22.5 tumors growing in NSG mice (Bar: 50 μm) (PNG 3777 kb)
Rights and permissions
About this article

Cite this article
Sahores, A., Figueroa, V., May, M. et al. Increased High Molecular Weight FGF2 in Endocrine-Resistant Breast Cancer. HORM CANC 9, 338–348 (2018). https://doi.org/10.1007/s12672-018-0339-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-018-0339-4