
Abstract
Integrins are heterodimeric structural components of the plasma membrane whose ligands include a large number of extracellular matrix (ECM) proteins. The ligands contain Arg–Gly–Asp (RGD) sequences that enable recognition of ECM proteins by as many as eight integrins, but other distinguishing features of the proteins permit the integrins to generate intracellular signals specific to the ECM molecules. Recently, integrin αvβ3 has been shown to have a panel of previously unappreciated small molecule receptor sites for thyroid hormone and hormone analogues, for dihydrotestosterone, and for resveratrol, a polyphenol that has certain estrogen-like features. These binding sites are close to the RGD recognition site of αvβ3. The thyroid hormone receptor site on the extracellular domain of αvβ3 contains two domains with discrete functions in terms of intracellular protein trafficking and gene expression. Occupancy of the receptor by a deaminated thyroid hormone analogue, tetraiodothyroacetic acid (tetrac), prevents cell responses to agonist thyroid hormones (l-thyroxine; 3, 5, 3′-triiodo-l-thyronine) and modulates expression of a number of cancer cell survival pathway genes in an up- or downregulation pattern coherent to induction of cell death. The small molecule thyroid hormone receptor on the integrin also regulates activity of five vascular growth factor receptors and/or their ligands, providing control of angiogenesis via specific pharmacologic regulation of this thyroid hormone receptor. The resveratrol receptor induces programmed cancer cell death via p53, even when the latter has undergone specific mutations. There is also evidence for the presence of several receptors on integrin αvβ3 for authentic steroids, including a dihydrotestosterone site that supports proliferation of breast cancer cells that lack nuclear androgen and estrogen receptors. The existence of these small molecule hormone receptors on an integrin with a remarkably complex functional profile defines novel pharmacologic options via individual small molecule receptor manipulation for control of cancer cell behavior. This refinement of up-down control at the level of discrete receptors is not a function of the use of αvβ3 antibody or RGD peptides that occlude regions of the integrin.
Similar content being viewed by others


Integrins are signal transducing structural proteins of the plasma membrane [1, 2]. Composed of representative α and β monomers, more than 20 integrin heterodimers have been described [1]. The integrins have generous extracellular domains and small domains that are transmembrane and intracellular; the latter are responsible for interactions with signal transducing kinase pathways and with the cytoskeleton [3]. Extracellular matrix (ECM) proteins are specific ligands of the extracellular domain of integrins, generating ligand-specific outside-in signals that are relevant to a variety of cell functions, including motility, cell attachment, cell division, and gene transcription. The large molecule ECM protein ligands of integrins include vitronectin, fibronectin, fibrinogen, and osteopontin [1]. These ligands contain an exposed Arg–Gly–Asp (RGD) tripeptide sequence. αvβ3 is an integrin important to angiogenesis and tumor cell biology [4]; it is one of eight integrins to have an RGD (sequence) recognition site that facilitates ECM protein–integrin interaction [1]. Integrin αvβ3 also specifically interacts with adjacent vascular growth factor receptors on the cell surface [5, 6] and, in some cases, with growth factors themselves that are present in ECM. The interactions (“crosstalk”) with nearby growth factor receptors permit the integrin to modulate the activity of these receptors.
Until recently, the naturally occurring ligands of αvβ3 have been viewed exclusively to be proteins. Small molecule ligands of the integrins have been manufactured that contain the RGD sequence and enable pharmacologic disruption of integrin–protein binding or of crosstalk of the integrin with adjacent vascular growth factor receptors to achieve anti-angiogenesis. In the past decade, however, we described thyroid hormone-induced angiogenesis in a variety of models that is integrin αvβ3 dependent and demonstrated its initiation at a high-affinity receptor for thyroid hormone on this integrin [7, 8]. Thyroid hormone, i.e., l-thyroxine (T4) and 3,5,3′-triiodo-l-thyronine (T3) with molecular weights of 777 and 650 Da, respectively, are thus small molecule naturally occurring ligands for this integrin and are specific for αvβ3; that is, they do not bind to other integrins. The αvβ3 receptor for iodothyronines is wholly different structurally and functionally from the well-studied intracellular, nuclear receptors for thyroid hormone (TRs) [9]. The integrin receptor for thyroid hormone is involved in angiogenesis in the nonmalignant and cancer settings, in tumor cell proliferation, in tumor cell chemosensitization [8], and in the inflammatory process [10]. Further, the receptor distinguishes among thyroid hormone analogues, enabling anti-angiogenesis, as well as pro-angiogenesis [8]. Following the description of the iodothyronine receptor on αvβ3, it has become clear that other nonpeptide hormones and the estrogen-like stilbene, resveratrol, have discrete receptor pockets on cell surface integrin αvβ3 (Fig. 1). These receptors, like those for thyroid hormone, are distinct from previously described binding sites for these hormone or hormone-like molecules within the cell.

