
Abstract
Fibroblast growth factor receptor (FGFR) signaling is involved in multiple biological processes, including cell proliferation, survival, differentiation, migration, and apoptosis during embryonic development and adult tissue homeostasis. Given its role in the activation of critical signaling pathways, aberrant FGFR signaling has been implicated in multiple cancer types. A comprehensive search of PubMed and congress abstracts was conducted to identify reports on FGFR pathway components in breast cancer. In breast cancers, FGFR1 and FGFR4 gene amplification and single nucleotide polymorphisms in FGFR2 and FGFR4 have been detected. Commonly, these FGFR aberrations and gene amplifications lead to increased FGFR signaling and have been linked with poor prognosis and resistance to breast cancer treatments. Here, we review the role of FGFR signaling and the impact of FGFR genetic amplifications/aberrations on breast tumors. In addition, we summarize the most recent preclinical and clinical data on FGFR-targeted therapies in breast cancer. Finally, we highlight the ongoing clinical trials of the FGFR-targeted agents dovitinib, AZD4547, lucitanib, BGJ398, and JNJ-42756493, which are selected for patients with FGFR pathway-amplified breast cancer. Aberrant FGFR pathway amplification may drive some breast cancers. Inhibition of FGFR signaling is being explored in the clinic, and data from these trials may refine our ability to select patients who would best respond to these treatments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
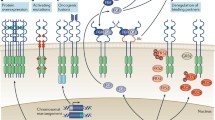
Role of FGFR signaling
The fibroblast growth factor receptor (FGFR) family comprises four transmembrane tyrosine kinase receptors (FGFR1-4). Each FGFR has an extracellular ligand-binding domain containing three immunoglobulin (Ig)-like domains and an acidic box, a single-pass transmembrane domain, and an intracellular tyrosine kinase domain [1]. Twenty-two FGF ligands bind to the second and third Ig-like domains of the different FGFRs and their splice variants, creating ligand-binding specificity [2–4]. Ligand binding induces receptor dimerization, enabling transphosphorylation of intracellular kinases, and phosphorylation of intracellular signaling proteins.
FGFR signaling is involved in cell proliferation, survival, differentiation, migration, and apoptosis during embryonic development and adult tissue homeostasis [1]. Signaling is regulated in part by the spatial and temporal expression patterns of the different ligands and receptors, leading to distinct cell type-specific functions. For example, during embryogenesis, FGFR signaling plays a key role in the development of the nervous system, limbs, midbrain, lungs, and mammary glands. In contrast, FGFR signaling in the adult drives tissue repair, angiogenesis, and inflammation.
FGFR genetic aberrations in breast cancer
Multiple genetic aberrations in FGFRs leading to increased pathway activation have been identified in breast cancer. The most common aberration is FGFR1 gene amplification (one of several putative oncogenes on 8p11-12), which is present in 7.5–17 % of all breast cancer and in 16–27 % of luminal B-type breast cancer (although variability in these numbers is expected, given the different methods used to assess amplification) [5–13]. In addition to FGFR1 gene amplification, elevated mRNA is detected in 22 % of tumor cell lines and breast-tumor samples [14], and gene amplification is robustly associated with FGFR1 overexpression [7, 10, 13]. Amplification of other FGFRs has also been observed. For example, amplification of FGFR2 (10q26) was identified in 4 % of triple-negative breast cancers (TNBCs) [15], and two TNBC cell lines (SUM52PE, MFM223) with FGFR2 amplification showed constitutive activation of FGFR2. Interestingly, survival of SUM52PE and MFM223 was dependent on FGFR2 expression and tyrosine kinase activity. FGFR4 amplification has been detected in ≈10 % of breast cancer samples and was associated with estrogen receptor (ER) and progesterone receptor (PR) positivity and lymph node metastases [16]. In a separate study, elevated FGFR4 mRNA levels were detected in 32 % of breast cancer samples [14].
Single nucleotide polymorphisms (SNPs) have also been detected in FGFR genes in breast tumors. For example, SNPs were identified in FGFR2 intron 2 [17–22] and, in some studies, were more common in ER-positive (ER+) tumors [18, 22]. Intron 2 of FGFR2 contains putative transcription factor binding sites, and one of the SNPs, rs2981578, increased Oct-1/Runx2 and C/EBPβ transcription factor binding, which led to increased FGFR2 expression [21]. In the transmembrane region of FGFR4, a SNP involving the conversion of Gly to Arg was identified, and FGFR4Arg388-expressing MDA-MB-231 mammary tumor cells showed increased motility in comparison with cells expressing the FGFR4Gly388 allele [23]. Additionally, introduction of the FGFR4Arg388 allele in a transgenic mammary tumor model induced more rapid tumor formation and progression than did the FGFR4Gly388 allele [24] and was shown to promote tumor progression through membrane type-1 matrix metalloproteinase-induced extracellular matrix degradation involving the epithelial-to-mesenchymal transition at the tumor/stromal border [25].