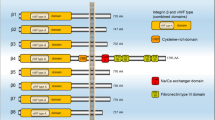
Fit of small molecule ligands into receptor pockets proximal to the RGD (Arg–Asp–Gly) recognition site of integrin αvβ3, with diagrams of downstream consequences of binding site occupancy. Upper panel, crystallographic projection of hormone (thyroid hormone, steroids) and resveratrol ligands into specific receptor sites about the RGD recognition site. This region of the “head” of heterodimeric αvβ3 is composed of contributions from αv (left, yellow) and β3 (right, violet) monomers. The ligands are T4 (red), trans-resveratrol (dark blue) and estradiol (green). RGD cyclic peptide is light blue. The orienting integrin amino acid residues identified are Asp150 (D150) and Asp218 (D218) from αv and Tyr122 (Y22) and Asn215 (N215) from β3. The projections are drawn primarily from ref. [73]. Lower panel, schematic drawing of small molecule ligand pockets of heterodimeric αvβ3, showing selected functional cellular consequences of binding of thyroid hormone (T4) [8], resveratrol [47, 48, 53], and sex steroid (estradiol or, in certain tumor cells, DHT [31]). The steroid and thyroid hormone pockets do not interact. The proximity of the resveratrol and thyroid hormone pockets allows subtle interactions. Thus, T4 blocks p53-dependent apoptosis in cancer cells, but tetraiodothyroacetic acid (tetrac) does not affect resveratrol action; tetrac does eliminate the ability of added T4 to inhibit resveratrol-dependent apoptosis (see text)
In this review we examine these plasma membrane small molecule receptor sites and the functions so far assigned to them. It will become clear that these receptor sites on the integrin are discrete, able to discern small modifications to their ligands, very complex in terms of downstream actions, and important to survival of the cancer cells or vascular cells they serve. Targeting such individual receptor sites on αvβ3—rather than a more general approach to the integrin with antibody or confounding of the RGD recognition site—may offer strategic advantages in controlling tumor cells or modulating angiogenesis.
Thyroid Hormone Receptor Site on αvβ3
Evidence that thyroid hormone has cellular or systemic effects that are independent of the classical nuclear TR molecules has existed for several decades [8]. Such effects were demonstrated in vitro in plasma membrane preparations or purified sarcoplasmic reticulum preparations. They were also shown in intact cells that lacked TR or were sufficiently rapid in onset to exclude TR-mediated effects on gene transcription. Evidence that an integrin might be involved in mediating nongenomic actions of the hormone emerged from studies of actin polymerization and integrin–laminin interactions [11, 12] and of bone resorption [13] that were affected by the inclusion of RGD peptides. Such studies were interpreted to document participation of ECM proteins in the hormone-induced actions, not to infer an initiation site for hormone effect. It was also shown in one of these studies [11] that T4 was capable of regulating the clustering of integrins on the plasma membrane of astrocytes and, we presume, of other types of cells.
In 2005, we demonstrated that the pro-angiogenic activity of physiologic levels of thyroid hormone in the chick chorioallantoic membrane (CAM) model [14] was in fact dependent upon hormone interaction with αvβ3 and the presence of a high-affinity thyroid hormone receptor on the integrin [7]. This report was the first to document that a naturally occurring small molecule—in this case, a nonpeptide hormone—utilized αvβ3 to initiate a biologic function. As noted above, αvβ3 had already been extensively implicated in regulation of angiogenesis. The thyroid hormone studies showed that the novel receptor could distinguish among T4, T3, and the deaminated derivative of T4, tetraiodothyroacetic acid (tetrac). The latter inhibited the binding and pro-angiogenic actions of T4 and T3 initiated at the integrin. Agonist thyroid hormone moieties such as T4 when covalently bound to agarose—and thus incapable of cell entry—reproduced the angiogenic effect of the hormones. This agarose-bound state of iodothyronines modeled nanoparticulate formulations of the hormone discussed below. The affinity of the receptor on αvβ3 was higher for T4 than for T3. Diiodothyropropionic acid [15] and the non-iodinated agonist thyroid hormone analogue, GC-1 [16], have also been shown to induce angiogenesis at the integrin.
X-ray crystallographic modeling of the interaction of thyroid hormone and hormone analogues with αvβ3 placed the hormone receptor at the base of the head of the integrin [17, 18] and near the RGD recognition site. The binding site groove or pocket is composed of contributions from both the αv and β3 monomers (Fig. 1), explaining failure to find binding of radiolabeled hormone to integrin monomers internalized in αvβ3-bearing cells [19]. Indeed, the integrin does not appear to be a cell uptake mechanism for thyroid hormone. The fit of T4, T3, and tetrac into the binding groove is by crystallographic modeling distinctive for each molecule [17, 18].
The receptor site for thyroid hormone on αvβ3 has been shown by us to contain two domains [20]. The S1 domain binds T3 exclusively and when occupied by its ligand activates signal transducing phosphatidylinositol 3-kinase (PI3K) via Src kinase. The S2 domain binds both T4 and T3 and activates mitogen-activated protein kinase (specifically, extracellular regulated kinase 1/2, ERK1/2). The specialization of the domains is remarkable [20]. For example, S1 occupancy by T3 leads to enhanced expression of the hypoxia-inducible factor-1α gene and results in importation by the nucleus of cytoplsmic TRα1. S2 liganding of T4 and T3 results in the complex process of cell proliferation and drives cytoplasmic TRβ1 into the nucleus. RGD peptide blocks actions of T3 at S1, but not at S2; the peptide inhibits effects of T4 at S2. Tetrac has global inhibitory activity at S1 and S2 and, as will be noted below, tetrac also has discrete actions at the receptor that are unrelated to the absence or presence of T3 and T4. All of the complex integrin-based actions of thyroid hormone and analogues such as tetrac are blocked by antibody to αvβ3.
The binding of T4 and T3 to the integrin also provided insight into nongenomic actions of thyroid action on a plasma membrane ion transporter. The stimulatory effect of T3 on the Na+/H+ exchanger (NHE1) in myoblasts [21] was subsequently shown to be inhibited by triac—the deaminated analogue of T3—and antibody to αvβ3 [22]. This thyroid hormone-supported enzyme is an important component in the intracellular alkalinization of cancer cells and acidification of the microenvironment of such cells [23, 24].
Thyroid hormone and hormone analogues such as tetrac that bind to the receptor on αvβ3 and initiate nongenomic actions also are taken up by cells. Intracellular T3 has genomic actions within the nucleus that are distinct from integrin-initiated effects; intracellular actions of T3 also increase translational activity of endoplasmic reticulum and stimulate mitochondrial metabolism [9]. T4 at the plasma membrane may also be converted by deiodinase 2 into T3—which is accessible to the cell interior—and T4, without prior deiodination, when taken up by cells contributes to stabilization of the actin cytoskeleton [12]. Finally, tetrac within the cell is a low-potency thyromimetic [25], rather than an antagonist of T4 and T3 as it is at the integrin. These observations caused us to reformulate tetrac and agonist thyroid hormones as nanoparticles that cannot gain entry to the cell. Hormone and hormone analogues are covalently bound via the outer ring hydroxyl to the surface of the nanoparticle [26]. The formulations are designed to cause the hormone or hormone analogue to protrude sufficiently from the nanoparticle to act reproducibly at the receptor on αvβ3. The nanoparticles, themselves, are biodegradable.
The ability of tetrac formulations to inhibit the pro-angiogenic properties of T4 and T3 was initially understood only in the context of inhibition of hormone binding. However, subsequent angiogenic studies of tetrac conducted in the CAM model in the absence of agonist T4 and T3 revealed that tetrac blocked the angiogenic activity of basic fibroblast growth factor (bFGF; FGF2) [27], vascular endothelial growth factor [27], platelet-derived growth factor and epidermal growth factor (SA Mousa, unpublished observations). The ability of tetrac to disrupt actions of four or more growth factors is unique to-date. It is attributed to the crosstalk that exists between integrin αvβ3 and the adjacent cell surface receptors for these factors. The anti-angiogenic actions of tetrac formulations at the thyroid hormone receptor on this integrin transcend crosstalk with vascular growth factors, however, in that tetrac can also impair local release of vascular growth factors, such as bFGF [14], and impair transcription of bFGF [14]. Tetrac formulations also impair endothelial cell migration toward a fibronectin cue in a Boyden chamber apparatus (SA Mousa, unpublished observations). These multiple anti-angiogenic properties contribute to the experimental anti-tumor effects of tetrac formulations (see below) and are of potential value in settings of excessive angiogenesis in the absence of cancer, e.g., erythematous skin diseases or other noncancerous inflammatory states [10]. These multiple actions also testify to the complex roles that integrin αvβ3 plays in processes such as angiogenesis.
In the absence of tetrac, T4 and T3 have pro-angiogenic properties initiated at αvβ3 that, in the noncancerous setting, may be clinically desirable. Such applications may include wound-healing—in which the original observations were attributed primarily to an effect on keratinocytes [28]—and rescue of ischemic tissue [29]. Generous expression of αvβ3 by rapidly dividing blood vessel cells suggested that tetrac formulations would be effective in experimental cancer models as an anti-angiogenesis agent. Tumor cells also exhibit increased expression of the integrin. Early testing of actions of tetrac formulations against a variety of human cancer cells in vitro revealed anti-proliferative effects that included induction of apoptosis by several pathways and suppression of transcription of genes in multiple cell survival pathways [8, 30]. Among the genes whose expression is affected by tetrac formulations that act at αvβ3 are cyclins, caspases, catenin pathway components, thrombospondin 1 and epidermal growth factor receptor. The range of activities testifies to the complex downstream signaling pathways that originate at integrin αvβ3. Tetrac and nanoparticulate tetrac significantly decreased tumor-associated vascularity and tumor cell viability in a variety of xenografts. The agents have been shown to be effective against xenografts of several human lung cancer cell lines [31, 32], breast cancer [26], pancreatic cancer cells [33], medullary thyroid carcinoma [34], follicular thyroid cancer [35], and renal cell carcinoma [36].
Acting at the integrin, tetrac formulations are also capable of disordering other defense mechanisms of cancer cells. Tetrac and nanotetrac radiosensitize human cancer cells [37]. The mechanism is inhibition of cellular repair of double-strand DNA breaks induced by radiation [38]. Tetrac has also been shown by us to chemosensitize human breast cancer (MCF7-Dx) cells that are resistant to doxorubicin [39]. In addition to this action on cell response to doxorubicin, tetrac can enhance sensitivity of cancer cells to etoposide and cisplatin. The molecular basis of this αvβ3-mediated action is not yet defined, but liganding of ECM osteopontin by the integrin has been shown to enhance P-glycoprotein (P-gp) (multidrug-resistance, MDR) pump activity that is essential to enhanced efflux of chemotherapeutic agents from cancer cells [40]. It is possible that tetrac interferes with this osteopontin–integrin signaling process, resulting in the extended intracellular residence time of anticancer drugs that we have described for doxorubicin. If this were indeed the case, it would be another example of the influence of the hormone on the interaction of the integrin with a principal ECM ligand. The initial example, cited above, is influence of thyroid hormone on migration of αvβ3-bearing endothelial cells toward an ECM protein cue. It should also be noted that agonist thyroid hormone, T3, can affect the activity of P-gp and expression of the gene for the pump [41].
Antibody to αvβ3 obscures the receptor site for thyroid hormone and hormone analogues. Disabling the integrin with antibody is to be distinguished from selective exploitation with tetrac formulations of a small molecule receptor site for thyroid hormone on αvβ3 from which coherent up- and downregulation of selected genes relevant to cancer cell survival may be achieved. Chemosensitization and radiosensitization are endpoints that to-date are not achieved with antibody disabling of αvβ3.