Point mutations are rare; however, they have been detected in human breast tumors, including S125L in FGFR1 and R203C in FGFR2 [26]. An activating point mutation in FGFR4 (Y367C), which causes constitutive receptor dimerization and activation of MAPK, was identified in the MDA-MB-453 breast cancer cell line [27].
Prognostic significance of FGFR aberrancy
Various studies have investigated the role of the FGFR pathway as a predictive/prognostic marker, with several suggesting that aberrant FGFR expression is associated with an increased risk of breast cancer and is correlated with a poor prognosis. For example, FGFR1 and FGFR2 are preferentially amplified in ER+ breast cancer and TNBC, respectively [28], and FGFR1 amplification was an independent predictor of overall survival and poor outcome in patients with ER+ breast cancer [9, 29]. However, these results may be confounded by other potential oncogenes on 8p11–12, which may also contribute [30–33]. Therefore, amplification status of 8p11-12 may not be a sufficient screening mechanism for clinical trials of FGFR-targeted drugs. Other genes on different chromosomes may also have an impact. For example, co-amplification of cyclin D1 (CCND1; chromosome 11) and FGFR1 were associated with a significant reduction in patient survival [34].
The prognostic role of FGFR2 has also been reported. Indeed, increased expression of FGFR2 is associated with poor overall survival and disease-free survival [35]. Following the detection of FGFR2 and FGFR3 constitutive activation in hormone-independent murine mammary tumors, FGFR expression levels were determined in tumor samples from patients with breast cancer (n = 58) [36]. This analysis detected a strong association between FGFR2 and FGFR3, with weaker associations between FGFR2 or FGFR3 and ER. The study also found that FGFR4 expression correlated with human epidermal growth factor receptor 2 (HER2) overexpression. Other studies have shown that the FGFR2 SNPs rs2981582, rs1219648, rs2420946, and rs2981579 were significantly associated with an increased risk of breast cancer [37, 38]. Women with all three SNPs had a 1.36-fold greater chance of getting breast cancer than did those with no or no more than two SNPs, and the risk was greater in women with ER+ and/or PR+ cancer, premenopausal women, and women who gave birth to their first child at a later age.
The FGFR4Arg388 allele has been correlated with significantly reduced disease-free survival [23], and, in a meta-analysis and pooled analysis of patients with cancer, including breast, a significant association between the FGFR4Arg388 allele and nodal involvement was identified [39]. Additionally, carriers of the FGFR4Arg388 allele had an increased hazard of poor overall survival compared with carriers of the FGFR4Gly388 allele.
Role of FGFR in resistance mechanisms
Various studies support the role of the FGFR pathway in endocrine resistance. For example, in patients with ER+ breast cancer treated with tamoxifen, high FGFR4 mRNA levels were associated with poor clinical benefit and shorter progression-free survival [40]. Additionally, evidence suggests that resistance to HER2-targeted therapy may result from a switch in dependency on the ER/HER2 signaling pathway to the FGFR signaling pathway [41]. In that study, FGFR2 was highly amplified, FGFR2 was overexpressed, and HER2 expression was reduced in a lapatinib-resistant cell line (UACC812/LR). The survival of UACC812/LR cells appeared dependent on FGFR2, because inhibition of FGFR2 induced cell apoptosis.
Several studies suggest that FGFR signaling mediates resistance to hormonal therapies through activation of the MAPK and PI3K pathways. For example, in FGFR1-amplified breast cancer cell lines, FGFR signaling caused persistent MAPK activation followed by cyclin D1 expression, which led to tamoxifen resistance [13]. This activated signaling occurred in both an ER-dependent and -independent manner, and the FGFR1-induced tamoxifen resistance was reversible following treatment with FGFR1-directed siRNA. Additionally, constitutive activation of FGFR3 in MCF-7 cells reduced sensitivity to tamoxifen in an ER-independent manner through stimulation of the MAPK and PI3K pathways [42]. In this study, activation of phospholipase Cγ was critical for activation of MAPK and PI3K and subsequently for tamoxifen resistance.
FGFR signaling may also mediate resistance to chemotherapy in breast cancer. In patients with node-positive breast cancer receiving chemotherapy as an adjuvant therapy, presence of the FGFR4Arg388 allele was significantly associated with poor disease-free survival and overall survival [43]. Additionally, FGFR4 is upregulated in MDA-MB-453 breast cancer cells that are resistant to doxorubicin and cyclophosphamide, and FGFR4 overexpression leads to upregulation of the anti-apoptotic molecule Bcl-xl through MAPK activation [27].