Resveratrol Receptor Site on αvβ3
Resveratrol is a naturally occurring polyphenol with anti-cancer properties that have been documented experimentally, but not clinically [42], and the agent is the focus of a large literature relating to other biologic effects [43]. Resveratrol has limited oral bioavailability and is metabolized rapidly, has multiple intracellular targets, including sirtuins [44, 45], and has been reported to have estrogenic activities [46]. Where its cellular actions are initiated has not been clear. In studying its anti-cancer activity [47–50], we described a cell surface receptor pocket for this polyphenol on integrin αvβ3 [51] that is linked to p53-dependent apoptosis in cancer cells (Fig. 1). The receptor is largely situated on the β3 monomer and the downstream pathway to p53 activation from the integrin is via ERK1/2. p53-dependent apoptosis may be induced by resveratrol in cancer cells with mutant p53 [48], as long as Ser-15 is intact. Resveratrol causes phosphorylation of p53 at serines 15, 20, and 392 [49]. Several resveratrol analogues have been shown to bind to the receptor [52]. Tetrac has no effect on apoptosis induced by resveratrol at αvβ3 [53]. T4 [53] and estrogen [49] both block activity of resveratrol-induced apoptosis at the integrin; these hormones do not inhibit ERK 1/2 that is intrinsic to resveratrol action at the integrin and the inhibitory actions of T4 and estradiol are initiated within the cell and nucleus at p53. T4 blocks resveratrol-induced, integrin-dependent Ser-15 phosphorylation of p53 that is required for apoptosis; estrogen blocks phosphorylation of p53 at multiple serines that is resveratrol-induced and suppresses acetylation [49] promoted by the polyphenol.
Against this background, it may not be surprising that the anti-cancer effects of resveratrol that were demonstrated in cultured cells have not been reproduced in the limited clinical trials that have been conducted [42]. That is, circulating levels of endogenous thyroid hormone may be sufficient in study patient populations to inhibit resveratrol-induced cancer cell apoptosis. This antagonistic effect of thyroid hormone on resveratrol action is eliminated by tetrac formulations which, as noted above, act at αvβ3 to permit pro-apoptotic activity of resveratrol to be expressed.
A second mechanism exists by which the anti-neoplastic actions of resveratrol might be suppressed clinically. Constitutive cyclooxygenase-2 (COX-2) expression in tumor cells is a cancer biomarker [54]. However, a pool of inducible intracellular COX-2 has recently been described in the cancer cell nucleus [55] and this is pro-apoptotic via a p53-dependent mechanism. Resveratrol is an inducer of this second pool of COX-2 [55] via αvβ3. Thus, the use in the clinical sector of medications such as nonsteroidal anti-inflammatory agents which reduce COX-2 production may be desirable in the setting of constitutive COX-2 production, but undesirable in blocking induction of the pro-apoptotic nuclear pool of the enzyme by resveratrol in anti-cancer trials of the polyphenol. There are of course other possible explanations for the unfavorable initial clinical trial experience with resveratrol, including drug pharmacokinetics and pharmacodynamics that are different in human subjects from the animal models in which efficacy was established.
Monoclonal antibody to αvβ3 blocks the action of resveratrol at its integrin receptor, as does RGD peptide [51]. Hence, the use of anti-αvβ3 or of designed cyclic RGD peptide derivatives as anti-cancer drugs may not permit utilization of this small molecule receptor on the integrin.
Sex Steroid Receptor Site(s) on αvβ3; Glucocorticoid Action on αvβ3
In studies of human breast cancer (MDA-MB-231) cells, we found that a non-aromatizable androgen, dihydrotestosterone (DHT), stimulated cell proliferation in vitro [56]. Anti-αvβ3 and RGD peptide blocked the DHT effect, inferring the existence on the integrin of a receptor for the androgen that was proximal to the RGD recognition site. This cell line lacks estrogen receptor-α (ERα) gene. In separate experiments involving ERα-expressing breast cancer (MCF-7) cells, DHT also induced cell proliferation, but αvβ3 antibody did not affect the action of DHT and the action of the latter was found to require the presence of ER. That is, DHT action was inhibited by the ER antagonist, ICI 182,780 (fulvestrant, Faslodex®) and by siRNA knockdown of ERα [56]. The possibility that nuclear androgen receptor (AR) mediated the actions of DHT in either MDA-MB-231 or MCF-7 cells was excluded in these studies. We can conclude that in MDA-MB-231 breast cancer cells, the extracellular domain of integrin αvβ3 exposes a receptor site that is capable of binding the androgen, DHT, but that DHT may promote breast cancer cell division by more than one mechanism and without participation of the classical AR.
X-ray crystallographic modeling of the “head” of αvβ3 has also revealed a pocket that is capable of binding estrogen [18]. However, binding studies have not been conducted to verify the existence of a functional estrogen receptor on αvβ3 and an extensive literature exists on the binding of estradiol by the plasma membrane [57] that has been attributed to insertion of functional ERα into the plasma membrane [57, 58].
While extranuclear effects of nuclear steroid receptors have been described [59, 60], it should be noted that progesterone [61] and dexamethasone [62] also have been reported to modulate the osteopontin–αvβ3 interaction. Such observations may indicate existence of binding of these steroids by the integrin, but control of ECM protein-αvβ3 interactions may exist on an “inside-out” basis by which a signal from an intracellular steroid may be transduced into a conformational change in the extracellular domain of the integrin. As noted earlier in the thyroid hormone section, the osteopontin–αvβ3 interaction is a component of the control of function of the plasma membrane MDR pump, P-gp.
Summary
That an important integrin such as αvβ3—a principal component of cell-ECM matrix protein interactions—may participate in a variety of plasma-membrane-initiated effects of nonpeptide hormones has only recently been appreciated. It is clear that via a specific and complex hormone receptor site, αvβ3 mediates a number of nongenomic effects of thyroid hormone that are particularly relevant to cancer cells and to angiogenesis, whether or not the latter is cancer relevant. Remarkably, a thyroid hormone derivative, tetrac and its formulations, not only blocks the agonist actions of T4 and T3 at the cell membrane, but has an important set of effects on cancer cells that do not require the presence of agonist thyroid hormone. These effects include disordering of multiple gene expression-based cancer cell survival pathways, inhibition of the spontaneous repair by cancer cells of double-strand DNA breaks that occur naturally or clinically are radiation-induced, and the crosstalk between the integrin and nearby vascular growth factor receptors that are critical to tumor-related angiogenesis.
Receptor sites for steroids have been described on αvβ3, but with substantially less detail than is available for thyroid hormone. Estrogen-resembling resveratrol has a receptor site on αvβ3 that is one explanation for the p53-dependent pro-apoptotic activity of this polyphenol demonstrated in cancer cells. Much attention has previously been focused on actions of resveratrol that require or are reduced in extent by cellular uptake and metabolism of the agent. The existence of the integrin αvβ3 receptor site provides a basis for understanding a cancer-related, rapid action of resveratrol that is independent of cellular uptake of the agent. Crystallographic modeling evidence mentioned here suggests the existence of a receptor site for traditional estrogens on the integrin, but no search for functionality of this putative site has been conducted and, as noted above, ERα may be inserted in the plasma membrane to explain nongenomic effects of estrogen. On the other hand, the existence of a receptor for DHT on αvβ3 has been substantiated in breast cancer cells. This receptor permits stimulation of proliferation by DHT of certain cancer cells in the absence of AR and ER. When ER is present in breast cancer cells, DHT action on proliferation may be mediated by this receptor. Progesterone and dexamethasone are other steroids whose actions on the osteopontin–αvβ3 interaction warrant studies of the possible existence of specific steroid-αvβ3 interactions.
The integrin receptor pockets for nonpeptide hormones or hormone-like molecules are in the “head” of αvβ3 and near the RGD recognition site (Fig. 1). A number of these small molecule effects of thyroid hormone analogues—e.g., tetrac formulations—and of resveratrol that have been described experimentally may have clinical implications. Specifically inactivating the RGD site with peptide analogues or general blockade of the integrin with anti-αvβ3 may eliminate or curtail the substantial panel of cancer and/or angiogenesis management options at the integrin that small molecule receptor sites offer. These include pro-apoptotic and “anti-anti-apoptotic” effects, p53-dependent apoptosis in the presence of mutations of p53, disorganization of the cell cycle, inhibition of activities of as many as four vascular growth factors, and radiosensitization. Certainly, anti-cancer or anti-angiogenesis strategies that are focused on RGD peptide and αvβ3 antibody require surveys of effects on molecular mechanisms of actions of small molecules that receptors for these molecules on αvβ3 offer.
A critical functional issue in studies of small molecule ligands of αvβ3 is the types of cells that express the integrin. Rapidly dividing blood vessel (endothelial) cells [5], tumor cells [4] and osteoclasts [63, 64] generously express the integrin. We have studied a variety of human tumor cell lines and have found all to express sufficient αvβ3 to be tetrac- and/or T4-responsive [8, 31, 34, 36]. Human dermal microvascular endothelial cells in microtubular assays [27, 65] and the rapidly dividing blood vessel cells of the chick chorioallantoic membrane (CAM) [14, 16] express the integrin. Quiescent epithelial cells express little or no αvβ3 [4]. Dividing osteoblasts may also contain accessible αvβ3 [66]. Smooth muscle cells [67], skeletal muscle myoblasts [22] and platelets express low-but-functional levels of the integrin [68] and platelet function—agglutination—requires αvβ3 [69]. Activated macrophages contain the functional integrin [70] and this observation, coupled with angiogenesis, may explain the presence of the integrin in the area of cardiac repair after experimental myocardial infarction [71]. Imaging agents that target αvβ3 also indicate that normal colon, brain, salivary glands, and thyroid gland contain very low-but-detectable levels of the integrin [72], the significance of which is not yet known. Thus, rapidly dividing cells—tumor cells or certain nonmalignant cells, particularly in blood vessels—express substantial amounts of the integrin, whereas quiescent cells do not. These observations rationalize the interest in αvβ3 as a chemotherapeutic target and tumor imaging target.