Targeting the FGFR pathway in preclinical studies
The evidence supporting a role of the FGFR pathway in breast cancer has led to preclinical investigation of FGFR pathway-targeting agents, and several approaches have been explored. For example, FP-1039 is an FGF ligand trap consisting of the extracellular domain of FGFR1 fused to the Fc region of IgG [44]. In xenograft models using JIMT-1, an FGFR1-amplified and -overexpressing cell line [13], FP-1039 blocked tumor formation. In another approach, monoclonal antibody GP369 targets the epithelial-expressed FGFR2-IIIb receptor isoform, whose overexpression can drive tumorigenesis [45, 46]. In a xenograft model with MFM-223 breast cancer cells (a cell line with 287 genomic copies of FGFR2), GP369 induced tumor stasis.
More commonly, the FGFR pathway has been targeted using small molecule inhibitors. SU5402, an FGFR1 small molecule inhibitor, reduced cell survival in several breast cancer cell lines; the sensitivity was greatest in MDA-MB-134 cells [47].
PD173074 is a selective FGFR1 and FGFR3 inhibitor that has shown activity in numerous breast cancer cell lines. For example, it inhibited FGFR tyrosine kinase activity and autophosphorylation and caused G1 growth arrest in MDA-MB-415, MDA-MB-453, and SUM52 [48]. The study also showed that FGFR inhibition led to the downregulation of cyclin D1/2 expression, inhibition of cyclin D/cdk4 activity, and reduced pRB phosphorylation. In the FGFR1-, FGFR2-, and FGFR3-expressing cell line 4T1, PD173074 demonstrated concentration-dependent apoptosis, inhibited tumor growth and metastases to the lung, and increased CD4 and CD8 T cell infiltration to the spleen and tumors in xenograft models [49]. Other studies have shown that PD173074 treatment inhibited MAPK and PI3K/AKT signaling and induced cell-cycle arrest and apoptosis in 47 % of TNBC cell lines and significantly reduced the growth of CAL51 basal-like breast cancer cell line-containing xenografts [15, 50]. Finally, decreased phosphorylation of FGFR2 and induction of apoptosis was observed in a lapatinib-resistant breast cancer cell line (UACC812/LR) following treatment with PD173074 [41].
Brivanib alaninate (BMS-582664) inhibits VEGFR2, VEGFR3, and FGFR3 (and to a lesser extent FGFR1 and FGFR2) [51] and has caused decreased receptor autophosphorylation, inhibited FGF2-induced tyrosine kinase activity, and reduced phosphorylation of ERK and AKT in breast cancer cell lines with amplification of FGFR1 [52]. Additionally, brivanib inhibited the growth of human H3396 breast-tumor xenografts [53] and elicited tumor growth regression in tamoxifen-sensitive (MCRF-7 E2) and tamoxifen-stimulated (MCF-7 Ral, MCF-7 Tam) breast cancer cell lines [54].
Lucitanib (E-3810)—a small molecule inhibitor of VEGFR1-3, FGFR1-2, and colony stimulating factor 1 receptor—has demonstrated anti-angiogenic and anti-tumor activity in preclinical models [55]. Additionally, in combination with paclitaxel, lucitanib caused long-lasting tumor regressions in xenografts of the TNBC cell line MDA-MB-231 [56].
Dovitinib (TKI258) is a small molecule inhibitor of FGFR1-3, VEGFR1-3, c-KIT, fms-related tyrosine kinase 3 (FLT3), platelet-derived growth factor receptor (PDGFR) β, c-KIT, and FLT3 [57]. In 4T1 and the FGFR2- and FGFR3-expressing cell line 67NR, dovitinib decreased FGFR substrate 2 (FRS2) phosphorylation; decreased the activity of ERK1/2, AKT, and phospholipase Cγ; and blocked proliferation [58]. Dovitinib also significantly reduced primary tumor outgrowth in xenograft models with these cell lines and reduced tumor-induced lung metastasis in the 4T1 model [58]. In other tumor models using these cell lines, combining dovitinib with the PI3K/mTOR inhibitor BEZ235 or the pan-ErbB inhibitor AEE788 blocked the FGFR/FRS2/Erk and PI3K/Akt/mTOR pathways and further inhibited tumor growth and blocked tumor spread [59]. In experiments with FGFR-amplified cell lines, dovitinib decreased pFRS2 and pERK/MAPK and inhibited cell growth in MDA-MB-134 (FGFR1-amplified) and SUM52 (FGFR2-amplified) lines [60]. Furthermore, dovitinib caused tumor regressions in HBCx-2 (8 FGFR1 copies) and HBCx-3 (10 FGFR2 copies) xenograft models.
ACTB-1003—a multitargeted kinase inhibitor of FGFR1, VEGFR2, Tie-2, RSK, and p70S6K—demonstrated potent, low nanomolar activity in a variety of tumor models, including breast cancer [61]. Ponatinib, an oral multitargeted kinase inhibitor of Bcr-Abl as well as FGFR1-4 [62–64], inhibited cell growth in MDA-MB-134 and SUM52PE and in MFM-223—a cell line that also has FGFR2 amplification [65].
Clinical trials with FGFR pathway-targeting agents in breast cancer
Data from the various preclinical studies clearly demonstrate the role of the FGFR pathway in breast cancer and have led to clinical trials exploring patients who may best benefit from targeted treatments (Table 1). Indeed, many of the clinical trials testing FGFR tyrosine kinase inhibitors in breast cancer are requiring molecular prescreening prior to study entry to enable correlation between response and FGFR pathway activation or are only enrolling patients with FGFR pathway activation.
In a phase 2 trial of dovitinib (NCT00958971), patients with metastatic HER2− breast cancer were grouped based on hormone receptor (HR) status and FGFR1 amplification (as assayed by silver in situ hybridization) [60]. Dovitinib was reasonably well tolerated in the heavily pretreated patients (n = 81), and unconfirmed partial response and/or stable disease at ≥24 weeks were observed in 25 % of the 20 evaluable patients with HR+ FGFR1-amplified breast cancer (3 % in the 31 evaluable patients with FGFR1 nonamplified, HR− disease). In preplanned analyses, amplifications of FGFR1/2 by quantitative polymerase chain reaction (PCR) were found to be associated with anti-tumor activity in patients with HR+. These data led to the initiation of a phase 2 study of dovitinib in combination with fulvestrant in postmenopausal patients with locally advanced or metastatic HER2−, HR+ breast cancer who have progressed during or following endocrine therapy (NCT01528345) [66]. Of interest, the study requires molecular prescreening prior to study entry, with patients grouped based on FGFR pathway amplification status (FGFR1, FGFR2, or FGF3 by quantitative PCR). Additional ongoing studies include a phase 2 study of dovitinib as salvage therapy in patients with relapsed stage IV HER2− inflammatory breast cancer (NCT01262027) and a phase 1/2 study of dovitinib in combination with aromatase inhibitors in patients with metastatic breast cancer with evidence of resistance to prior aromatase inhibitor therapy (NCT01484041). The latter study also requires tumor availability for determination of FGFR1 amplification for study inclusion.
AZD4547 is an inhibitor of FGFR1-3 and VEGFR2 [67]. A phase 1/2 study of AZD4547 in combination with the estrogen receptor antagonist fulvestrant in patients with ER+ breast cancer with high levels of FGFR1 is currently ongoing (NCT01202591). Tumor biopsy must be available to confirm FGFR1 polysomy or amplification for study inclusion. Another ongoing phase 1/2 study is comparing AZD4547 in combination with letrozole or anastrozole versus exemestane in patients with ER+ breast cancer who relapsed during adjuvant endocrine therapy with anastrozole or letrozole (NCT01791985). Enrollment in the expansion phase of this study requires tumor biopsy for confirmation of FGFR1 status by fluorescence in situ hybridization (FISH). Additionally, an ongoing phase 2 study is evaluating AZD4547 in patients with FGFR1- or FGFR2-amplified tumors (NCT01795768) [68]. Patients with breast cancer must have FGFR1 amplification and are required to provide archival or fresh tumor biopsy for confirmation of FGFR amplification, with a second biopsy planned on days 10–14.
A phase 1/2 study testing lucitanib in patients with advanced solid tumors is currently ongoing (NCT01283945) [69]. In dose expansion, patients with FGFR1 or 11q12-14 amplification per FISH or comparative genomic hybridization array were enrolled; of nine evaluable patients with breast cancer, four achieved PR sustained over four to six courses, three achieved stable disease, and two had progressive disease [70]. A phase 2 study of lucitanib is also ongoing in patients with ER+ FGFR1-amplified and -nonamplified metastatic breast cancer (NCT02053636).
The pan-FGFR inhibitor BGJ398 is being tested in patients with tumors containing FGFR1 or FGFR2 amplifications or FGFR3 mutations (NCT01004224). In this study, reductions in tumor volume were observed with single-agent BGJ398 in some patients with breast cancer [71]. BGJ398 is also being studied in a phase 1b trial in combination with the α-selective PI3K inhibitor BYL719 in patients whose tumors have PIK3CA mutations and FGFR1-3 alterations (NCT01928459).