References
Plow EF, Haas TA, Zhang L, Loftus J, Smith JW (2000) Ligand binding to integrins. J Biol Chem 275:21785–21788
Campbell ID, Humphries MJ (2011) Integrin structure, activation and interactions. Cold Spring Harb Perspect Biol 3:a004994
Huttenlocher A, Horwitz AR (2011) Integrins in cell migration. Cold Spring Harb Perspect Biol 3(9):a005074
Desgrosellier JS, Cheresh DA (2010) Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 10:9–22
Somananth PR, Ciocea A, Byzova TV (2009) Integrin and growth factor receptor alliance in angiogenesis. Cell Biochem Biophys 53:53–64
Eliceiri BP (2001) Integrin and growth factor receptor crosstalk. Circ Res 89:1104–1110
Bergh JJ, Lin HY, Lansing L, Mohamed SN, Davis FB, Mousa S, Davis PJ (2005) Integrin αvβ3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinology 146:2864–2871
Davis PJ, Davis FB, Mousa SA, Luidens MK, Lin HY (2011) Membrane receptor for thyroid hormone: physiologic and pharmacologic implications. Annu Rev Pharmacol Toxicol 51:99–115
Cheng SY, Leonard JL, Davis PJ (2010) Molecular aspects of thyroid hormone actions. Endocr Rev 31:139–170
Davis PJ, Glinsky GV, Lin HY, Incerpi S, Davis FB, Mousa SA, Tang HY, Hercbergs A, Luidens MK (2013) Molecular mechanisms of actions of formulations of the thyroid hormone analogue, tetrac, on the inflammatory response. Endocr Res 38:112–118
Farwell AP, Tranter MP, Leonard JL (1995) Thyroxine-dependent regulation of integrin-laminin interactions in astrocytes. Endocrinology 136:3909–3915
Leonard JL, Farwell AP (1997) Thyroid hormone-regulated actin polymerization in brain. Thyroid 7:147–151
Hoffman SJ, Vasko-Moser J, Miller WH, Lark MW, Gowen M, Stroup G (2002) Rapid inhibition of thyroxine-induced bone resorption in the rat by an orally active vitronectin receptor antagonist. J Pharmacol Exp Ther 302:205–211
Davis FB, Mousa SA, O'Connor L, Mohamed S, Lin HY, Cao HJ, Davis PJ (2004) Proangiogeneic action of thyroid hormone is fibroblast growth factor-dependent and is initiated at the cell surface. Circ Res 94:1500–1506
Mousa SA, O'Connor L, Davis FB, Davis PJ (2006) Proangiogenesis action of the thyroid hormone analog 3, 5-diiodothyropropionic acid (DITPA) is initiated at the cell surface and is integrin-mediated. Endocrinology 1476:1602–1607
Mousa SA, O'Connor LJ, Bergh JJ, Davis FB, Scanlan TS, Davis PJ (2005) The proangiogenic action of thyroid hormone analogue GC-1 is initiated at an integrin. J Cardiovasc Pharmacol 46:356–360
Cody V, Davis PJ, Davis FB (2007) Molecular modeling of the thyroid hormone interactions with αvβ3 integrin. Steroids 72:166–170
Lin HY, Cody V, Davis FB, Hercbergs AA, Luidens MK, Mousa SA, Davis PJ (2011) Identification and functions of the plasma membrane receptor for thyroid hormone analogues. Discov Med 11:337–347
Lin HY, Su Y-F, Hsieh M-T, Lin S, Meng R, London D, Lin C, Tang H-Y, Hwang J, Davis FB, Mousa SA, Davis PJ (2013) Nuclear monomeric integrin αv in cancer cells is a co-activator regulated by thyroid hormone. FASEB J 27:3209–3216
Lin HY, Sun M, Tang HY, Lin C, Luidens MK, Mousa SA, Incerpi S, Drusano GL, Davis FB, Davis PJ (2009) l-Thyroxine vs. 3, 5, 3′-triiodo-l-thyronine and cell proliferation: activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Am J Physiol Cell Physiol 296:C980–C991
Incerpi S, Luly P, De Vito P, Farias RN (1999) Short-term effects of thyroid hormone on the Na/H antiport in l-6 myoblasts: high molecular specificity for 3, 5, 3′-triiodo-l-thyronine. Endocrinology 140:683–689
D'Arezzo S, Incerpi S, Davis FB, Acconcia F, Marino M, Farias RN, Davis PJ (2004) Rapid nongenomic effects of 3, 5, 3′-triiodo-l-thyronine on the intracellular pH of l-6 myoblasts are mediated by intracellular calcium mobilization and kinase pathways
Amith SR, Fliegel L (2013) Regulation of the Na+/H+ exchanger (NHE1) in breast cancer metastasis. Cancer Res 73:1259–1264
Stock C, Ludwig FT, Schwab A (2012) Is the multifunctional Na(+)/H(+) exchanger isoform 1 a potential therapeutic target in cancer? Curr Med Chem 19:647–660
Moreno M, de Lange P, Lombardi A, Silvestri E, Lanni A, Goglia F (2008) Metabolic effects of thyroid hormone derivatives. Thyroid 18:239–253
Bharali DJ, Yalcin M, Davis PJ, Mousa SA (2013) Tetraiodothyroacetic acid-conjugated PLGA nanoparticles: a nanomedicine approach to treat drug-resistant breast cancer. Nanomedicine (in press)
Mousa SA, Bergh JJ, Dier E, Rebbaa A, O'Connor LJ, Yalcin M, Aljada A, Dyskin E, Davis FB, Lin HY, Davis PJ (2008) Tetraiodothyroacetic acid, a small molecule integrin ligand, blocks angiogenesis induced by vascular endothelial growth factor and basic fibroblast growth factor. Angiogenesis 11:183–190
Safer JD, Crawford TM, Holick MF (2004) Topical thyroid hormone accelerates wound healing in mice. Endocrinology 145:4425–4430