In a dose-escalation study, the pan-FGFR inhibitor JNJ-42756493 also demonstrated reductions in tumor volume in a patient with breast cancer (NCT01962532) [72]. In a second phase 1 trial in patients with advanced or refractory solid tumors or lymphoma, the dose-expansion phase will include a cohort of patients with breast cancer with FGFR1, FGFR2, or FGFR4 gene amplifications (NCT01703481).
Other FGFR-targeted agents have been explored in clinical trials, but without requiring FGFR testing at study entry. For example, orantinib (TSU-68)—an inhibitor of VEGFR2, PDGFR, and FGFR—had no activity as a single agent in patients with metastatic breast cancer progressing despite treatment with a prior anthracycline-containing regimen and taxane [73]. However, in patients with anthracycline resistance, orantinib in combination with docetaxel achieved an overall response rate of 21 % [74]. Additional ongoing studies not requiring FGFR testing at entry include a study of brivanib alaninate in combination with chemotherapy in patients with advanced cancer (NCT00798252) and a study of docetaxel with or without nintedanib (BIBF 1120)—an inhibitor of FGFR1-3, VEGFR1-3, PDGFRα, PDGFRβ, and FLT3 [75]—as second-line chemotherapy in patients with HER2− metastatic or locally recurrent breast cancer (NCT01658462).
Bioassays
Challenges remain in identifying the patient population that may benefit from FGFR pathway-targeted therapy. For example, definitions of FGFR pathway amplification vary both in the methods used and the threshold copy number. For example, amplicons may not accurately reflect the level of amplification of the component genes and be confounded by other potential oncogenes in the amplicon. Additionally, measurement of gene amplification may not reflect protein expression or activity. Conversely, detection of activating mutations may not be relevant unless the gene is expressed. The best method is yet unknown, but analyses comparing screening methods with efficacy can be informative. For example, in a phase 2 study with dovitinib, tumor reduction was greater in patients with FGF pathway amplification identified by quantitative PCR than in those with FGF pathway amplification identified by in situ hybridization [60]. However, no consensus has been reached, and ongoing trials are using different methods for detecting FGFR pathway activation/aberration, which will complicate cross-study data interpretation. Careful analyses of the biomarker data from clinical trials may improve the ability to identify the optimal patient population that will benefit from FGFR-targeted therapies.
Conclusions
The FGFR pathway is involved in multiple cellular processes, and research has shown that aberrant pathway amplification is observed in multiple tumor types, including breast cancer. As research advances toward targeted treatment of cancer, it is increasingly important to understand how the FGFR pathway is driving the disease and, more importantly, how it can be targeted. Multiple agents that target FGFR pathway components are being investigated. Results from these trials are also expected to help refine our ability to identify patients with FGFR pathway-amplified tumors who may best respond to these treatments.
References
Haugsten EM, Wiedlocha A, Olsnes S et al (2010) Roles of fibroblast growth factor receptors in carcinogenesis. Mol Cancer Res 8:1439–1452
Johnson DE, Williams LT (1993) Structural and functional diversity in the FGF receptor multigene family. Adv Cancer Res 60:1–41
Zhang H, Wu F, Tao YM et al (2009) Down-regulated expression of UNC5b related to hepatocellular carcinoma angiogenesis. Zhonghua Wai Ke Za Zhi 47:1569–1573
Itoh N, Ornitz DM (2011) Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J Biochem 149:121–130
Adnane J, Gaudray P, Dionne CA et al (1991) BEK and FLG, two receptors to members of the FGF family, are amplified in subsets of human breast cancers. Oncogene 6:659–663
Jacquemier J, Adelaide J, Parc P et al (1994) Expression of the FGFR1 gene in human breast-carcinoma cells. Int J Cancer 59:373–378
Courjal F, Cuny M, Simony-Lafontaine J et al (1997) Mapping of DNA amplifications at 15 chromosomal localizations in 1875 breast tumors: definition of phenotypic groups. Cancer Res 57:4360–4367
Letessier A, Sircoulomb F, Ginestier C et al (2006) Frequency, prognostic impact, and subtype association of 8p12, 8q24, 11q13, 12p13, 17q12, and 20q13 amplifications in breast cancers. BMC Cancer 6:245
Elbauomy Elsheikh S, Green AR, Lambros MB et al (2007) FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis. Breast Cancer Res 9:R23
Andre F, Job B, Dessen P et al (2009) Molecular characterization of breast cancer with high-resolution oligonucleotide comparative genomic hybridization array. Clin Cancer Res 15:441–451
Kadota M, Sato M, Duncan B et al (2009) Identification of novel gene amplifications in breast cancer and coexistence of gene amplification with an activating mutation of PIK3CA. Cancer Res 69:7357–7365
Moelans CB, de Weger RA, Monsuur HN et al (2010) Molecular profiling of invasive breast cancer by multiplex ligation-dependent probe amplification-based copy number analysis of tumor suppressor and oncogenes. Mod Pathol 23:1029–1039
Turner N, Pearson A, Sharpe R et al (2010) FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res 70:2085–2094
Penault-Llorca F, Bertucci F, Adelaide J et al (1995) Expression of FGF and FGF receptor genes in human breast cancer. Int J Cancer 61:170–176
Turner N, Lambros MB, Horlings HM et al (2010) Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene 29:2013–2023
Jaakkola S, Salmikangas P, Nylund S et al (1993) Amplification of fgfr4 gene in human breast and gynecological cancers. Int J Cancer 54:378–382
Easton DF, Pooley KA, Dunning AM et al (2007) Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447:1087–1093
Garcia-Closas M, Hall P, Nevanlinna H et al (2008) Heterogeneity of breast cancer associations with five susceptibility loci by clinical and pathological characteristics. PLoS Genet 4:e1000054
Huijts PE, Vreeswijk MP, Kroeze-Jansema KH et al (2007) Clinical correlates of low-risk variants in FGFR2, TNRC9, MAP3K1, LSP1 and 8q24 in a Dutch cohort of incident breast cancer cases. Breast Cancer Res 9:R78
Hunter DJ, Kraft P, Jacobs KB et al (2007) A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet 39:870–874
Meyer KB, Maia AT, O’Reilly M et al (2008) Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol 6:1098–1103
Stacey SN, Manolescu A, Sulem P et al (2008) Common variants on chromosome 5p12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet 40:703–706
Bange J, Prechtl D, Cheburkin Y et al (2002) Cancer progression and tumor cell motility are associated with the FGFR4 Arg(388) allele. Cancer Res 62:840–847
Seitzer N, Mayr T, Streit S et al (2010) A single nucleotide change in the mouse genome accelerates breast cancer progression. Cancer Res 70:802–812
Sugiyama N, Varjosalo M, Meller P et al (2010) Fibroblast growth factor receptor 4 regulates tumor invasion by coupling fibroblast growth factor signaling to extracellular matrix degradation. Cancer Res 70:7851–7861
Greenman C, Stephens P, Smith R et al (2007) Patterns of somatic mutation in human cancer genomes. Nature 446:153–158
Roidl A, Foo P, Wong W et al (2010) The FGFR4 Y367C mutant is a dominant oncogene in MDA-MB453 breast cancer cells. Oncogene 29:1543–1552
Katoh M (2010) Genetic alterations of FGF receptors: an emerging field in clinical cancer diagnostics and therapeutics. Expert Rev Anticancer Ther 10:1375–1379
Karlsson E, Waltersson MA, Bostner J et al (2011) High-resolution genomic analysis of the 11q13 amplicon in breast cancers identifies synergy with 8p12 amplification, involving the mTOR targets S6K2 and 4EBP1. Genes Chromosom Cancer 50:775–787
Garcia MJ, Pole JC, Chin SF et al (2005) A 1 Mb minimal amplicon at 8p11-12 in breast cancer identifies new candidate oncogenes. Oncogene 24:5235–5245
Gelsi-Boyer V, Orsetti B, Cervera N et al (2005) Comprehensive profiling of 8p11-12 amplification in breast cancer. Mol Cancer Res 3:655–667
Yang ZQ, Streicher KL, Ray ME et al (2006) Multiple interacting oncogenes on the 8p11-p12 amplicon in human breast cancer. Cancer Res 66:11632–11643
Bernard-Pierrot I, Gruel N, Stransky N et al (2008) Characterization of the recurrent 8p11–12 amplicon identifies PPAPDC1B, a phosphatase protein, as a new therapeutic target in breast cancer. Cancer Res 68:7165–7175
Cuny M, Kramar A, Courjal F et al (2000) Relating genotype and phenotype in breast cancer: an analysis of the prognostic significance of amplification at eight different genes or loci and of p53 mutations. Cancer Res 60:1077–1083