El-Eter E, Rabee H, Alkayali A, Mousa SA (2007) Role of thyroid hormone analogs in angiogenesis and the development of collaterals in rabbit hind limb ischemia model. J Thromb Thrombolysis 5(suppl 1):375
Glinskii AB, Glinsky GV, Lin HY, Tang HY, Sun M, Davis FB, Luidens MK, Mousa SA, Hercbergs AH, Davis PJ (2009) Modification of survival pathway gene expression in human breast cancer cells by tetraiodothyroacetic acid (tetrac). Cell Cycle 8:3554–3562
Meng R, Tang HY, Westfall J, London D, Cao JH, Mousa SA, Luidens M, Hercbergs A, Davis FB, Davis PJ, Lin HY (2011) Crosstalk between integrin αvβ3 and estrogen receptor-α is involved in thyroid hormone-induced proliferation in human lung carcinoma cells. PLoS One 6(11):e27547
Mousa SA, Yalcin M, Bharali DJ, Meng R, Tang HY, Lin HY, Davis FB, Davis PJ (2012) Tetraiodothyroacetic acid and its nanoformulation inhibit thyroid hormone stimulation of non-small cell lung cancer cells in vitro and in xenografts. Lung Cancer 76:39–45
Yalcin M, Lin HY, Sudha T, Bharali DJ, Meng R, Tang HY, Davis FB, Stain SC, Davis PJ, Mousa SA (2013) Response of human pancreatic cancer cell xenografts to tetraiodothyroacctic acid nanoparticles. Horm Cancer 4:176–185
Yalcin M, Dyskin E, Lansing L, Bharali DJ, Mousa SS, Bridoux A, Hercbergs AH, Lin HY, Davis FB, Glinsky GV, Glinskii A, Ma J, Davis PJ, Mousa SA (2010) Tetraiodothyroacetic acid (tetrac) and nanoparticulate tetrac arrest growth of medullary carcinoma of the thyroid. J Clin Endocrinol Metab 95:1972–1980
Yalcin M, Bharali DJ, Dyskin E, Dier E, Lansing L, Mousa SS, Davis FB, Davis PJ, Mousa SA (2010) Tetraiodothyroacetic acid and tetraiodothyroacetic acid nanoparticle effectively inhibit the growth of human follicular thyroid cell carcinoma. Thyroid 20:281–286
Yalcin M, Bharali DJ, Lansing L, Dyskin E, Mousa SS, Hercbergs A, Davis FB, Davis PJ, Mousa SA (2009) Tetraiodothyroacetic acid (tetrac) and tetrac nanoparticles inhibit growth of human renal cell carcinoma xenografts. Anticancer Res 29:3825–3831
Hercbergs A, Davis PJ, Davis FB, Ciesielski MJ, Leith JT (2009) Radiosensitization of GL261 cells by tetraiodothyroacetic acid (tetrac). Cell Cycle 8:2586–2591
Hercbergs A, Lin HY, Davis FB, Davis PJ, Leith JT (2011) Radiosensitization and production of DNA double-strand breaks in U87MG brain tumor cells induced by tetraiodothyroacetic acid (tetrac). Cell Cycle 10:352–357
Rebbaa A, Chu F, Davis FB, Davis PJ, Mousa SA (2008) Novel function of the thyroid hormone analog tetraiodothyroacetic acid: a cancer chemosensitizing and anti-cancer agent. Angiogenesis 11:269–276
Hsieh IS, Huang WH, Liou HC, Chuang WJ, Yan RS, Fu WM (2013) Upregulation of drug transporter expression by osteopontin in prostate cancer cells. Mol Pharmacol 83:968–977
Nishio N, Katsura T, Inui K (2008) Thyroid hormone regulates the expression and function of P-glycoprotein in Caco-2 cells. Pharm Res 25:1037–1042
Tome-Carneiro J, Larrosa M, Gonzalez-Sarrias A, Tomas-Barberian FA, Garcia-Conesa MT, Espin JC (2013) Resveratrol and clinical trials: the crossroad from in vitro studies to human evidence. Curr Pharm Des 72:69–82
Baur JA, Sinclair DA (2006) Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov 5:493–506
Morris BJ (2013) Seven sirtuins for seven deadly diseases of aging. Free Radic Biol Med 56:133–171
Villalba JM, de Cabo R, Alcain FJ (2012) A patent review of sirtuin activators: an update. Expert Opin Ther Pat 22:355–367
Gehm GD, McAndrews JM, Chien PY, Jameson JL (1997) Resveratrol, a polyphenol compound found in grapes and wine, is an agonist for the estrogen receptor. Proc Natl Acad Sci U S A 94:14138–14143
Shih A, Davis FB, Lin HY, Davis PJ (2002) Resveratrol induces apoptosis in thyroid cancer cell lines via a MAPK- and p53-dependent mechanism. J Clin Endocrinol Metab 87:1223–1232
Lin HY, Shih A, Davis FB, Tang HY, Martino LJ, Bennett JA, Davis PJ (2002) Resveratrol induced serine phosphorylation of p53 causes apoptosis in a mutant p53 prostate cancer cell line. J Urol 168:748–755
Zhang S, Cao HJ, Davis FB, Tang HY, Davis PJ, Lin HY (2004) Oestrogen inhibits resveratrol-induced post-translational modification of p53 and apoptosis in breast cancer cells. Br J Cancer 91:l178–l185
Shih A, Zhang S, Cao HJ, Boswell S, Wu YH, Tang HY, Lennartz MR, Davis FB, Davis PJ, Lin HY (2004) Inhibitory effect of epidermal growth factor on resveratrol-induced apoptosis in prostate cancer cells is mediated by protein kinase C-alpha. Mol Cancer Ther 3:1355–1364
Lin HY, Lansing L, Merillon JM, Davis FB, Tang HY, Shih A, Vitrac X, Krisa S, Keating T, Cao HJ, Bergh J, Quackenbush S, Davis PJ (2006) Integrin alphavbeta3 contains a receptor site for resveratrol. FASEB J 20:1742–1744