Sun S, Jiang Y, Zhang G et al (2012) Increased expression of fibroblastic growth factor receptor 2 is correlated with poor prognosis in patients with breast cancer. J Surg Oncol 105:773–779
Cerliani JP, Vanzulli SI, Pinero CP et al (2012) Associated expressions of FGFR-2 and FGFR-3: from mouse mammary gland physiology to human breast cancer. Breast Cancer Res Treat 133:997–1008
Liang J, Chen P, Hu Z et al (2008) Genetic variants in fibroblast growth factor receptor 2 (FGFR2) contribute to susceptibility of breast cancer in Chinese women. Carcinogenesis 29:2341–2346
Peng S, Lu B, Ruan W et al (2011) Genetic polymorphisms and breast cancer risk: evidence from meta-analyses, pooled analyses, and genome-wide association studies. Breast Cancer Res Treat 127:309–324
Frullanti E, Berking C, Harbeck N et al (2011) Meta and pooled analyses of FGFR4 Gly388Arg polymorphism as a cancer prognostic factor. Eur J Cancer Prev 20:340–347
Meijer D, Sieuwerts AM, Look MP et al (2008) Fibroblast growth factor receptor 4 predicts failure on tamoxifen therapy in patients with recurrent breast cancer. Endocr Relat Cancer 15:101–111
Azuma K, Tsurutani J, Sakai K et al (2011) Switching addictions between HER2 and FGFR2 in HER2-positive breast tumor cells: FGFR2 as a potential target for salvage after lapatinib failure. Biochem Biophys Res Commun 407:219–224
Tomlinson DC, Knowles MA, Speirs V (2012) Mechanisms of FGFR3 actions in endocrine resistant breast cancer. Int J Cancer 130:2857–2866
Thussbas C, Nahrig J, Streit S et al (2006) FGFR4 Arg388 allele is associated with resistance to adjuvant therapy in primary breast cancer. J Clin Oncol 24:3747–3755
Harding TC, Long L, Palencia S (2013) Blockade of nonhormonal fibroblast growth factors by FP-1039 inhibits growth of multiple types of cancer. Sci Transl Med 5:178ra39
Kabbarah O, Lerner L, Siddiquee Z et al (2009) Abstract #1348: fibroblast growth factor receptor-2 cancer models for biomarker discovery and therapeutic response prediction, in AACR Annual Meeting: Abstracts 1348
Bai A, Meetze K, Vo NY et al (2010) GP369, an FGFR2-IIIb-specific antibody, exhibits potent antitumor activity against human cancers driven by activated FGFR2 signaling. Cancer Res 70:7630–7639
Reis-Filho JS, Simpson PT, Turner NC et al (2006) FGFR1 emerges as a potential therapeutic target for lobular breast carcinomas. Clin Cancer Res 12:6652–6662
Koziczak M, Holbro T, Hynes NE (2004) Blocking of FGFR signaling inhibits breast cancer cell proliferation through downregulation of D-type cyclins. Oncogene 23:3501–3508
Ye T, Wei X, Yin T et al (2014) Inhibition of FGFR signaling by PD173074 improves antitumor immunity and impairs breast cancer metastasis. Breast Cancer Res Treat 143:435–446
Sharpe R, Pearson A, Herrera-Abreu MT et al (2011) FGFR signalling promotes the growth of triple-negative and basal-like breast cancer cell lines both in vitro and in vivo. Clin Cancer Res 17:5275–5286
Bhide RS, Cai ZW, Zhang YZ et al (2006) Discovery and preclinical studies of (R)-1-(4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-5-methylpyrrolo[2,1-f][1,2,4]triazin-6-yloxy)propan-2-ol (BMS-540215), an in vivo active potent VEGFR-2 inhibitor. J Med Chem 49:2143–2146
Shiang CY, Qi Y, Wang B et al (2010) Amplification of fibroblast growth factor receptor-1 in breast cancer and the effects of brivanib alaninate. Breast Cancer Res Treat 123:747–755
Bhide RS, Lombardo LJ, Hunt JT et al (2010) The antiangiogenic activity in xenograft models of brivanib, a dual inhibitor of vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1 kinases. Mol Cancer Ther 9:369–378
Patel RR, Sengupta S, Kim HR et al (2010) Experimental treatment of oestrogen receptor (ER) positive breast cancer with tamoxifen and brivanib alaninate, a VEGFR-2/FGFR-1 kinase inhibitor: a potential clinical application of angiogenesis inhibitors. Eur J Cancer 46:1537–1553
Bello E, Colella G, Scarlato V et al (2011) E-3810 is a potent dual inhibitor of VEGFR and FGFR that exerts antitumor activity in multiple preclinical models. Cancer Res 71:1396–1405
Bello E, Taraboletti G, Colella G et al (2013) The tyrosine kinase inhibitor E-3810 combined with paclitaxel inhibits the growth of advanced-stage triple-negative breast cancer xenografts. Mol Cancer Ther 12:131–140