Hsieh TC, Wong C, John Bennett D, Wu JM (2011) Regulation of p53 and cell proliferation by resveratrol and its derivatives in breast cancer cells: an in silico and biochemical approach targeting integrin αvβ3. Int J Cancer 129:2732–2743
Lin HY, Tang HY, Keating T, Wu YH, Shih A, Hammond D, Sun M, Hercbergs A, Davis FB, Davis PJ (2008) Resveratrol is pro-apoptotic and thyroid hormone is anti-apoptotic in glioma cells: both actions are integrin and ERK mediated. Carcinogenesis 29:62–69
Harris RE (2009) Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate and lung. Inflammopharmacology 17:55–67
Tang HY, Shih A, Cao HJ, Davis FB, Davis PJ, Lin HY (2006) Resveratrol-induced cyclooxygenase-2 facilitates p53-dependent apoptosis in human breast cancer cells. Mol Cancer Ther 5:2034–2042
Lin HY, Sun M, Lin C, Tang HY, London D, Shih A, Davis FB, Davis PJ (2009) Androgen-induced human breast cancer cell proliferation is mediated by discrete mechanisms in estrogen receptor-alpha-positive and –negative breast cancer cells. J Steroid Biochem Mol Biol 113:182–188
Levin ER (2009) Plasma membrane estrogen receptors. Trends Endocrinol Metab 20:477–482
Kelly MJ, Ronnekleiv OK (2012) Membrane-initiated actions of estradiol that regulate reproduction, energy balance and body temperature. Front Neuroendocrinol 33:376–387
Hammes SR, Levin ER (2011) Minireview: recent advances in extranuclear steroid receptor actions. Endocrinology 152:4489–4495
Dressing GE, Goldberg JE, Charles NJ, Schwertfeger KL, Lange CA (2011) Membrane progesterone receptor expression in mammalian tissues: a review of regulation and physiological implications. Steroids 76:11–17
Lessey BA (2003) Two pathways of progesterone action in the human endometrium: implications for implantation and contraception. Steroids 68:809–815
Filla MS, Schwinn MK, Nosie AK, Clark RW, Peters DM (2011) Dexamethasone-associated cross-linked actin network formation in human trabecular meshwork cells involves β3 integrin signaling. Invest Ophthalmol Vis Sci 52:2952–2959
Zheleznyak A, Wadas TJ, Sherman CD, Wilson JM, Kostenuik PJ, Weilbaecher KN, Anderson CJ (2012) Integrin α(v)β3 as a PET imaging biomarker for osteoclast number in mouse models of negative and positive osteoclast regulation. Mol Imaging Biol 14:500–508
Nakamura I, le Duong T, Rodan SB, Rodan GA (2007) Involvement of alpha(v)beta3 integrins in osteoclast function. J Bone Miner Metab 25:337–344
Mousa SA, Davis FB, Mohamed S, Davis PJ, Feng X (2006) Pro-angiogenesis action of thyroid hormone and analogs in a three-dimensional in vitro microvascular endothelial sprouting model. Int Angiol 25:407–413
Mas-Moruno C, Dorfner PM, Manzenrieder F, Neubauer S, Reuning U, Burgkart R, Kessler H (2013) Behavior of primary human osteoblasts on trimmed and sandblasted Ti6A14V surfaces functionalized with integrin αvβ3-selective cyclic RGD peptides. J Biomed Mater Res A 101:87–97
Mao X, Said R, Louis H, Max JP, Bourhim M, Challande P, Wahl D, Li Z, Regnault V, Lacolley P (2012) Cyclic stretch-induced thrombin generation by rat vascular smooth muscle cells is mediated by the integrin αvβ3 pathway. Cardiovasc Res 96(3):513–523
Kasirer-Friede A, Kahn ML, Shattil SJ (2007) Platelet integrins and immunoreceptors. Immunol Rev 218:247264
Mousa SS, Davis FB, Davis PJ, Mousa SA (2010) Human platelet aggregation and degranulation is induced in vitro by l-thyroxine, but not by 3,5,3′-triiodo-l-thyronine or diiodothyropropionic acid (DITPA). Clin Appl Thromb Hemost 16:288–293
Wilder RL (2002) Integrin alpha V beta 3 as a target for treatment of rheumatoid arthritis and related rheumatic diseases. Ann Rheum Dis 61(Suppl 2):ii96–ii99
Laitinen I, Notni J, Phle K, Rudelius M, Farrell E, Nekolla SG, Henriksen G, Neubauer S, Kessler H, Wesler HJ, Schwaiger M (2013) Comparison of cyclic RGD peptides for αvβ3 integrin detection in a rat model of myocardial infarction. EJNMMI Res 3(1):38
Mittra ES, Goris ML, Iagaru AH, Kardan A, Burton L, Berganos R, Chang E, Liu S, Shen B, Chin FT, Chen X, Gambhir SS (2011) Pilot pharmacokinetic and dosimetric studies of (18)F-FPPRGD2: a PET radiopharmaceutical agent for imaging α(v)β(3) integrin levels. Radiology 260:182–191
Freindorf M, Furlani TR, Kong J, Cody V, Davis FB, Davis PJ (2012) Combined QM/MM study of thyroid and steroid hormone analogue interactions with αvβ3 integrin. J Biomed Biotechnol 2012:959057
Acknowledgments
The authors appreciate the support of Candace K. Weir, Margaret D. Rudy, and the late M. Frank Rudy for some of the studies reported in this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Davis, P.J., Mousa, S.A., Cody, V. et al. Small Molecule Hormone or Hormone-Like Ligands of Integrin αVβ3: Implications for Cancer Cell Behavior. HORM CANC 4, 335–342 (2013). https://doi.org/10.1007/s12672-013-0156-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-013-0156-8