Lee SH, Lopes de Menezes D, Vora J et al (2005) In vivo target modulation and biological activity of CHIR-258, a multitargeted growth factor receptor kinase inhibitor, in colon cancer models. Clin Cancer Res 11:3633–3641
Dey JH, Bianchi F, Voshol J et al (2010) Targeting fibroblast growth factor receptors blocks PI3K/AKT signaling, induces apoptosis, and impairs mammary tumor outgrowth and metastasis. Cancer Res 70:4151–4162
Issa A, Gill JW, Heideman MR et al (2013) Combinatorial targeting of FGF and ErbB receptors blocks growth and metastatic spread of breast cancer models. Breast Cancer Res 15:R8
André F, Bachelot T, Campone M et al (2013) Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res 19:3693–3702
Patel K, Fattaey A, Burd A (2010) ACTB-1003: an oral kinase inhibitor targeting cancer mutations (FGFR), angiogenesis (VEGFR2, TEK), and induction of apoptosis (RSK and p70S6K). ASCO Meeting Abstracts 28:e13665
O’Hare T, Shakespeare WC, Zhu X et al (2009) AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 16:401–412
Huang WS, Metcalf CA, Sundaramoorthi R et al (2010) Discovery of 3-[2-(imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-Abelson (BCR-ABL) kinase including the T315I gatekeeper mutant. J Med Chem 53:4701–4719
Zhou T, Commodore L, Huang WS et al (2011) Structural mechanism of the Pan-BCR-ABL inhibitor ponatinib (AP24534): lessons for overcoming kinase inhibitor resistance. Chem Biol Drug Des 77:1–11
Gozgit JM, Wong MJ, Moran L et al (2012) Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol Cancer Ther 11:690–699
Andre F, Neven P, Musolino A et al (2013) Dovitinib plus fulvestrant in postmenopausal endocrine resistant HER2-/HR + breast cancer: a phase II, randomized, placebo-controlled study. ASCO Meeting Abstracts 31:TPS651
Gavine PR, Mooney L, Kilgour E et al (2012) AZD4547: an orally bioavailable, potent and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res 72:2045–2056
Smyth EC, Turner NC, Popat S et al (2013) FGFR: proof-of-concept study of AZD4547 in patients with FGFR1 or FGFR2 amplified tumours. ASCO Meeting Abstracts 31:TPS2626
Soria J, De Braud FG, Cereda R et al (2011) First-in-man study of E-3810, a novel VEGFR and FGFR inhibitor, in patients with advanced solid tumors. ASCO Meeting Abstracts 29:TPS149
Dienstmann R, Andre F, Soria J et al (2012) Significant antitumor activity of E-3810, a novel FGFR and VEGFR inhibitor, in patients with FGFR1 amplified breast cancer. Ann Oncol, ESMO Annual Meeting Abstracts 23:319O
Sequist LV, Cassier P, Varga A et al (2014) Phase I study of BGJ398, a selective pan-FGFR inhibitor in genetically preselected advanced solid tumors. AACR Annual Meeting: Abstract CT326
Dienstmann R, Bahleda R, Adamo B et al (2014) First in human study of JNJ-42756493, a potent pan fibroblast growth factor receptor (FGFR) inhibitor in patients with advanced solid tumors. AACR Annual Meeting: Abstract CT325
Suzuki Y, Saeki T, Aogi K et al (2013) A multicenter phase II study of TSU-68, a novel oral multiple tyrosine kinase inhibitor, in patients with metastatic breast cancer progressing despite prior treatment with an anthracycline-containing regimen and taxane. Int J Clin Oncol 18:590–597
Toi M, Saeki T, Iwata H et al (2014) A multicenter phase II study of TSU-68, an oral multiple tyrosine kinase inhibitor, in combination with docetaxel in metastatic breast cancer patients with anthracycline resistance. Breast Cancer 21:20–27
Hilberg F, Roth GJ, Krssak M et al (2008) BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res 68:4774–4782
Acknowledgments
Medical editorial assistance was provided by Peter J. Simon, PhD. Financial support for editorial assistance was provided by Novartis Pharmaceuticals Corporation.
Conflicts of interest
Dr. Andre discloses a Consultant/Advisory Role with Novartis and AstraZeneca and a Funding relationship with Novartis and AstraZeneca. Dr. Cortes discloses Remuneration from Celgene, Roche, Eisai, and Novartis and a Consultant/Advisory Role with Celgene and Roche.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
André, F., Cortés, J. Rationale for targeting fibroblast growth factor receptor signaling in breast cancer. Breast Cancer Res Treat 150, 1–8 (2015). https://doi.org/10.1007/s10549-015-3301-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-015-3301-y